
Optical Genome Mapping as a Diagnostic Tool in Pediatric Acute Myeloid Leukemia


 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Origin of the Samples Analyzed
2.2. Isolation of Ultra-High Molecular Weight DNA
2.3. Quantification of UHMW DNA
2.4. Labeling of UHMW gDNA and Chip Loading
2.5. Assemblies and Variant Calling
2.6. Data Comparison
2.7. Validation of Newly Detected SVs by Long-Distance Touchdown PCR and Sequencing
2.8. Applying Newly Detected SVs for Monitoring of Minimal Residual Disease (MRD)
3. Results
3.1. Comparison of Karyotyping and FISH Results Versus OGM Results
3.1.1. Cases with Identical Results
3.1.2. Cases with SVs Detected by Karyotyping Only
3.1.3. Cases with SVs Detected by OGM Only
3.1.4. Cases with SVs Only Detected by Karyotyping and Other SVs Only Detected by OGM within the Same Case
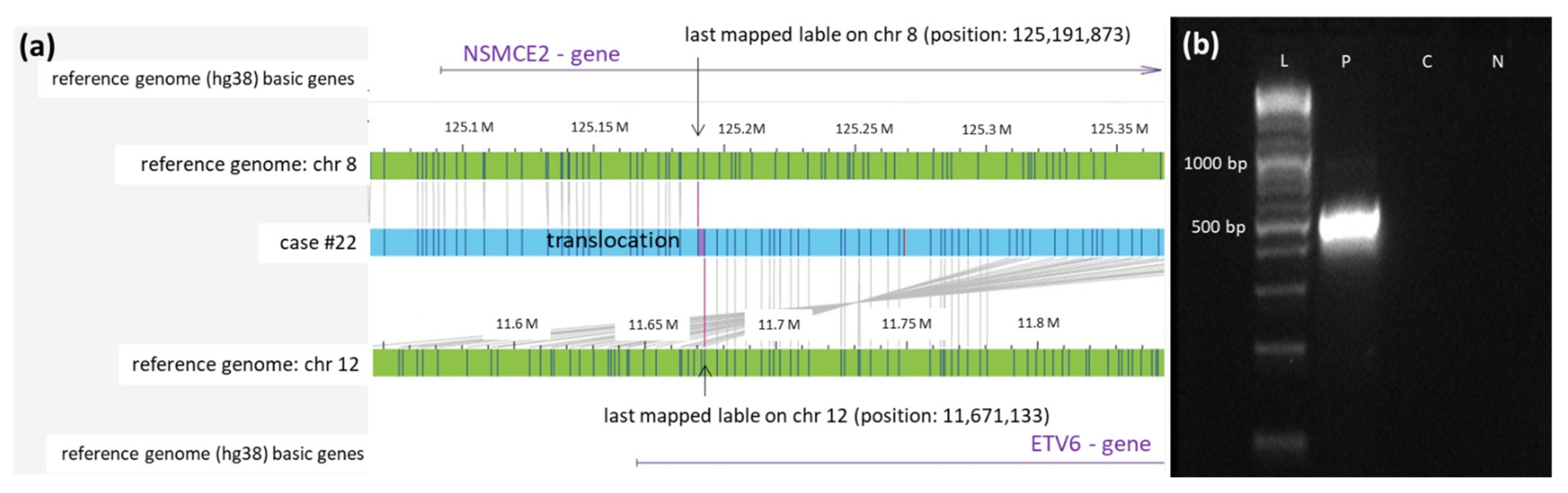
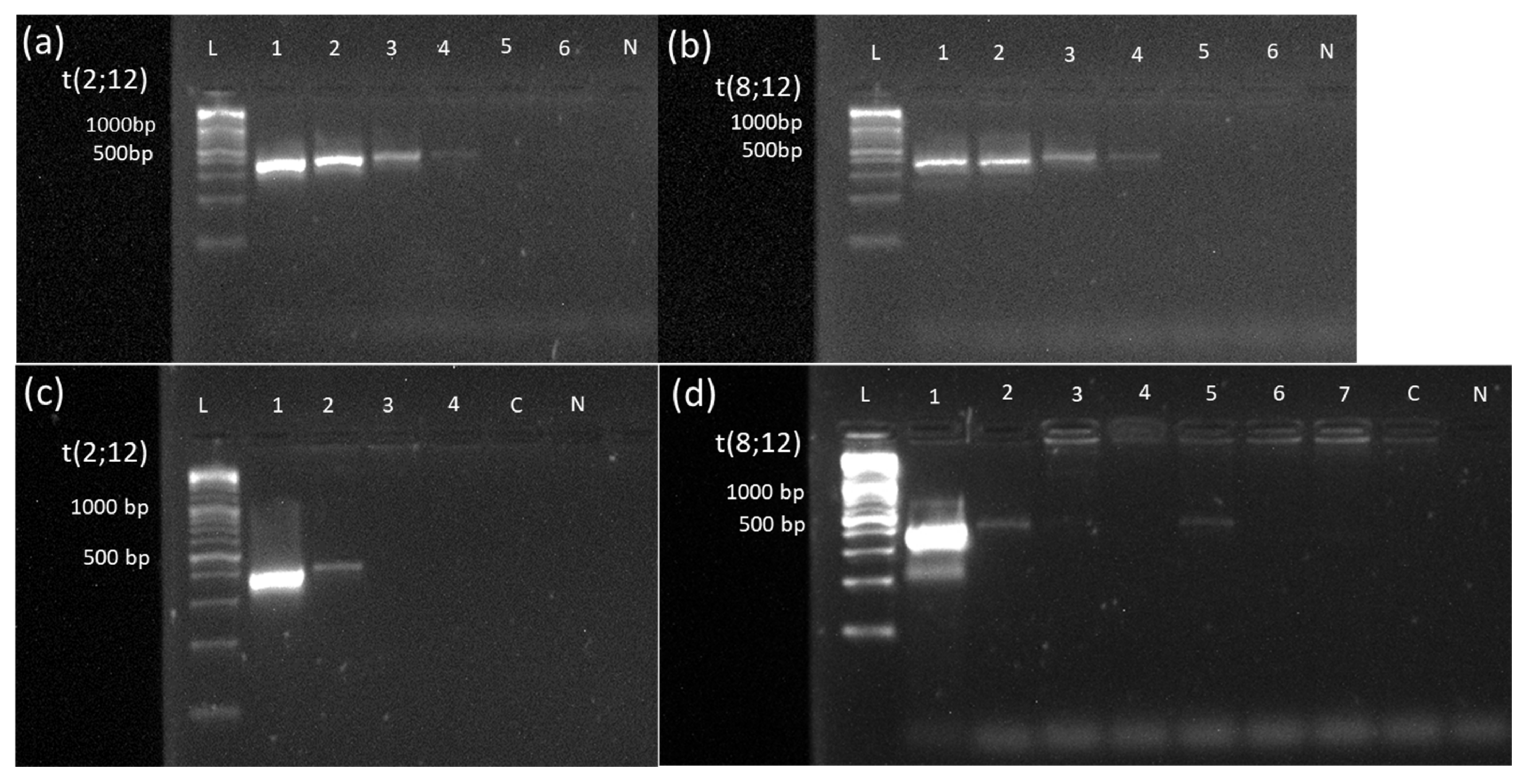
3.2. Successful Validation of 3 Novel OGM Findings by Breakpoint-Spanning PCR
3.3. Stratification of Pediatric AML by Cytogenetics Versus OGM
3.4. MRD Monitoring Based on Validated Novel OGM Findings
4. Discussion
4.1. Practicability
4.2. Higher Resolution
4.3. Subclones
4.4. Blast Count
4.5. Cultural Artifacts
4.6. OGM Has No Markers on Defined Chromosome Segments
4.7. Newly Discovered SVs
