
A Novel Machine Learning 13-Gene Signature: Improving Risk Analysis and Survival Prediction for Clear Cell Renal Cell Carcinoma Patients

 ,
, 
 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Literature Search Using PubMed
2.2. Data
2.3. Data Pre-Processing
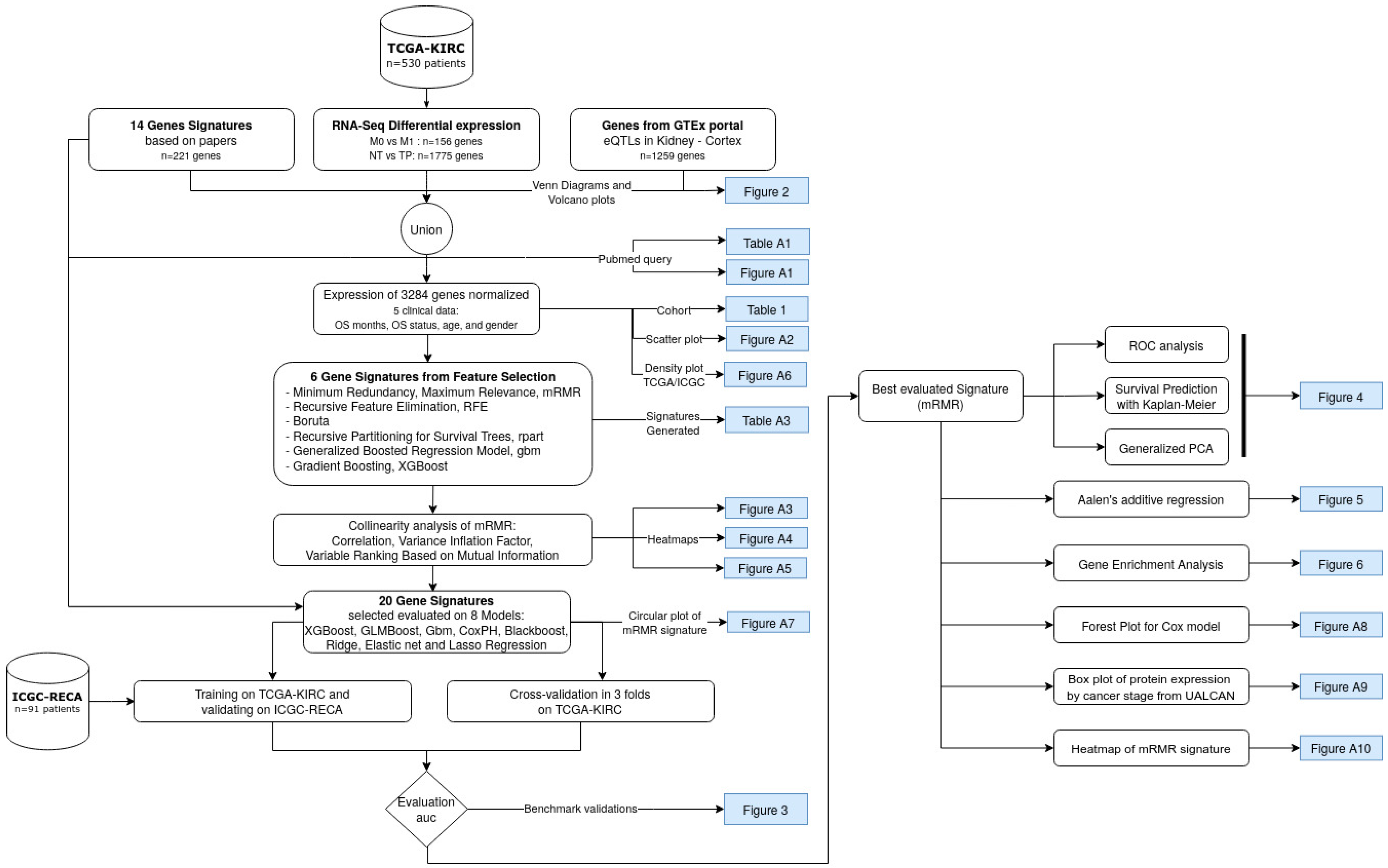
2.4. Feature Selection with Bioinformatics Analyses and Machine Learning
- Filtering methods of feature importance: Extreme Gradient Boosting (XGBoost), Generalized Boosted Regression Model (GBM), and Recursive Partitioning for Survival Trees (Rpart).
- Wrapper methods: Minimum Redundancy Maximum Relevance (mRMR); Recursive Feature Elimination (RFE); and Boruta.
2.5. Model Evaluation and Statistical Analysis
3. Results
3.1. Clinical Characteristics of the ccRCC Cohorts
3.2. mMRM Gene Selection
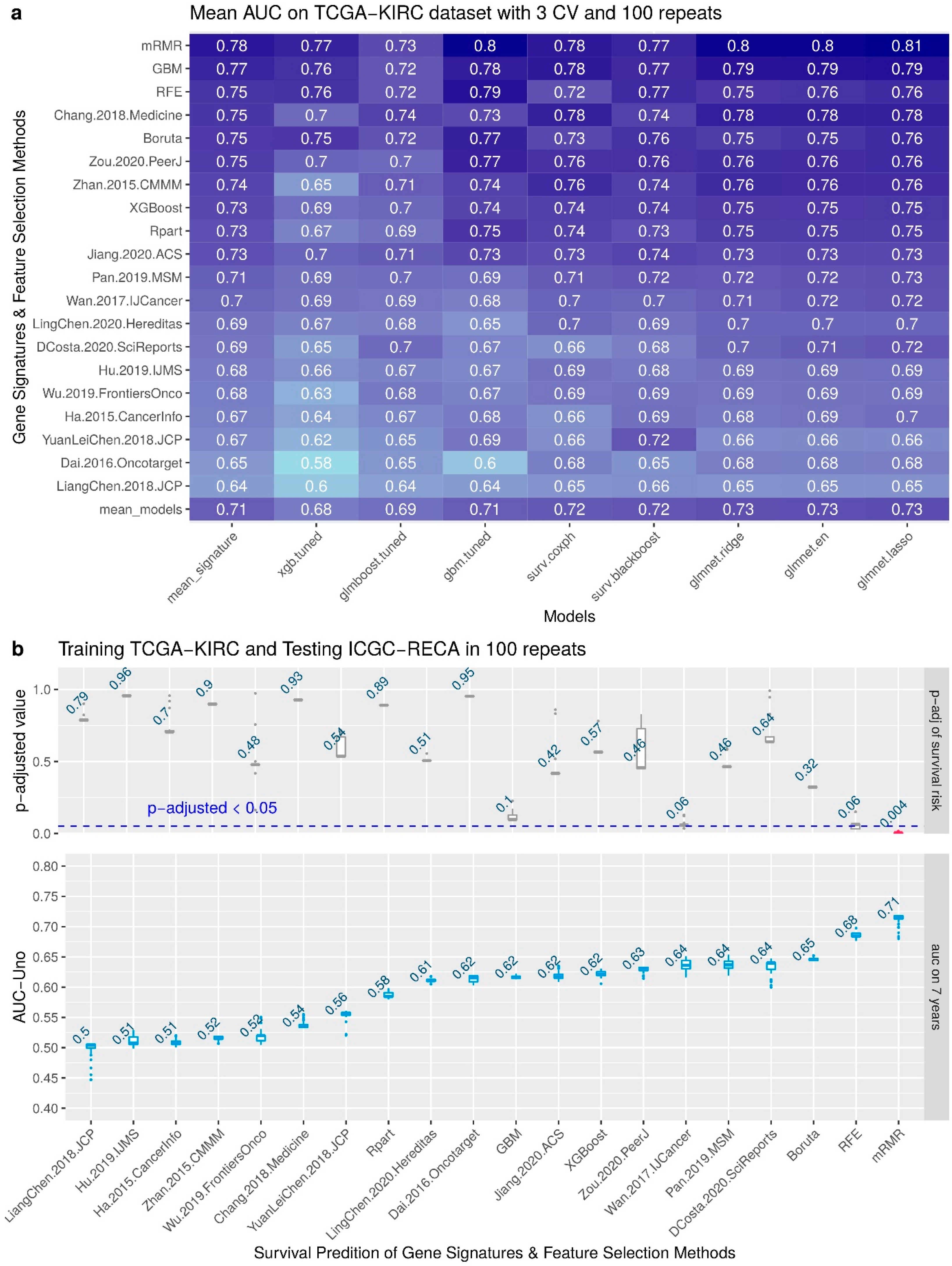
3.3. Performance of the Feature Selection Models for Internal and External Validations
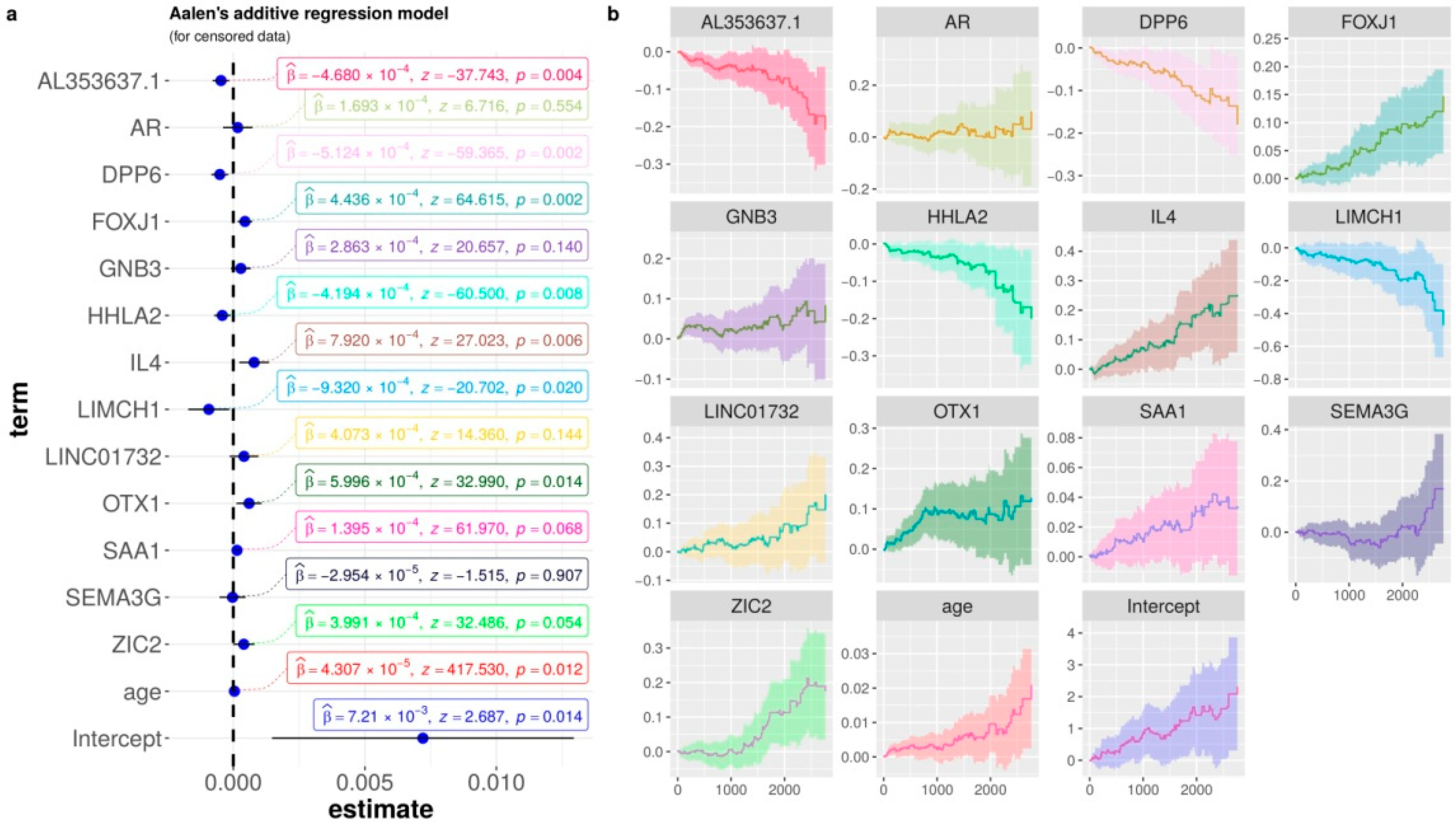
3.4. Biological Interpretation: Gene Contributions for Survival Risk and Enrichment Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AJCC | American Joint Committee on Cancer |
| TCGA | The Cancer Genome Atlas |
| ICGC | International Cancer Genome Consortium |
| KIRC | Kidney Renal Clear Cell Carcinoma |
| RECA | Renal Cell Cancer |
| ccRCC | clear cell Renal cell carcinoma |
| mRMR | Minimum Redundancy Maximum Relevance |
| AUC | Area Under the Curve |
| ROC | Receiving Operator Characteristics |
| PCA | Principal Component Analysis |
| RFE | Recursive Feature Elimination |
| GBM | Generalized Boosted Regression Model |
| Rpart | Recursive Partitioning and Regression Trees |
| XGBoost | eXtreme Gradient Boosting |
| CoxPH | Cox proportional hazards regression model |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title and Code in Figure 3 | Gene Signature |
|---|---|
| Prognostic gene signature identification using causal structure learning: applications in kidney cancer [122] Code: Ha.2014.CaInfo | ETV5, CREB3L1, GMPS, RBM15, SEPT6, TTL, ARID1A, ERCC5, TFG, FLT3, SLC34A2, FAM46C, PER1, DDB2, NACA, MLLT10, HMGA1, TCF12, RUNX1, CANT1, REL, ZNF331, JAZF1, ASPSCR1, PLAG1, NOTCH1, TAL2, ERCC2, SMARCA4, DNMT3A, HOXA11, GNAS, CHEK2, HLF, GNAQ, ETV6, SET, KIF5B, TRRAP, CDKN2C, VHL, RPL22, CHN1, STAT3, CDK4, CD274, KTN1, CYLD, BRD3, TRIM33 |
| A Five-Gene Signature Predicts Prognosis in Patients with Kidney Renal Clear Cell Carcinoma [8] Code: Zhan.2015.CMMM | CKAP4, ISPD, MAN2A2, OTOF, SLC40A1 |
| A four-gene signature predicts survival in clear-cell renal-cell carcinoma [120] Code: Dai.2016.Oncotarget | PTEN, PIK3C2A, ITPA, BCL3 |
| Identification and validation of an eight-gene expression signature for predicting high Fuhrman grade renal cell carcinoma [17] Code: Wan.2017.IJCancer | ATOH8, ATP1A3, C10orf4, C17orf79, CHMP4C, CNGA1, EDA, FBXL3, GMDS, ISL2, KISS1, KLF2, MYADML2, NCRNA00116, OAZ1, ODZ3, PLA2G15, PPP1R1A, RAB40A, RRAS, SPOCK1, SQSTM1, TXNDC16, VAMP3 |
