
Endothelial Cell Metabolism in Vascular Functions


Abstract
:Simple Summary
Abstract
1. Introduction
2. The Origin of the Endothelial Cells
3. Angiogenesis in Cancer
4. Overview of Cellular Metabolism
5. The Basis of Endothelial Cell Activation
6. Metabolic State of the Endothelial Cell in the Adult Vasculature
7. Endothelial Cell Metabolism, Angiogenesis and Inflammation
8. Endothelial Cell Metabolism and Viral Infection
9. Involvement of Plasma Membrane Microdomains to Endothelial Cell Metabolism
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Al-Soudi, A.; Kaaij, M.H.; Tas, S.W. Endothelial cells: From innocent bystanders to active participants in immune responses. Autoimmun. Rev. 2017, 16, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, K.; Zieger, B. Endothelial cells and coagulation. Cell Tissue Res. 2022, 387, 391–398. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Xia, P.; Wang, H.; Tu, J.; Liang, X.; Zhang, X.; Li, L. The endothelial tip-stalk cell selection and shuffling during angiogenesis. J. Cell Commun. Signal. 2019, 13, 291–301. [Google Scholar] [CrossRef]
- Machado, M.J.C.; Watson, M.G.; Devlin, A.H.; Chaplain, M.; McDougall, S.R.; Mitchell, C.A. Dynamics of Angiogenesis During Wound Healing: A Coupled In Vivo and In Silico Study. Microcirculation 2011, 18, 183–197. [Google Scholar] [CrossRef]
- Aird, W.C. Endothelium as an organ system. Crit. Care Med. 2004, 32, S271–S279. [Google Scholar] [CrossRef]
- Florey, L. The endothelial cell. BMJ 1966, 2, 487–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. Phenotypic Heterogeneity of the Endothelium. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic Heterogeneity of the Endothelium. Circ. Res. 2007, 100, 174–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. Endothelial Cell Heterogeneity. Cold Spring Harb. Perspect. Med. 2021, 2, a006429. [Google Scholar] [CrossRef]
- Kalucka, J.; de Rooij, L.P.M.H.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.; Taverna, F.; Teuwen, L.-A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e20. [Google Scholar] [CrossRef] [PubMed]
- Bruns, R.R.; Palade, G.E. Studies on blood capillaries. I. General organization of blood capillaries in muscle. J. Cell Biol. 1968, 37, 244–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwell, D.; Mishra, A.; Hall, C.N.; O’Farrell, F.M.; Dalkara, T. What is a pericyte? J. Cereb. Blood Flow Metab. 2016, 36, 451–455. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Mäe, M.A.; Muhl, L.; Sun, Y.; Pietilä, R.; Nahar, K.; Liébanas, E.V.; Fagerlund, M.J.; Oldner, A.; Liu, J.; et al. Pericyte-specific vascular expression of SARS-CoV-2 receptor ACE2—Implications for microvascular inflammation and hypercoagulopathy in COVID-19. bioRxiv 2020. [Google Scholar] [CrossRef]
- Török, O.; Schreiner, B.; Schaffenrath, J.; Tsai, H.-C.; Maheshwari, U.; Stifter, S.A.; Welsh, C.; Amorim, A.; Sridhar, S.; Utz, S.G.; et al. Pericytes regulate vascular immune homeostasis in the CNS. Proc. Natl. Acad. Sci. USA 2021, 118, e2016587118. [Google Scholar] [CrossRef]
- Figueiredo, A.M.; Villacampa, P.; Diéguez-Hurtado, R.; Lozano, J.J.; Kobialka, P.; Cortazar, A.R.; Martinez-Romero, A.; Angulo-Urarte, A.; Franco, C.; Claret, M.; et al. Phosphoinositide 3-Kinase–Regulated Pericyte Maturation Governs Vascular Remodeling. Circulation 2020, 142, 688–704. [Google Scholar] [CrossRef]