4.8. Newly detected SVs used as MRD marker
4.9. Changes in Risk Classification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rasche, M.; Zimmermann, M.; Borschel, L.; Bourquin, J.P.; Dworzak, M.; Klingebiel, T.; Lehrnbecher, T.; Creutzig, U.; Klusmann, J.H.; Reinhardt, D. Successes and challenges in the treatment of pediatric acute myeloid leukemia: A retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 2018, 32, 2167–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwaan, C.M.; Kolb, E.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; De Bont, E.S.; De Moerloose, B.; Dworzak, M.; Gibson, B.E.; et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balgobind, B.V.; Hollink, I.H.I.M.; Arentsen-Peters, S.T.C.J.M.; Zimmermann, M.; Harbott, J.; Beverloo, H.B.; Von Bergh, A.R.M.; Cloos, J.; Kaspers, G.J.L.; De Haas, V.; et al. Integrative analysis of type-I and type-II aberrations underscores the genetic heterogeneity of pediatric acute myeloid leukemia. Haematologica 2011, 96, 1478–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdmann, F.; Grabow, D.; Spix, C. German Childhood Cancer Registry—Annual Report. Available online: https://www.kinderkrebsregister.de/dkkr/ergebnisse/jahresberichte/jahresbericht-2019.html (accessed on 12 January 2022).