| Comprehensive assessment gene signatures for clear cell renal cell carcinoma prognosis [9] Code: Chang.2018.Medicine | INTS8, GTPBP2, ANK3, SLC16A12, LIMCH1, Hsa-mir-374a |
| A five-gene signature may predict sunitinib sensitivity and serve as prognostic biomarkers for renal cell carcinoma [123] Code: YuanLeiChen.2018.JCP | BIRC5, CD44, MUC1, TF, CCL5 |
| A Gene Signature of Survival Prediction for Kidney Renal Cell Carcinoma by Multi-Omic Data Analysis [18] Code: Hu.2019.IJMS | BID, CCNF, DLX4, FAM72D, PYCR1, RUNX1, TRIP13 |
| Prognostic value of a gene signature in clear cell renal cell carcinoma [10] Code: LiangChen.2018.JCP | CENPW, FOXM1, NUF2 |
| Identification of a 5-Gene Signature Predicting Progression and Prognosis of Clear Cell Renal Cell Carcinoma [12] Code: Pan.2019.MSM | OTX1, FOXE1, FAM83A, HMGA2, KRT6A, DPYSL5, ANXA8, MATN4, ROS1, CSMD3, MAGEC3, AMER2, CPLX2, PI3, KRT13, ERVV-2, ERVFRDE1, ANKFN1, VTN, NFE4, ZNF114 |
| Construction and Validation of a 9-Gene Signature for Predicting Prognosis in Stage III Clear Cell Renal Cell Carcinoma [13] Code: Wu.2019.FrontiersOnco | ATP6V1C2, PCSK1N, PREX1, ANK3, HLA-DRA, SELENBP1, TYRP1, GABRA2, SERPINA5 |
| Construction and validation of a seven-gene signature for predicting overall survival in patients with kidney renal clear cell carcinoma via an integrated bioinformatics analysis [11] Code: Jiang.2020.ACS | PODXL, SLC16A12, ZIC2, ATP2B3, KRT75, C20orf141, CHGA |
| A 14 immune-related gene signature predicts clinical outcomes of kidney renal clear cell carcinoma [19] Code: Zou.2020.PeerJ | TXLNA, SEMA3G, AR, BID, IL20RB, CCR10, BMP8A, SEMA3A, CCL7, GDF1, KLRC2, LHB, FGF17, IL4 |
| A seven-gene signature model predicts overall survival in kidney renal clear cell carcinoma [2] Code: LingChen.2020.Hereditas | APOLD1, C9orf66, G6PC, PPP1R1A, CNN1G, TIMP1, TUBB2B |
| Identification of gene signature for treatment response to guide precision oncology in clear-cell renal cell carcinoma [121] Code: DCosta.2020.SciReports | ANGPT4, EDN1, VEGFA, ESM1, FLT1, KDR, CD34, PECAM1, NOTCH1, EDNRB, STIM2, FYN, VWF, GJA1, MCF2L, PPM1F, PTPRB, HEY1, ETS1, EXOC3L2, TBXA2R, TCF4, S1PR1, SLC9A3R2, NES, NFATC1, NOS3, PDE2A, CORO1A, CCR5, CXCR3, PTK2B, WAS, CD72, IL16, FYB1, FASLG, FERMT3, FOXP3, XCL2, CD3E, CD7, LAX1, CD38, LCP1, LCP2, ITK, LAT, LCK, GRK2, CCL4, CCL5, CD2, PRF1, TIGIT, GZMA, GZMB, CD8A, CTLA4, EOMES, PDCD1, PYHIN1, SLA2, LTA, PSMB8, PSMB9 |
| Code | Method | N. Genes | Gene Signature |
|---|---|---|---|
| GBM | Filtering with Generalized Boosted Regression Models for Cox Proportional Hazard | 30 | AC084117.1, CRHBP, LINC00973, ITPKA, IGFN1, C14orf37, OTX1, LINC02446, HOTTIP, NEIL3, ZIC5, CCDC154, IL4, AC008663.1, FER1L4, DUSP5P1, AL078604.2, KRT6A, SPATC1L, RTL1, LINC01597, CRABP1, RASGRP3, C3orf85, AL034399.1, TRIM4, LINC00475, ADAMTS14, DPP6 |