- Uemura, M.T.; Maki, T.; Ihara, M.; Lee, V.M.Y.; Trojanowski, J.Q. Brain Microvascular Pericytes in Vascular Cognitive Impairment and Dementia. Front. Aging Neurosci. 2020, 12, 80. [Google Scholar] [CrossRef] [Green Version]
- Ferland-McCollough, D.; Slater, S.; Richard, J.; Reni, C.; Mangialardi, G. Pericytes, an overlooked player in vascular pathobiology. Pharmacol. Ther. 2016, 171, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Friederici, H. The tridimensional ultrastructure of fenestrated capillaries. J. Ultrastruct. Res. 1968, 23, 444–456. [Google Scholar] [CrossRef]
- Griffin, C.T.; Gao, S. Building discontinuous liver sinusoidal vessels. J. Clin. Investig. 2017, 127, 790–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, J.-P.; Springer, T.A. High endothelial venules (HEVs): Specialized endothelium for lymphocyte migration. Immunol. Today 1995, 16, 449–457. [Google Scholar] [CrossRef]
- Liu, M.; Kluger, M.S.; D’Alessio, A.; García-Cardeña, G.; Pober, J.S. Regulation of Arterial-Venous Differences in Tumor Necrosis Factor Responsiveness of Endothelial Cells by Anatomic Context. Am. J. Pathol. 2008, 172, 1088–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquier, J.; Ghiabi, P.; Chouchane, L.; Razzouk, K.; Rafii, S.; Rafii, A. Angiocrine endothelium: From physiology to cancer. J. Transl. Med. 2020, 18, 52. [Google Scholar] [CrossRef]
- Grasman, J.M.; Kaplan, D.L. Human endothelial cells secrete neurotropic factors to direct axonal growth of peripheral nerves. Sci. Rep. 2017, 7, 409. [Google Scholar] [CrossRef]
- Rafii, S.; Butler, J.M.; Ding, B.-S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ferkowicz, M.J.; Johnson, S.A.; Shelley, W.C.; Yoder, M.C. Endothelial Cells in the Early Murine Yolk Sac Give Rise to CD41-expressing Hematopoietic Cells. Stem Cells Dev. 2005, 14, 44–54. [Google Scholar] [CrossRef]
- Ferkowicz, M.J.; Yoder, M.C. Blood island formation: Longstanding observations and modern interpretations. Exp. Hematol. 2005, 33, 1041–1047. [Google Scholar] [CrossRef]
- Shalaby, F.; Rossant, J.; Yamaguchi, T.P.; Gertsenstein, M.; Wu, X.-F.; Breitman, M.L.; Schuh, A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376, 62–66. [Google Scholar] [CrossRef]
- Eichmann, A.; Yuan, L.; Moyon, D.; Lenoble, F.; Pardanaud, L.; Bréant, C. Vascular development: From precursor cells to branched arterial and venous networks. Int. J. Dev. Biol. 2005, 49, 259–267. [Google Scholar] [CrossRef]
- Risau, W.; Flamme, I. Vasculogenesis. Annu. Rev. Cell Dev. Biol. 1995, 11, 73–91. [Google Scholar] [CrossRef]
- Poole, T.J.; Coffin, J.D. Vasculogenesis and angiogenesis: Two distinct morphogenetic mechanisms establish embryonic vascular pattern. J. Exp. Zool. 1989, 251, 224–231. [Google Scholar] [CrossRef]
- Coffin, J.; Poole, T. Embryonic vascular development: Immunohistochemical identification of the origin and subsequent morphogenesis of the major vessel primordia in quail embryos. Development 1988, 102, 735–748. [Google Scholar] [CrossRef]
- Risau, W.; Sariola, H.; Zerwes, H.; Sasse, J.; Ekblom, P.; Kemler, R.; Doetschman, T. Vasculogenesis and angiogenesis in embryonic-stem-cell-derived embryoid bodies. Development 1988, 102, 471–478. [Google Scholar] [CrossRef]
- Klagsbrun, M.; D’Amore, P.A. Regulators of Angiogenesis. Annu. Rev. Physiol. 1991, 53, 217–239. [Google Scholar] [CrossRef]