- Coustan-Smith, E.; Campana, D. Should evaluation for minimal residual disease be routine in acute myeloid leukemia? Curr. Opin. Hematol. 2013, 20, 86–92. [Google Scholar] [CrossRef]
- Quessada, J.; Cuccuini, W.; Saultier, P.; Loosveld, M.; Harrison, C.J.; Lafage-Pochitaloff, M. Cytogenetics of Pediatric Acute Myeloid Leukemia: A Review of the Current Knowledge. Genes 2021, 12, 924. [Google Scholar] [CrossRef]
- Nguyen-Khac, F.; Bidet, A.; Veronese, L.; Daudignon, A.; Penther, D.; Troadec, M.-B.; Lefebvre, C.; Lafage-Pochitaloff, M. Recommendations for cytogenomic analysis of hematologic malignancies: Comments from the Francophone Group of Hematological Cytogenetics (GFCH). Leukemia 2020, 34, 1711–1713. [Google Scholar] [CrossRef]
- Rack, K.A.; van den Berg, E.; Haferlach, C.; Beverloo, H.B.; Costa, D.; Espinet, B.; Foot, N.; Jeffries, S.; Martin, K.; O’Connor, S.; et al. European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms: Reponse to the comments from the Francophone Group of Hematological Cytogenetics (GFCH). Leukemia 2020, 34, 2262–2264. [Google Scholar] [CrossRef] [Green Version]
- Rack, K.A.; Van Den Berg, E.; Haferlach, C.; Beverloo, H.B.; Costa, D.; Espinet, B.; Foot, N.; Jeffries, S.; Martin, K.; O’Connor, S.; et al. European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms. Leukemia 2019, 33, 1851–1867. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Schroeder, M.C.; O’Laughlin, M.; Wilson, R.; Macmillan, S.; Bohannon, A.; Kruchowski, S.; Garza, J.; Du, F.; Hughes, A.E.O.; et al. Genome Sequencing as an Alternative to Cytogenetic Analysis in Myeloid Cancers. N. Engl. J. Med. 2021, 384, 924–935. [Google Scholar] [CrossRef]
- Schwartz, D.C.; Li, X.; Hernandez, L.I.; Ramnarain, S.P.; Huff, E.J.; Wang, Y.K. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science 1993, 262, 110–114. [Google Scholar] [CrossRef]
- LLevy, B.; Baughn, L.B.; Chartrand, S.; LaBarge, B.; Claxton, D.; Lennon, A.; Akkari, Y.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. A National Multicenter Evaluation of the Clinical Utility of Optical Genome Mapping for Assessment of Genomic Aberrations in Acute Myeloid Leukemia. medRxiv 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.11.07.20227728v1 (accessed on 10 February 2022).