| Rpart | Filtering with Recursive partitioning for survival trees | 30 | TROAP, KIF18B, AURKB, LINC00973, AC003092.1, G6PC, ZNF181, MYBL2, FOXM1, NUF2, POU4F1, APOM, AR, NPHS1, AC018638.2, MERTK, AC098679.1, AL353637.1, IYD, C17orf80, SLC12A3, CDCA2, LINC02362, SRD5A3, EIF3F, AC138393.1, MCC, WFIKKN1, ALDOB, APOL5 |
| XGBoost | Filtering with XGBoost for Survival Analysis | 30 | LINC00973, LINC01271, CHAT, SPIC, AL355796.1, DLK1, ZIC5, LINC01700, ENTPD6, ATOH8, C14orf37, WNT7B, THEG, AC084117.1, ADA2, DCSTAMP, AL450311.2, A3GALT2, CNTNAP3B, TBC1D27, BIRC7, LINC00943, LINC01529, OR4C6, FAM47E, BCL3, AC105118.1, AL359736.1, SLC44A3, LINP1 |
| Boruta | Wrapper Boruta with XGBoost for Survival Data | 43 | Age, ZIC2, CHAT, AMH, OTX1, BARX1, TROAP, CKAP4, ITPKA, NUF2, KRT75, KIF18B, SLC18A3, AL355796.1, RPL10P19, LINC02154, LINC00973, IL4, HOTAIRM1, Z84485.1, LINC02362, CASP9, CCNF, RTL1, BID, CHGA, RANBP3L, ZIC5, SLC16A12, SPATC1L, CD44, KRI1, RUFY4, AC073324.1, AC091812.1, AC156455.1, AGAP6, AC128685.1, SEMA3G, IGFN1, KLRC2, ANXA8, AURKB |
| RFE | Wrapper with Recursive Feature Elimination | 89 | A3GALT2, AC006450.2, AC073324.1, AC093520.1, AC103925.1, AC120498.6, AC128685.1, AC156455.1, ADAMTS14, AL355796.1, AL592494.1, AL606519.1, AMH, ANK3, ANXA8, AP000697.1, AP001029.1, AURKB, BARX1, BIRC5, C20orf141, CCNF, CDC42P2, CENPW, CHAT, CHGA, CKAP4, CRHBP, DLX4, DMRT3, DUSP5P1, G6PC, GOLGA6L2, GOLGA6L7P, HAMP, HAO1, HOTAIRM1, HP, IGFN1, IGHJ3P, IL20RB, IL4, ISL2, ITPKA, KIF18B, KLRC2, KRT75, KRT78, LINC00051, LINC00460, LINC00524, LINC00896, LINC00973, LINC01234, LINC01501, LINC01655, LINC01700, LINC01956, LINC02154, LINC02362, NEIL3, NFE4, NUF2, OTX1, PAEP, PGLYRP2, PI3, PITX1, PLG, PTPRB, RALYL, RPL10P19, RTL1, SAA1, SAA2, SAA4, SIM2, SLC16A12, SLC18A3, TGM3, TRIP13, TROAP, VSX1, WFDC10B, Z84485.1, ZIC2, ZIC5, ZPLD1 |
| mRMR | Ensemble of Min-redundancy and Max-relevance with survival data | 65 | AR, AL353637.1, DPP6, FOXJ1, GNB3, HHLA2, IL4, LIMCH1,LINC01732, OTX1, SAA1, SEMA3G, ZIC2 |










References
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal Cell Carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiang, Z.; Chen, X.; Zhu, X.; Peng, X. A Seven-Gene Signature Model Predicts Overall Survival in Kidney Renal Clear Cell Carcinoma. Hereditas 2020, 157, 38. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Shan, H.; Miao, M.Z.; Jiang, Z.; Meng, Y.; Chen, R.; Zhang, L.; Liu, Y. Identification of the Key Genes and Pathways Involved in the Tumorigenesis and Prognosis of Kidney Renal Clear Cell Carcinoma. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Society, A.C. Facts & Figures: 2020 Edition. 2020. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2020.html (accessed on 1 March 2022).