- Folkman, J.; Klagsbrun, M. Angiogenic factors. Science 1987, 235, 442–447. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Cleaver, O. Tubulogenesis during blood vessel formation. Semin. Cell Dev. Biol. 2011, 22, 993–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zovein, A.; Luque, A.; Turlo, K.A.; Hofmann, J.J.; Yee, K.M.; Becker, M.S.; Fassler, R.; Mellman, I.; Lane, T.F.; Iruela-Arispe, M.L. β1 Integrin Establishes Endothelial Cell Polarity and Arteriolar Lumen Formation via a Par3-Dependent Mechanism. Dev. Cell 2010, 18, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–966. [Google Scholar] [CrossRef]
- Keighron, C.; Lyons, C.J.; Creane, M.; O’Brien, T.; Liew, A. Recent advances in endothelial progenitor cells toward their use in clinical translation. Front. Med. 2018, 5, 354. [Google Scholar] [CrossRef] [Green Version]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef]
- Asahara, T.; Takahashi, T.; Masuda, H.; Kalka, C.; Chen, D.; Iwaguro, H.; Inai, Y.; Silver, M.; Isner, J.M. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999, 18, 3964–3972. [Google Scholar] [CrossRef] [Green Version]
- Asahara, T.; Masuda, H.; Takahashi, T.; Kalka, C.; Pastore, C.; Silver, M.; Kearne, M.; Magner, M.; Isner, J.M. Bone Marrow Origin of Endothelial Progenitor Cells Responsible for Postnatal Vasculogenesis in Physiological and Pathological Neovascularization. Circ. Res. 1999, 85, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. New Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Montemagno, C.; Pagès, G. Resistance to Anti-angiogenic Therapies: A Mechanism Depending on the Time of Exposure to the Drugs. Front. Cell Dev. Biol. 2020, 8, 584. [Google Scholar] [CrossRef]
- Wang, F.-T.; Sun, W.; Zhang, J.-T.; Fan, Y.-Z. Cancer-associated fibroblast regulation of tumor neo-angiogenesis as a therapeutic target in cancer (Review). Oncol. Lett. 2019, 17, 3055–3065. [Google Scholar] [CrossRef] [Green Version]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef]
- Rose, A.J. Rose Amino Acid Nutrition and Metabolism in Health and Disease. Nutrients 2019, 11, 2623. [Google Scholar] [CrossRef] [Green Version]
- Dashty, M. A quick look at biochemistry: Carbohydrate metabolism. Clin. Biochem. 2013, 46, 1339–1352. [Google Scholar] [CrossRef]
- Skrede, S.; Sørensen, H.N.; Larsen, L.N.; Steineger, H.H.; Høvik, K.; Spydevold, S.; Horn, R.; Bremer, J. Thia fatty acids, metabolism and metabolic effects. Biochim. Biophys. Acta Lipids Lipid Metab. 1997, 1344, 115–131. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Thompson, C.B. Cellular Metabolism and Disease: What Do Metabolic Outliers Teach Us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Zecchin, A.; Stapor, P.C.; Goveia, J.; Carmeliet, P. Metabolic pathway compartmentalization: An underappreciated opportunity? Curr. Opin. Biotechnol. 2015, 34, 73–81. [Google Scholar] [CrossRef]
- Martin, W. Evolutionary origins of metabolic compartmentalization in eukaryotes. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 847–855. [Google Scholar] [CrossRef] [Green Version]
- Willms-Kretschmer, K.; Flax, M.H.; Cotran, R.S. The fine structure of the vascular response in hapten-specific delayed hypersensitivity and contact dermatitis. Lab. Investig. 1967, 17, 334–349. [Google Scholar]
- Collins, T.; Korman, A.J.; Wake, C.T.; Boss, J.M.; Kappes, D.J.; Fiers, W.; Ault, K.A.; Gimbrone, M.A.; Strominger, J.L.; Pober, J.S. Immune interferon activates multiple class II major histocompatibility complex genes and the associated invariant chain gene in human endothelial cells and dermal fibroblasts. Proc. Natl. Acad. Sci. USA 1984, 81, 4917–4921. [Google Scholar] [CrossRef] [Green Version]