- Gerding, W.M.; Tembrink, M.; Nilius-Eliliwi, V.; Mika, T.; Dimopoulos, F.; Ladigan-Badura, S.; Eckhardt, M.; Pohl, M.; Wünnenberg, M.; Farshi, P.; et al. Optical genome mapping reveals additional prognostic information compared to conventional cytogenetics in AML / MDS patients. Int. J. Cancer 2022. [Google Scholar] [CrossRef] [PubMed]
- Lestringant, V.; Duployez, N.; Penther, D.; Luquet, I.; Derrieux, C.; Lutun, A.; Preudhomme, C.; West, M.; Ouled-Haddou, H.; Devoldere, C.; et al. Optical genome mapping, a promising alternative to gold standard cytogenetic approaches in a series of acute lymphoblastic leukemias. Genes Chromosomes Cancer 2021, 60, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Lühmann, J.L.; Stelter, M.; Wolter, M.; Kater, J.; Lentes, J.; Bergmann, A.K.; Schieck, M.; Göhring, G.; Möricke, A.; Cario, G.; et al. The Clinical Utility of Optical Genome Mapping for the Assessment of Genomic Aberrations in Acute Lymphoblastic Leukemia. Cancers 2021, 13, 4388. [Google Scholar] [CrossRef]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Faber, Z.J.; Chen, X.; Gedman, A.L.; Boggs, K.; Cheng, J.; Ma, J.; Radtke, I.; Chao, J.R.; Walsh, M.P.; Song, G.; et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat. Genet. 2016, 48, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Creutzig, U.; Van Den Heuvel-Eibrink, M.M.; Gibson, B.; Dworzak, M.N.; Adachi, S.; De Bont, E.; Harbott, J.; Hasle, H.; Johnston, D.; Kinoshita, A.; et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: Recommendations from an international expert panel. Blood 2012, 120, 3187–3205. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Hills, R.K.; Moorman, A.V.; Grimwade, D.J.; Hann, I.; Webb, D.K.H.; Wheatley, K.; De Graaf, S.S.N.; Van Den Berg, E.; Burnett, A.K.; et al. Cytogenetics of Childhood Acute Myeloid Leukemia: United Kingdom Medical Research Council Treatment Trials AML 10 and 12. J. Clin. Oncol. 2010, 28, 2674–2681. [Google Scholar] [CrossRef] [PubMed]
- Database of Genome Variants. Available online: http://dgv.tcag.ca/dgv/app/home (accessed on 27 January 2022).
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Korbie, D.J.; Mattick, J.S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat. Protoc. 2008, 3, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Castelli, I.; Astolfi, A.; Bertuccio, S.N.; Indio, V.; Togni, M.; Belotti, T.; Serravalle, S.; Tarantino, G.; Zecca, M.; et al. Genomic complexity and dynamics of clonal evolution in childhood acute myeloid leukemia studied with whole-exome sequencing. Oncotarget 2016, 7, 56746–56757. [Google Scholar] [CrossRef] [PubMed]
- Creutzig, U.; Zimmermann, M.; Reinhardt, D.; Rasche, M.; von Neuhoff, C.; Alpermann, T.; Dworzak, M.; Perglerová, K.; Zemanova, Z.; Tchinda, J.; et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer 2016, 122, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Bionano Genomics. Molecule Quality Report. Available online: https://bionanogenomics.com/wp-content/uploads/2018/04/30223-Saphyr-Molecule-Quality-Report-Guidelines.pdf (accessed on 7 February 2022).
- Bionano Genomics. Bionano Solve Theory of Operation: Structural Variant Calling. Available online: https://bionanogenomics.com/wp-content/uploads/2018/04/30110-Bionano-Solve-Theory-of-Operation-Structural-Variant-Calling.pdf (accessed on 7 February 2022).