- Padala, S.A.; Barsouk, A.; Thandra, K.C.; Saginala, K.; Mohammed, A.; Vakiti, A.; Rawla, P.; Barsouk, A. Epidemiology of Renal Cell Carcinoma. World J. Oncol. 2020, 11, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Kann, B.H.; Hosny, A.; Aerts, H.J.W.L. Artificial Intelligence for Clinical Oncology. Cancer Cell 2021, 39, 916–927. [Google Scholar] [CrossRef]
- Chibon, F. Cancer Gene Expression Signatures—The Rise and Fall? Eur. J. Cancer 2013, 49, 2000–2009. [Google Scholar] [CrossRef]
- Zhan, Y.; Guo, W.; Zhang, Y.; Wang, Q.; Xu, X.-J.; Zhu, L. A Five-Gene Signature Predicts Prognosis in Patients with Kidney Renal Clear Cell Carcinoma. Comput. Math. Methods Med. 2015, 2015, 842784. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.; Bing, Z.; Tian, J.; Zhang, J.; Li, X.; Ge, L.; Ling, J.; Yang, K.; Li, Y. Comprehensive Assessment Gene Signatures for Clear Cell Renal Cell Carcinoma Prognosis. Medicine 2018, 97, e12679. [Google Scholar] [CrossRef]
- Chen, L.; Luo, Y.; Wang, G.; Qian, K.; Qian, G.; Wu, C.-L.; Dan, H.C.; Wang, X.; Xiao, Y. Prognostic Value of a Gene Signature in Clear Cell Renal Cell Carcinoma. J. Cell. Physiol. 2019, 234, 10324–10335. [Google Scholar] [CrossRef]
- Jiang, H.; Chen, H.; Chen, N. Construction and Validation of a Seven-Gene Signature for Predicting Overall Survival in Patients with Kidney Renal Clear Cell Carcinoma via an Integrated Bioinformatics Analysis. Anim. Cells Syst. 2020, 24, 160–170. [Google Scholar] [CrossRef]
- Pan, Q.; Wang, L.; Zhang, H.; Liang, C.; Li, B. Identification of a 5-Gene Signature Predicting Progression and Prognosis of Clear Cell Renal Cell Carcinoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 4401–4413. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jin, S.; Gu, W.; Wan, F.; Zhang, H.; Shi, G.; Qu, Y.; Ye, D. Construction and Validation of a 9-Gene Signature for Predicting Prognosis in Stage III Clear Cell Renal Cell Carcinoma. Front. Oncol. 2019, 9, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalantzakos, T.J.; Sullivan, T.B.; Gloria, T.; Canes, D.; Moinzadeh, A.; Rieger-Christ, K.M. MiRNA-424-5p Suppresses Proliferation, Migration, and Invasion of Clear Cell Renal Cell Carcinoma and Attenuates Expression of O-GlcNAc-Transferase. Cancers 2021, 13, 5160. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, Y.; Reddy, C.K. Machine Learning for Survival Analysis: A Survey. ACM Comput. Surv. 2019, 51, 1–36. [Google Scholar] [CrossRef]
- Cox, D.R. Regression Models and Life-Tables. J. R. Stat. Soc. Ser. B 1972, 34, 187–220. [Google Scholar] [CrossRef]
- Wan, F.; Zhu, Y.; Han, C.; Xu, Q.; Wu, J.; Dai, B.; Zhang, H.; Shi, G.; Gu, W.; Ye, D. Identification and Validation of an Eight-Gene Expression Signature for Predicting High Fuhrman Grade Renal Cell Carcinoma. Int. J. Cancer J. Int. Du Cancer 2017, 140, 1199–1208. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Zeng, W.; Liu, X. A Gene Signature of Survival Prediction for Kidney Renal Cell Carcinoma by Multi-Omic Data Analysis. Int. J. Mol. Sci. 2019, 20, 5720. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Hu, C. A 14 Immune-Related Gene Signature Predicts Clinical Outcomes of Kidney Renal Clear Cell Carcinoma. PeerJ 2020, 8, e10183. [Google Scholar] [CrossRef]
- Peng, H.; Long, F.; Ding, C. Feature Selection Based on Mutual Information Criteria of Max-Dependency, Max-Relevance, and Min-Redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive Molecular Characterization of Clear Cell Renal Cell Carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- GDC TCGA Kidney Clear Cell Carcinoma (KIRC). 2022. Available online: https://xenabrowser.net/datapages/ (accessed on 1 March 2022).
- Zhang, J.; Bajari, R.; Andric, D.; Gerthoffert, F.; Lepsa, A.; Nahal-Bose, H.; Stein, L.D.; Ferretti, V. The International Cancer Genome Consortium (ICGC) Data Portal. Nat. Biotechnol. 2019, 37, 367–369. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium. Renal Cell Cancer; EU/FR (RECA): Paris, France, 2022; Available online: https://dcc.icgc.org/projects/RECA-EU (accessed on 1 March 2022).
- Gao, G.F.; Parker, J.S.; Reynolds, S.M.; Silva, T.C.; Wang, L.-B.; Zhou, W.; Akbani, R.; Bailey, M.; Balu, S.; Berman, B.P.; et al. Before and After: Comparison of Legacy and Harmonized TCGA Genomic Data Commons’ Data. Cell Syst. 2019, 9, 24–34.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M. Building Predictive Models in R Using the Caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Consortium, Gte. The GTEx Consortium Atlas of Genetic Regulatory Effects across Human Tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Consortium, Gte. Human Genomics. The Genotype-Tissue Expression (GTEx) Pilot Analysis: Multitissue Gene Regulation in Humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Spooner, A.; Chen, E.; Sowmya, A.; Sachdev, P.; Kochan, N.A.; Trollor, J.; Brodaty, H. A Comparison of Machine Learning Methods for Survival Analysis of High-Dimensional Clinical Data for Dementia Prediction. Sci. Rep. 2020, 10, 20410. [Google Scholar] [CrossRef]
- Simon, N.; Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Cox’s Proportional Hazards Model via Coordinate Descent. J. Stat. Softw. 2011, 39, 1–13. [Google Scholar] [CrossRef]
- Ding, C.; Peng, H. Minimum Redundancy Feature Selection from Microarray Gene Expression Data. J. Bioinform. Comput. Biol. 2005, 3, 185–205. [Google Scholar] [CrossRef]
- Jay, N.D.; De Jay, N.; Papillon-Cavanagh, S.; Olsen, C.; El-Hachem, N.; Bontempi, G.; Haibe-Kains, B. MRMRe: An R Package for Parallelized MRMR Ensemble Feature Selection. Bioinformatics 2013, 29, 2365–2368. [Google Scholar] [CrossRef] [Green Version]
- Lang, M.; Binder, M.; Richter, J.; Schratz, P.; Pfisterer, F.; Coors, S.; Au, Q.; Casalicchio, G.; Kotthoff, L.; Bischl, B. Mlr3: A Modern Object-Oriented Machine Learning Framework in R. J. Open Source Softw. 2019, 4, 1903. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix. 2021. Available online: https://cran.r-project.org/web/packages/corrplot/index.html (accessed on 1 March 2022).
- Kratzer, G.; Furrer, R. Varrank: An R Package for Variable Ranking Based on Mutual Information with Applications to Observed Systemic Datasets. arXiv 2018, arXiv:1804.07134. [Google Scholar]
- Blanche, P.; Kattan, M.W.; Gerds, T.A. The C-Index Is Not Proper for the Evaluation of t-Year Predicted Risks. Biostatistics 2019, 20, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Uno, H.; Cai, T.; Tian, L.; Wei, L.J. Evaluating Prediction Rules Fort-Year Survivors with Censored Regression Models. J. Am. Stat. Assoc. 2007, 102, 527–537. [Google Scholar] [CrossRef]
- Potapov, S.; Adler, W.; Schmid, M. SurvAUC: Estimators of Prediction Accuracy for Time-to-Event Data. In Proceedings of the R User Conference, Nashville, TN, USA, 12–15 June 2012. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using “Ggplot2”. 2021. Available online: https://cran.r-project.org/web/packages/survminer/index.html (accessed on 1 March 2022).
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Boutros, P.C. VennDiagram: A Package for the Generation of Highly-Customizable Venn and Euler Diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R Package for Visually Combining Expression Data with Functional Analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. Open J. Anim. Sci. 2020, 11, 4. [Google Scholar]
- Therneau, T.M. A Package for Survival Analysis in R. 2022. Available online: https://cran.r-project.org/web/packages/survival/index.html (accessed on 1 March 2022).
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Harrison, E.; Drake, T.; Ots, R. Finalfit: Quickly Create Elegant Regression Results Tables and Plots When Modelling. 2022. Available online: https://github.com/ewenharrison/finalfit (accessed on 1 March 2022).
- Kolde, R. Pheatmap: Pretty Heatmaps. 2019. Available online: https://cran.r-project.org/web/pheatmap/survival/index.html (accessed on 1 March 2022).
- Patil, I. Visualizations with Statistical Details: The “ggstatsplot” Approach. J. Open Source Softw. 2021, 6, 3167. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; Liu, B.; Huang, Y.; Yu, G.; Lv, C. Prognostic Value of Immune-Related Genes in Clear Cell Renal Cell Carcinoma. Aging 2019, 11, 11474–11489. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yang, J.; Chen, Y. Identification of a Four Immune-Related Genes Signature Based on an Immunogenomic Landscape Analysis of Clear Cell Renal Cell Carcinoma. J. Cell. Physiol. 2020, 235, 9834–9850. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, E.; Zhuang, H.; Xie, L.; Feng, X.; Liu, J.; Yu, Y. Construction of a Novel Gene-Based Model for Prognosis Prediction of Clear Cell Renal Cell Carcinoma. Cancer Cell Int. 2020, 20, 27. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.W.; Park, H.; Seo, S.P.; Byun, Y.J.; Piao, X.M.; Kim, S.M.; Kim, W.T.; Yun, S.J.; Jang, W.; Shon, H.S.; et al. Methylation Signature for Prediction of Progression Free Survival in Surgically Treated Clear Cell Renal Cell Carcinoma. J. Korean Med. Sci. 2019, 34, e144. [Google Scholar] [CrossRef]
- Jia, Z.; Wan, F.; Zhu, Y.; Shi, G.; Zhang, H.; Dai, B.; Ye, D. Forkhead-Box Series Expression Network Is Associated with Outcome of Clear-Cell Renal Cell Carcinoma. Oncol. Lett. 2018, 15, 8669–8680. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Piao, Y.; Dong, X.; Jin, Z. Forkhead Box J1 Expression Is Upregulated and Correlated with Prognosis in Patients with Clear Cell Renal Cell Carcinoma. Oncol. Lett. 2015, 10, 1487–1494. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Pan, Q.; Xiao, R.; Yu, Y.; Lu, W.; Wang, L. A cluster of metabolism-related genes predict prognosis and progression of clear cell renal cell carcinoma. Sci. Rep. 2020, 10, 12949. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Zhu, B.; Ma, L.; Xing, Q. A Novel Nine Apoptosis-Related Genes Signature Predicting Overall Survival for Kidney Renal Clear Cell Carcinoma and its Associations with Immune Infiltration. Front. Mol. Biosci. 2021, 8, 567730. [Google Scholar] [CrossRef]
- Kang, M.A.; Lee, J.; Ha, S.H.; Lee, C.M.; Kim, K.M.; Jang, K.Y.; Park, S.H. Interleukin4Rα (IL4Rα) and IL13Rα1 Are Associated with the Progress of Renal Cell Carcinoma through Janus Kinase 2 (JAK2)/Forkhead Box O3 (FOXO3) Pathways. Cancers 2019, 11, 1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.-S.; Chae, S.-C.; Lee, J.-H.; Zhang, Q.; Chung, H.-T. Identification of Single Nucleotide Polymorphisms in FOXJ1 and Their Association with Allergic Rhinitis. J. Hum. Genet. 2006, 51, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-S.; Zhang, Q.; Lim, M.-K.; Sheen, D.-H.; Shim, S.-C.; Kim, J.-Y.; Lee, S.-S.; Yun, K.-J.; Moon, H.-B.; Chung, H.-T.; et al. Association of FOXJ1 Polymorphisms with Systemic Lupus Erythematosus and Rheumatoid Arthritis in Korean Population. Exp. Mol. Med. 2007, 39, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, S.; Peng, S.L. Cutting Edge: Foxj1 Protects against Autoimmunity and Inhibits Thymocyte Egress. J. Immunol. 2005, 175, 7805–7809. [Google Scholar] [CrossRef] [Green Version]
- Xian, S.; Shang, D.; Kong, G.; Tian, Y. FOXJ1 Promotes Bladder Cancer Cell Growth and Regulates Warburg Effect. Biochem. Biophys. Res. Commun. 2018, 495, 988–994. [Google Scholar] [CrossRef]
- Chen, H.-W.; Huang, X.-D.; Li, H.-C.; He, S.; Ni, R.-Z.; Chen, C.-H.; Peng, C.; Wu, G.; Wang, G.-H.; Wang, Y.-Y.; et al. Expression of FOXJ1 in Hepatocellular Carcinoma: Correlation with Patients’ Prognosis and Tumor Cell Proliferation. Mol. Carcinog. 2013, 52, 647–659. [Google Scholar] [CrossRef]
- Liu, K.; Fan, J.; Wu, J. Forkhead Box Protein J1 (FOXJ1) Is Overexpressed in Colorectal Cancer and Promotes Nuclear Translocation of B-Catenin in SW620 Cells. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 856–866. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cai, X.; Xia, L.; Zhou, J.; Xin, J.; Liu, M.; Shang, X.; Liu, J.; Li, X.; Chen, Z.; et al. Decreased Expression of FOXJ1 Is a Potential Prognostic Predictor for Progression and Poor Survival of Gastric Cancer. Ann. Surg. Oncol. 2015, 22, 685–692. [Google Scholar] [CrossRef]