- Bakker, B.; van den Bos, H.; Lansdorp, P.M.; Foijer, F. How to count chromosomes in a cell: An overview of current and novel technologies. Bioessays 2015, 37, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Hook, E.B. Exclusion of Chromosomal Mosaicism: Tables of 90%, 95%, and 99% Confidence Limits and Comments on Use. Am. J. Hum. Genet. 1977, 29, 94–97. [Google Scholar]
- Dremsek, P.; Schwarz, T.; Weil, B.; Malashka, A.; Laccone, F.; Neesen, J. Optical Genome Mapping in Routine Human Genetic Diagnostics—Its Advantages and Limitations. Genes 2021, 12, 1958. [Google Scholar] [CrossRef]
- Liu, F.; Liu, Q.; Guo, N.; Zhang, G.M.; Deng, Y.F.; Hu, W.W.; Rao, H.L. Expression of myocyte enhancer factor 2B in mantle cell lymphoma and its clinical significance. Zhonghua Bing Li Xue Za Zhi 2020, 49, 40–46. [Google Scholar] [CrossRef]
- De Braekeleer, E.; Douet-Guilbert, N.; Morel, F.; Le Bris, M.J.; Basinko, A.; De Braekeleer, M. ETV6 fusion genes in hematological malignancies: A review. Leuk. Res. 2012, 36, 945–961. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Jacome, A.; Gutierrez-Martinez, P.; Schiavoni, F.; Tenaglia, E.; Martinez, P.; Rodríguez-Acebes, S.; Lecona, E.; Murga, M.; Méndez, J.; Blasco, M.A.; et al. NSMCE2 suppresses cancer and aging in mice independently of its SUMO ligase activity. Embo J. 2015, 34, 2604–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, M.J.L.; Ravandi, F.; Patel, K.P.; Short, N.J. Prognostic and therapeutic implications of measurable residual disease in acute myeloid leukemia. J. Hematol. Oncol. 2021, 14, 137. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Mondal, A.K.; Ananth, S.; Saul, D.; Shams, S.; Hastie, A.R.; Savage, N.M.; Kota, V.; Chaubey, A.; Kolhe, R. Clinical Utility of Combined Optical Genome Mapping and 523-gene Next Generation Sequencing Panel For Comprehensive Evaluation of Myeloid Cancers. medRxiv 2022. Available online: https://www.medrxiv.org/content/10.1101/2022.01.15.22269355v1.full (accessed on 7 February 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pat.# | Sex | Age (Years) | Blast Count/Source | FAB Type | Risk Group * |
|---|---|---|---|---|---|
| 1 | m | 17 | 48% BM | M2 Auer positive | intermediate |
| 2 | m | 10 | 22% BM | M2 | standard |
| 3 | f | 3 | 44% BM | sec. AML | high |
| 4 | m | 16 | 97% BM | M5 | high |
| 5 | f | 1 | 87% BM | M4 | intermediate |
| 6 | m | 1 | 26% BM | M7 | high |
| 7 | m | 16 | 65% BM | M2 Auer positive | standard |
| 8 | m | 13 | 38% BM | M2 Auer positive | standard |
| 9 | m | 17 | 88% BM | M2 | intermediate |
| 10 | f | 14 | 71% BM | M4 eo | standard |
| 11 | f | 2 | 90% BM | M7 | high |
| 12 | m | 3 | 43% BM | M5 | intermediate |
| 13 | f | 5 | 95% BM | M5 | high |
| 14 | m | 1 | 23% BM | M7 | high |