- Abedalthagafi, M.S.; Wu, M.P.; Merrill, P.H.; Du, Z.; Woo, T.; Sheu, S.-H.; Hurwitz, S.; Ligon, K.L.; Santagata, S. Decreased FOXJ1 Expression and Its Ciliogenesis Programme in Aggressive Ependymoma and Choroid Plexus Tumours. J. Pathol. 2016, 238, 584–597. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.M.; Nadkarni, G.N.; Tao, R.; Graff, M.; Fornage, M.; Buyske, S.; Matise, T.C.; Highland, H.M.; Wilkens, L.R.; Carlson, C.S.; et al. Genetics of Chronic Kidney Disease Stages Across Ancestries: The PAGE Study. Front. Genet. 2019, 10, 494. [Google Scholar] [CrossRef] [Green Version]
- Shirota, H.; Klinman, D.M.; Ito, S.-E.; Ito, H.; Kubo, M.; Ishioka, C. IL4 from T Follicular Helper Cells Downregulates Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.-E.; Shirota, H.; Kasahara, Y.; Saijo, K.; Ishioka, C. IL-4 Blockade Alters the Tumor Microenvironment and Augments the Response to Cancer Immunotherapy in a Mouse Model. Cancer Immunol. Immunother. 2017, 66, 1485–1496. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Xie, X.; Shi, X.; Li, S. Associations of Common IL-4 Gene Polymorphisms with Cancer Risk: A Meta-Analysis. Mol. Med. Rep. 2017, 16, 1927–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Borczuk, A.; Janakiram, M.; Ren, X.; Lin, J.; Assal, A.; Halmos, B.; Perez-Soler, R.; Zang, X. Wide Expression and Significance of Alternative Immune Checkpoint Molecules, B7x and HHLA2, in PD-L1–Negative Human Lung Cancers. Clin. Cancer Res. 2018, 24, 1954–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Chinai, J.M.; Buhl, S.; Scandiuzzi, L.; Ray, A.; Jeon, H.; Ohaegbulam, K.C.; Ghosh, K.; Zhao, A.; Scharff, M.D.; et al. HHLA2 Is a Member of the B7 Family and Inhibits Human CD4 and CD8 T-Cell Function. Proc. Natl. Acad. Sci. USA 2013, 110, 9879–9884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, J.M.; Cho, H.J.; Park, H.Y.; Lee, D.S.; Choi, I.H.; Kim, Y.N.; Jeong, C.H.; Kim, D.H.; Hwa Im, D.; Min, B.J.; et al. The Clinical Significance of HERV-H LTR -Associating 2 Expression in Cervical Adenocarcinoma. Medicine 2021, 100, e23691. [Google Scholar] [CrossRef]
- Boor, P.P.C.; Sideras, K.; Biermann, K.; Hosein Aziz, M.; Levink, I.J.M.; Mancham, S.; Erler, N.S.; Tang, X.; van Eijck, C.H.; Bruno, M.J.; et al. HHLA2 Is Expressed in Pancreatic and Ampullary Cancers and Increased Expression Is Associated with Better Post-Surgical Prognosis. Br. J. Cancer 2020, 122, 1211–1218. [Google Scholar] [CrossRef]
- Cheng, H.; Janakiram, M.; Borczuk, A.; Lin, J.; Qiu, W.; Liu, H.; Chinai, J.M.; Halmos, B.; Perez-Soler, R.; Zang, X. HHLA2, a New Immune Checkpoint Member of the B7 Family, Is Widely Expressed in Human Lung Cancer and Associated with EGFR Mutational Status. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Shimonosono, M.; Arigami, T.; Yanagita, S.; Matsushita, D.; Uchikado, Y.; Kijima, Y.; Kurahara, H.; Kita, Y.; Mori, S.; Sasaki, K.; et al. The Association of Human Endogenous Retrovirus-H Long Terminal Repeat-Associating Protein 2 (HHLA2) Expression with Gastric Cancer Prognosis. Oncotarget 2018, 9, 22069–22078. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhu, D.; Feng, J.; Zhou, Y.; Wang, Q.; Feng, H.; Zhang, J.; Jiang, J. Overexpression of HHLA2 in Human Clear Cell Renal Cell Carcinoma Is Significantly Associated with Poor Survival of the Patients. Cancer Cell Int. 2019, 19, 1–12. [Google Scholar] [CrossRef]
- Reidy, K.; Tufro, A. Semaphorins in Kidney Development and Disease: Modulators of Ureteric Bud Branching, Vascular Morphogenesis, and Podocyte-Endothelial Crosstalk. Pediatric Nephrol. 2011, 26, 1407–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Worzfeld, T. Semaphorins and Plexins in Kidney Disease. Nephron 2016, 132, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, G.; Mumblat, Y.; Smolkin, T.; Toledano, S.; Nir-Zvi, I.; Ziv, K.; Kessler, O. The Role of the Semaphorins in Cancer. Cell Adhes. Migr. 2016, 10, 652–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karayan-Tapon, L.; Wager, M.; Guilhot, J.; Levillain, P.; Marquant, C.; Clarhaut, J.; Potiron, V.; Roche, J. Semaphorin, Neuropilin and VEGF Expression in Glial Tumours: SEMA3G, a Prognostic Marker? Br. J. Cancer 2008, 99, 1153–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Malone, A.F.; Donnelly, E.L.; Kirita, Y.; Uchimura, K.; Ramakrishnan, S.M.; Gaut, J.P.; Humphreys, B.D. Single-Cell Transcriptomics of a Human Kidney Allograft Biopsy Specimen Defines a Diverse Inflammatory Response. J. Am. Soc. Nephrol. JASN 2018, 29, 2069–2080. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Liu, Z.; Zou, Z.; Tang, Y.; Zhou, C.; Yang, J.; Wei, X.; Lu, Y. The Correlation between the Immune and Epithelial-Mesenchymal Transition Signatures Suggests Potential Therapeutic Targets and Prognosis Prediction Approaches in Kidney Cancer. Sci. Rep. 2018, 8, 6570. [Google Scholar] [CrossRef] [Green Version]
- Balk, S.P.; Knudsen, K.E. AR, the Cell Cycle, and Prostate Cancer. Nucl. Recept. Signal. 2008, 6, nrs.06001. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Abdollah, F. Re: AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. Eur. Urol. 2015, 68, 162–163. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Sun, Y.; Zhai, W.; Ma, X.; Shen, D.; Du, S.; You, B.; Niu, Y.; Huang, C.-P.; Zhang, X.; et al. Androgen Receptor Modulates Metastatic Routes of VHL Wild-Type Clear Cell Renal Cell Carcinoma in an Oxygen-Dependent Manner. Oncogene 2020, 39, 6677–6691. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, Y.; Rao, Q.; Xu, H.; Li, L.; Chang, C. Androgen Receptor (AR) Suppresses MiRNA-145 to Promote Renal Cell Carcinoma (RCC) Progression Independent of VHL Status. Oncotarget 2015, 6, 31203–31215. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-H.; Kim, B.-C.; Jeong, S.-H.; Jeong, C.W.; Ku, J.H.; Kwak, C.; Kim, H.H. Histone Demethylase LSD1 Regulates Kidney Cancer Progression by Modulating Androgen Receptor Activity. Int. J. Mol. Sci. 2020, 21, 6089. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Sun, Y.; Tao, W.; Fei, X.; Chang, C. Androgen Receptor (AR) Promotes Clear Cell Renal Cell Carcinoma (CcRCC) Migration and Invasion via Altering the CircHIAT1/MiR-195-5p/29a-3p/29c-3p/CDC42 Signals. Cancer Lett. 2017, 394, 1–12. [Google Scholar] [CrossRef] [PubMed]