| 15 | m | 5 | 82% BM | Bi-lineage leukemia | not applicable ** |
| 16 | m | 16 | 88% BM | M3 Auer positive | not applicable ** |
| 17 | m | 7 | 26% BM | M2 | high |
| 18 | m | 11 | 48% BM | M2 | standard |
| 19 | f | 13 | 43% PB | M2 | high |
| 20 | f | 5 | 73% PB | sec. AML after Ewing’s sarcoma | high |
| 21 | f | 5 | 90% BM | M1 Auer positive | high |
| 22 | m | 5 | 90% BM | M1 Auer positive | high |
| 23 | m | 10 | 48% BM | M1 | intermediate |
| 24 | f | 13 | 91% BM | MPAL | not applicable ** |
| Pat. # | Karyotyping | FISH | OGM (hg38) 1 | OGM Predicted Karyotype |
|---|---|---|---|---|
| 2 | 46,XY,t(8;21)(q22;q22),del(9)(q21q31) [8]/46,XY[7] | nuc ish 8q22(ETOx3),21q22(AML1x3)(ETO con AML1x2)[65/100] | ogm[GRCh38]46,XY,t(8;21)(q22.1;q22.12)(92059784;34850575),9q21.11q31.1(67717842_102668165)x1 | 46,XY,t(8;21)(q22.1;q22.12),del(9)(q21.11q31.1) |
| 7 | 46,XY,t(8;21)(q22;q22)[13]/46,XY[2] | nuc ish 8q22(ETOx3),21q22(AML1x3)(ETO con AML1x2)[83/100] | ogm[GRCh38]46,XY,t(8;21)(q21.3;q22.12)(92067075;34843977) | 46,XY,t(8;21)(q21.3;q22.12) |
| 8 | 45,X,−Y,t(8;21)(q22;q22)[14]/46,XY[1] | nuc ish cen7(CEP7x2),7q31(D7S486x2[100/100],cen8(CEP8x2)[99/100),8q22(ETOx3),21q22(AML1x3)(ETO con AML1x2)[95/100] | ogm[GRCh38]45,X,t(8;21)(q21.3;q22.12)(92059784;34855785),Yp11.32q12(11554_57212132)x1~2 | 45,X,-Y,t(8;21)(q21.3;q22.12) |
| 17 | 45,XY,−7[14]/46,idem,+8[6] | nuc ish 8q22(RUNX1T1x3),21q22(RUNX1x2)[12/100] | ogm[GRCh38]45,XY,7p22.3q36.3(10487_159334984)x1,8p23.3q24.3(61806_145076125)x2~3 | 46,XY,-7,+8 2 |
| 18 | 46,XY[25] | negative | ogm[GRCh38]46,XY | 46,XY |
| 23 | 46,XY[25] | negative | ogm[GRCh38]46,XY | 46,XY |
| 1 | 47,+8[6]/45,XY,−11[3]/46,XY[13] | nuc ish 8q22(RUNX1T1x3),21q22(RUNX1x2)[13/100] nuc ish 11q23(MLLx2)[100/100] 3 | ogm[GRCh38]47,XY, 8p23.3q24.3(208898_145076125)x2~3 | 47,XY,+8 |
| 12 | 46,XY,t(9;11)(p21;q23)[16]/ 47,idem,+8[4]/46,XY[1] | nuc ish 11q23(MLLx2)(5′MLL sep 3′MLLx1)[15/100] | ogm[GRCh38]46,XY,t(9;11)(p21.3;q23.3)(118493942;20375121) | 46,XY,t(9;11)(p21.3;q23.3) |
| 20 | 46,XX,t(11;19)(q23;p13)[8]/ 47,idem,+dmin[7] | nuc ish 11q23(MLLx2)(5′MLL sep 3′MLLx1)[77/100] | ogm[GRCh38]46,XX,t(11;19)(q23.3;p13.3)(118479068;6205232) | 46,XX,t(11;19)(q23.3;p13.3) |
| 4 | 46,XY,del(10)(q21q22),del(11)(q23)[12]/46,XY[3] | nuc ish (MLLx1)(5′MLL sep 3′MLLx1)[95/100] | ogm[GRCh38]46,XY,10p12.31(20591034_21642851)x1,10q21.1(56175397_57570855)x1,10q21.1q21.2(58331941_62944232)x1,10q22.2(73474864_75389039)x1,10q23.1(82757631_85834864)x1,t(10;11)(q21.1;q23.2)(58331941;113938345),19p13.11(19153167_19162215)x1 | 46,XY,der(10;11)t(10;11)(q21.1;q23.2),del(19)(p13.11) |
| 9 | 46,XY[25] | negativ | ogm[GRCh38]46,XY,1p34.1p32.3(44994758_53861278)x1 | 46,XY,del(1)(p34.1p32.3) |
| 10 | 46,XX,inv(16)(p13q22)[10]/ 48,idem,+8,+22[9]/46,XX[1] | nuc ish 8q22(RUNX1T1x3),21q22(RUNX1x2)[36/100],16q22(CBFBx2)(5′CBFB sep 3′CBFBx1)[64/100] | ogm[GRCh38]48,XX,7q31.2q34(117134687_139402873)x1,8p23.3q24.3(61805_145076125)x2~3,16p13.11(15709259_16475295)x1,16p13.11q22.1(15706265_67104157)inv,22p13q13.33(10514803_50805587)x2~3 | 48,XX,+8,+22, del(7)(q31.2q34) 4,del(16)(p13.11),inv(16)(p13.11q22.1) |
| 13 | 46,XX,t(10;11)(p12;q23)[13]/46,XX[7] | nuc ish 11q23(MLLx2)(5′MLL sep 3′MLLx1)[96/100] | ogm[GRCh38]46,XX,t(10;11)(p12.31;q23.3)(21653601;108120278),11q22.3q23.3(108113047_118493942)x1 | 46,XX,t(10;11)(p12.31;q23.3),del(11)(q22.3q23.3) |
| 15 | 46,XY,?t(17;19)(q22;q13)[20] | nuc ish 12p13(ETV6x3),21q22(RUNX1x2)[85/100],16q22(CBFBx2)[99/100],17q21.1(RARAx2)[98/100],19p13(E2Ax2)[98/100] | ogm[GRCh38]46,XY,2p11.2(87120538_87736106)x2~3,t(2;12)(p24.3;p13.2)(12771965;11724938),8q23.1(108722194_109473788)x2~3,t(17;19)(q22;q13.32)(58275730;45095385) | 46,XY,dup(2)(p11.2),t(2;12)(p24.3;p13.2),dup(8)(q23.1),t(17;19)(q22;q13.32) |
| 16 | 46, XY,t(15;17)(q24;q21),inc[15] | nuc ish 17q21.1(5′RARAx3,3′RARAx2)(5′RARA con 3′RARAx2)[93/100] | ogm[GRCh38]46,XY,13q21.32q34(65889491_114352102)x2~3,t(15;17)(q24.1;q21.2)(74029809;40335716) | 46,XY,+13q 2,t(15;17)(q24.1;q21.2) |
| 19 | 46,XX,t(6;9)(p22;q34)[15] | nuc ish 6p22(DEKx3),9q34(NUP214x3)(DEK con NUP214x2)[85/100] | ogm[GRCh38]46,XX,t(6;9)(p22.3;q34.13)(18232692;131152428),11p12(41618837_42297919)x2~3 | 46,XX,t(6;9)(p22.3;q34.13),dup(11)(p12) |
| 21 | 46~48,XX,der(9)del(9)(p21)del(9) (q22q33),+21,+21[cp15]/46,XX[5] | nuc ish 8q22(RUNX1T1x2),21q22(RUNX1x3~4)[70/100],9q34(ABL1x2),22q11(BCRx2)[97/100] | * not described, because of chromothripsis | 48,XX,cth(9),+21,+21 |
| 24 | 46,XX[25] | negative | ogm[GRCh38]46,XX,2q36.3(229862838_229886192)ins,9p12p11.2(39560026_40813220)x2~3,9q21.11(66749836_67475278)x2~3 | 46,XX,ins(2)(q36.3),dup(9)(p12p11.2),dup(9)(q21.11) |
| 3 | 46,XX,t(6;11)(q26;q23)[10]/ 46,idem,del(1)(q24q41)[4]/46,XX[1] | nuc ish 11q23(MLLx2)(5′MLL sep 3′MLLx1)[84/100] | ogm[GRCh38]46,XX,2q36.3q37.2(229708729_235034836)x1,t(2;6)(q31.3;q22.31)(124062016;180428380),t(4;8)(p15.33;q24.21)(180428380;124062017),t(6;11)(q27;11q23.3)(167843969;118493942),t(8;14)(q11.21;q32.33)(47525976;105869036),8p23.1q24.21(10764562_129368956)x1 | 46,XX,del(2)(q36.3q37.2),t(2;6)(q31.3;q22.31),t(4;8)(p15.33;q24.21),t(6;11)(q27;q23.3),del(8)(p23.1q24.21),t(8;14)(q11.21;q32.33) |
| 5 | 46,XX,t(9;11)(p21,q23)[2]/ 46,idem,der(21)t(8;21)(q21;p13)[7]/ 46,XX[6] | nuc ish 8q22(RUNX1T1x3),21q22(RUNX1x2)[72/100],11q23(MLLx2)(5′MLL sep 3′MLLx1)[94/100] | ogm[GRCh38]46,XX,8q13.1q24.3(66259972_142032191)2~3,t(9p24.3;11q23.3)(21118146;118493942),t(14q24.3;17q21.32)(73466593;49017556),14q24.3q32.33(73473834_106873282)x1~2,17q21.32q25.3(49017556_83246392)x2~3 | 46,XX,+8q 5,t(9;11)(p24.3;q23.3),t(14;17)(q24.3;q21.32),del(14)(q24.3q32.33),dup(17)(q21.32q25.3) |