- You, B.; Sun, Y.; Luo, J.; Wang, K.; Liu, Q.; Fang, R.; Liu, B.; Chou, F.; Wang, R.; Meng, J.; et al. Androgen Receptor Promotes Renal Cell Carcinoma (RCC) Vasculogenic Mimicry (VM) via Altering TWIST1 Nonsense-Mediated Decay through LncRNA-TANAR. Oncogene 2021, 40, 1674–1689. [Google Scholar] [CrossRef]
- Larsen, K.B.; Lutterodt, M.C.; Møllgård, K.; Møller, M. Expression of the Homeobox Genes OTX2 and OTX1 in the Early Developing Human Brain. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2010, 58, 669–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Frigola, C.; Carreres, M.I.; Vegar, C.; Mason, C.; Herrera, E. Zic2 Promotes Axonal Divergence at the Optic Chiasm Midline by EphB1-Dependent and -Independent Mechanisms. Development 2008, 135, 1833–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinberg, I.; Millen, K.J. The ZIC Gene Family in Development and Disease. Clin. Genet. 2005, 67, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Marchini, S.; Poynor, E.; Barakat, R.R.; Clivio, L.; Cinquini, M.; Fruscio, R.; Porcu, L.; Bussani, C.; D’Incalci, M.; Erba, E.; et al. The Zinc Finger Gene ZIC2 Has Features of an Oncogene and Its Overexpression Correlates Strongly with the Clinical Course of Epithelial Ovarian Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 4313–4324. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-H.; Chen, M.-L.; Zhang, Q.; Zhang, Y.; An, X.; Luo, Y.-L.; Liu, X.-M.; Liu, S.-X.; Liu, Q.; Yang, T.; et al. ZIC2 Is Downregulated and Represses Tumor Growth via the Regulation of STAT3 in Breast Cancer. Int. J. Cancer. J. Int. Du Cancer 2020, 147, 505–518. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Li, L.; Chen, S.-L.; Yang, X.; Zhang, C.Z.; Cao, Y. A Zic2/Runx2/NOLC1 Signaling Axis Mediates Tumor Growth and Metastasis in Clear Cell Renal Cell Carcinoma. Cell Death Dis. 2021, 12, 319. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Zhen, Y.-Y.; Chien, K.-Y.; Lee, I.-C.; Lin, W.-C.; Chen, M.-Y.; Pai, L.-M. LIMCH1 Regulates Nonmuscle Myosin-II Activity and Suppresses Cell Migration. Mol. Biol. Cell 2017, 28, 1054–1065. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, T.; Kvarnbrink, S.; Holmlund, C.; Botling, J.; Micke, P.; Henriksson, R.; Johansson, M.; Hedman, H. LMO7 and LIMCH1 Interact with LRIG Proteins in Lung Cancer, with Prognostic Implications for Early-Stage Disease. Lung Cancer 2018, 125, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Cizkova, M.; Cizeron-Clairac, G.; Vacher, S.; Susini, A.; Andrieu, C.; Lidereau, R.; Bièche, I. Gene Expression Profiling Reveals New Aspects of PIK3CA Mutation in ERalpha-Positive Breast Cancer: Major Implication of the Wnt Signaling Pathway. PLoS ONE 2010, 5, e15647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halle, M.K.; Sødal, M.; Forsse, D.; Engerud, H.; Woie, K.; Lura, N.G.; Wagner-Larsen, K.S.; Trovik, J.; Bertelsen, B.I.; Haldorsen, I.S.; et al. A 10-Gene Prognostic Signature Points to LIMCH1 and HLA-DQB1 as Important Players in Aggressive Cervical Cancer Disease. Br. J. Cancer 2021, 124, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, 2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Expression of LIMCH1 in Renal Cancer—Interactive Survival Scatter Plot—The Human Protein Atlas. 2022. Available online: https://www.proteinatlas.org/ENSG00000064042-LIMCH1/pathology/renal+cancer (accessed on 1 March 2022).
- Clark, B.D.; Kwon, E.; Maffie, J.; Jeong, H.-Y.; Nadal, M.; Strop, P.; Rudy, B. DPP6 Localization in Brain Supports Function as a Kv4 Channel Associated Protein. Front. Mol. Neurosci. 2008, 1, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Cao, D.; Ren, Z.; Liu, Z.; Lv, S.; Zhu, J.; Li, L.; Lang, R.; He, Q. Dipeptidyl Peptidase like 6 Promoter Methylation Is a Potential Prognostic Biomarker for Pancreatic Ductal Adenocarcinoma. Biosci. Rep. 2020, 40, BSR20200214. [Google Scholar] [CrossRef]
- Choy, T.-K.; Wang, C.-Y.; Phan, N.N.; Khoa Ta, H.D.; Anuraga, G.; Liu, Y.-H.; Wu, Y.-F.; Lee, K.-H.; Chuang, J.-Y.; Kao, T.-J. Identification of Dipeptidyl Peptidase (DPP) Family Genes in Clinical Breast Cancer Patients via an Integrated Bioinformatics Approach. Diagnostics 2021, 11, 1204. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S. Quantitative Assessment of the Association between GNB3 C825T Polymorphism and Cancer Risk. JBUON J. Balk. Union Oncol. 2014, 19, 1092–1095. [Google Scholar]
- Fingas, C.D.; Katsounas, A.; Kahraman, A.; Siffert, W.; Jochum, C.; Gerken, G.; Nückel, H.; Canbay, A. Prognostic Assessment of Three Single-Nucleotide Polymorphisms (GNB3 825C>T, BCL2-938C>A, MCL1-386C>G) in Extrahepatic Cholangiocarcinoma. Cancer Investig. 2010, 28, 472–478. [Google Scholar] [CrossRef]
- Paleari, R.G.; Peres, R.M.R.; Florentino, J.O.; Heinrich, J.K.; Bragança, W.O.; Del Valle, J.C.T.; Zeferino, L.C.; Derchain, S.F.M.; Sarian, L.O. Reduced Prevalence of the C825T Polymorphism of the G-Protein Beta Subunit Gene in Women with Breast Cancer. Int. J. Biol. Markers 2011, 26, 234–240. [Google Scholar] [CrossRef]
- Santo, C.D.; De Santo, C.; Arscott, R.; Booth, S.; Karydis, I.; Jones, M.; Asher, R.; Salio, M.; Middleton, M.; Cerundolo, V. Invariant NKT Cells Modulate the Suppressive Activity of IL-10-Secreting Neutrophils Differentiated with Serum Amyloid A. Nat. Immunol. 2010, 11, 1039–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paret, C.; Schön, Z.; Szponar, A.; Kovacs, G. Inflammatory Protein Serum Amyloid A1 Marks a Subset of Conventional Renal Cell Carcinomas with Fatal Outcome. Eur. Urol. 2010, 57, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Expression of SAA1 in Renal Cancer—Interactive Survival Scatter Plot—The Human Protein Atlas. 2022. Available online: https://www.proteinatlas.org/ENSG00000173432-SAA1/pathology/renal+cancer (accessed on 1 March 2022).