| 6 | 48,XX,del(6)(q16q25),der(7)t(7;13) (p11;p11),+der(8)t(1;8)(q21;p11),+10 [17]/46,XX[4] | nuc ish 8q22(RUNX1T1x3),21q22(RUNX1x2)[67/100] | ogm[GRCh38]47,XX,1q21.1q44(143278152_248943333)x2~3,t(6;7)(q21;p15.2)(109354469;27165827),8q11.1q24.3(45972483_145076125)x2~3,10p15.3q26.3(18514_133785266)x2~3 | 47,XX,+1q,t(6;7)(q21;p15.2),+8q,+10 |
| 11 | 47,XX,+21[2]/49,idem,+6,+20[5]/ 92,XXYY[1]/46,XX[18] | nuc ish 3q26(EVIx4)[7/100],8q22(RUNX1T1x2),21q22(RUNX1x3)[37/100],11q23(MLLx4)[6/100],16q22(CBFBx4)[6/100]nuc ish 6q23(MYBx3)[19/100],20q12(D20S108x3)[16/100],21q22 (AML1x3)[21/100] 3 | ogm[GRCh38]46,XX,16p13.3q24.3(4331324_88868384)inv | 46,XX,inv(16)(p13.3q24.3) |
| 14 | 48,XY,+6,+6[1]/48,idem,del(3)(q13q26),add(11)(p14)[23]/46,XY[1] | - | ogm[GRCh38]48,XY,3q13.12q25.31(107775421_167698608)x1,3q25.31q26.1(156499437_167703086)inv,6p25.3q27(76216_170739897)x3~4,t(11;17)(p15.4;q24.2)(3733790;67956414) | 48,XY,del(3)(q13.12q25.31),inv(3)(q25.31q26.1),+6,+6,t(11;17)(p15.4;q24.2) |
| 22 | 45,XY,der(4)del(4)(p11p15)del(4) (q11q24),−7,add(13)(p11), t(17;19)(q22;q13)[20] | nuc ish 8q22(RUNX1T1x2),21q22(RUNX1x2)[100/100] | ogm[GRCh38]46,XY,1p36.13p35.2(14490385_32907733)x1,4p12(44197510_49078708)x1,4q12q13.1(51826792_63679087)x1,t(4;7)(p15.1;p21.3)(32503668;9394985),7q11.21q36.3(62995089_157624118)x1,t(8;12)(q24.13;12p13.2)(125189890;11672773),13q14.2q14.3(48278417_51484127)x1,15q15.1(40315717_41591804)x1~2,16p13.2p12.3(9863863_16429873)x1~2,t(17;19)(q22;q13.32)(58275730;45095385),22q12.2q12.3(28643629_34061240)x1~2 | 46,XY,del(1)(p36.13p35.2),del(4)(p12),del(4)(q12q13.1),t(4;7)(p15.1;p21.3),del(7)(q11.21q36.3),t(8;12)(q24.13;p13.2),del(13)(q14.2q14.3),del(15)(q15.1),del(16)(p13.2p12.3),t(17;19)(q22;q13.32),del(22)(q12.2q12.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suttorp, J.; Lühmann, J.L.; Behrens, Y.L.; Göhring, G.; Steinemann, D.; Reinhardt, D.; Neuhoff, N.v.; Schneider, M. Optical Genome Mapping as a Diagnostic Tool in Pediatric Acute Myeloid Leukemia. Cancers 2022, 14, 2058. https://doi.org/10.3390/cancers14092058
Suttorp J, Lühmann JL, Behrens YL, Göhring G, Steinemann D, Reinhardt D, Neuhoff Nv, Schneider M. Optical Genome Mapping as a Diagnostic Tool in Pediatric Acute Myeloid Leukemia. Cancers. 2022; 14(9):2058. https://doi.org/10.3390/cancers14092058
Chicago/Turabian StyleSuttorp, Julia, Jonathan Lukas Lühmann, Yvonne Lisa Behrens, Gudrun Göhring, Doris Steinemann, Dirk Reinhardt, Nils von Neuhoff, and Markus Schneider. 2022. "Optical Genome Mapping as a Diagnostic Tool in Pediatric Acute Myeloid Leukemia" Cancers 14, no. 9: 2058. https://doi.org/10.3390/cancers14092058