- Marshall, F.F. Serum Protein Profiling by SELDI Mass Spectrometry: Detection of Multiple Variants of Serum Amyloid Alpha in Renal Cancer Patients. J. Urol. 2005, 173, 1919–1920. [Google Scholar] [CrossRef]
- Guo, R.; Zou, B.; Liang, Y.; Bian, J.; Xu, J.; Zhou, Q.; Zhang, C.; Chen, T.; Yang, M.; Wang, H.; et al. LncRNA RCAT1 Promotes Tumor Progression and Metastasis via MiR-214-5p/E2F2 Axis in Renal Cell Carcinoma. Cell Death Dis. 2021, 12, 689. [Google Scholar] [CrossRef] [PubMed]
- Qi, N.; Chen, Y.; Gong, K.; Li, H. Concurrent Renal Cell Carcinoma and Urothelial Carcinoma: Long-Term Follow-up Study of 27 Cases. World J. Surg. Oncol. 2018, 16, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knez, V.M.; Barrow, W.; Lucia, M.S.; Wilson, S.; La Rosa, F.G. Clear Cell Urothelial Carcinoma of the Urinary Bladder: A Case Report and Review of the Literature. J. Med. Case Rep. 2014, 8, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotellini, M.; Fondi, C.; Paglierani, M.; Stomaci, N.; Raspollini, M.R. Clear Cell Carcinoma of the Bladder in a Patient with a Earlier Clear Cell Renal Cell Carcinoma: A Case Report with Morphologic, Immunohistochemical, and Cytogenetical Analysis. Appl. Immunohistochem. Mol. Morphol. AIMM Off. Publ. Soc. Appl. Immunohistochem. 2010, 18, 396–399. [Google Scholar] [CrossRef]
- van de Pol, J.A.A.; van den Brandt, P.A.; Schouten, L.J. Kidney Stones and the Risk of Renal Cell Carcinoma and Upper Tract Urothelial Carcinoma: The Netherlands Cohort Study. Br. J. Cancer 2018, 120, 368–374. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Lu, Y.; Wang, J.; Yang, L.; Han, Y.; Wang, Y.; Yan, D.; Ruan, Q.; Wang, S. A Four-Gene Signature Predicts Survival in Clear-Cell Renal-Cell Carcinoma. Oncotarget 2016, 7, 82712–82726. [Google Scholar] [CrossRef] [Green Version]
- D’Costa, N.M.; Cina, D.; Shrestha, R.; Bell, R.H.; Lin, Y.-Y.; Asghari, H.; Monjaras-Avila, C.U.; Kollmannsberger, C.; Hach, F.; Chavez-Munoz, C.I.; et al. Identification of Gene Signature for Treatment Response to Guide Precision Oncology in Clear-Cell Renal Cell Carcinoma. Sci. Rep. 2020, 10, 2026. [Google Scholar] [CrossRef] [Green Version]
- Ha, M.J.; Baladandayuthapani, V.; Do, K.-A. Prognostic Gene Signature Identification Using Causal Structure Learning: Applications in Kidney Cancer. Cancer Inform. 2015, 14, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-L.; Ge, G.-J.; Qi, C.; Wang, H.; Wang, H.-L.; Li, L.-Y.; Li, G.-H.; Xia, L.-Q. A Five-Gene Signature May Predict Sunitinib Sensitivity and Serve as Prognostic Biomarkers for Renal Cell Carcinoma. J. Cell. Physiol. 2018, 233, 6649–6660. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Guan, Y.; Wedge, D.C.; Ansari-Pour, N. Re-Evaluating Experimental Validation in the Big Data Era: A Conceptual Argument. Genome Biol. 2021, 22, 71. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Program. 2022. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 1 March 2022).
- TCGA/GDC Data Portal—Data Release 18.0. 2019. Available online: https://docs.gdc.cancer.gov/Data/Release_Notes/Data_Release_Notes/#data-release-180 (accessed on 1 March 2022).
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]






| Clinical Characteristics | Training Cohort TCGA-KIRC (n = 530) 1 | Validation Cohort ICGC-RECA (n = 91) | p Value 2 | |
|---|---|---|---|---|
| Overall survival (days) | Mean (SD) | 1343.2 (976.6) | 1511.6 (634.6) | 0.113 |
| Overall survival status, n./total n. (%) | Alive | 359/530 (67.7) | 61/91 (67.0) | 0.991 |
| Deceased | 171/530 (32.3) | 30/91 (33.0) | ||
| Age, years | Mean (SD) | 60.5 (12.0) | 60.5 (10.0) | 0.99 |
| Gender, n./total n. (%) | Female | 183/530 (34.5) | 39/91 (42.9) | 0.158 |
| Male | 347/530 (65.5) | 52/91 (57.1) | ||
| AJCC stage, n./Total (%) | T1 | 270/530 (50.9) | 54/91 (59.3) | 0.343 |
| T2 | 70/530 (13.2) | 13/91 (14.3) | ||
| T3 | 179/530 (33.8) | 22/91 (24.2) | ||
| T4 | 11/530 (2.1) | 2/91 (2.2) | ||
| Neoplasm, n. (%) | N0 | 79 (86.8) | 239 (45.1) | <0.001 |
| N1 | 2 (2.2) | 16 (3.0) | ||
| NX | 10 (11.0) | 275 (51.9) | ||
| Metastasis, n. (%) | M0 | 422/528 (79.9) | 81/91 (89.0) | 0.081 |
| M1 | 78/528 (14.8) | 9/91 (9.9) | ||
| MX | 28/528 (5.3) | 1/91 (1.1) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terrematte, P.; Andrade, D.S.; Justino, J.; Stransky, B.; de Araújo, D.S.A.; Dória Neto, A.D. A Novel Machine Learning 13-Gene Signature: Improving Risk Analysis and Survival Prediction for Clear Cell Renal Cell Carcinoma Patients. Cancers 2022, 14, 2111. https://doi.org/10.3390/cancers14092111
Terrematte P, Andrade DS, Justino J, Stransky B, de Araújo DSA, Dória Neto AD. A Novel Machine Learning 13-Gene Signature: Improving Risk Analysis and Survival Prediction for Clear Cell Renal Cell Carcinoma Patients. Cancers. 2022; 14(9):2111. https://doi.org/10.3390/cancers14092111
Chicago/Turabian StyleTerrematte, Patrick, Dhiego Souto Andrade, Josivan Justino, Beatriz Stransky, Daniel Sabino A. de Araújo, and Adrião D. Dória Neto. 2022. "A Novel Machine Learning 13-Gene Signature: Improving Risk Analysis and Survival Prediction for Clear Cell Renal Cell Carcinoma Patients" Cancers 14, no. 9: 2111. https://doi.org/10.3390/cancers14092111