- Pober, J.S.; Collins, T.; Gimbrone, M.A.; Cotran, R.S.; Gitlin, J.D.; Fiers, W.; Clayberger, C.; Krensky, A.M.; Burakoff, S.J.; Reiss, C.S. Lymphocytes recognize human vascular endothelial and dermal fibroblast Ia antigens induced by recombinant immune interferon. Nature 1983, 305, 726–729. [Google Scholar] [CrossRef]
- Pober, J.S.; Gimbrone, M.A.; Cotran, R.S.; Reiss, C.S.; Burakoff, S.J.; Fiers, W.; Ault, K.A. Ia expression by vascular endothelium is inducible by activated T cells and by human gamma interferon. J. Exp. Med. 1983, 157, 1339–1353. [Google Scholar] [CrossRef]
- Pober, J.S. Warner-Lambert/Parke-Davis award lecture. Cytokine-mediated activation of vascular endothelium. Physiology and pathology. Am. J. Pathol. 1988, 133, 426–433. [Google Scholar] [PubMed]
- Mantovani, A.; Sozzani, S.; Vecchi, A.; Introna, M.; Allavena, P. Cytokine Activation of Endothelial Cells: New Molecules for an Old Paradigm. Thromb. Haemost. 1997, 78, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Shaw, S. Leucocyte-endothelial interactions and regulation of leucocyte migration. Lancet 1994, 343, 831–836. [Google Scholar] [CrossRef]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Pober, J.S.; Min, W.; Bradley, J.R. Mechanisms of Endothelial Dysfunction, Injury, and Death. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 71–95. [Google Scholar] [CrossRef]
- Bach, F.; Robson, S.; Winkler, H.; Ferran, C.; Stuhlmeier, K.; Wrighton, C.; Hancock, W. Barriers to xenotransplantation. Nat. Med. 1995, 1, 869–873. [Google Scholar] [CrossRef]
- Sehnert, B.; Burkhardt, H.; Wessels, J.T.; Schröder, A.; May, M.J.; Vestweber, D.; Zwerina, J.; Warnatz, K.; Nimmerjahn, F.; Schett, G.; et al. NF-κB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-κB in immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 16556–16561. [Google Scholar] [CrossRef] [Green Version]
- Denk, A.; Goebeler, M.; Schmid, S.; Berberich, I.; Ritz, O.; Lindemann, D.; Ludwig, S.; Wirth, T. Activation of NF-κB via the IκB Kinase Complex Is Both Essential and Sufficient for Proinflammatory Gene Expression in Primary Endothelial Cells. J. Biol. Chem. 2001, 276, 28451–28458. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): A dual regulator for angiogenesis. Angiogenesis 2006, 9, 225–230. [Google Scholar] [CrossRef]
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H.; et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 2004, 5, 74–80. [Google Scholar] [CrossRef]
- Mäkinen, T.; Veikkola, T.; Mustjoki, S.; Karpanen, T.; Catimel, B.; Nice, E.C.; Wise, L.; Mercer, A.; Kowalski, H.; Kerjaschki, D.; et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 2001, 20, 4762–4773. [Google Scholar] [CrossRef] [Green Version]
- Heinolainen, K.; Karaman, S.; D’Amico, G.; Tammela, T.; Sormunen, R.; Eklund, L.; Alitalo, K.; Zarkada, G. VEGFR3 Modulates Vascular Permeability by Controlling VEGF/VEGFR2 Signaling. Circ. Res. 2017, 120, 1414–1425. [Google Scholar] [CrossRef]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380, 435–439. [Google Scholar] [CrossRef]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [Green Version]
- Davis, G.E.; Senger, D.R. Endothelial Extracellular Matrix. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef] [Green Version]
- Seano, G.; Chiaverina, G.; Gagliardi, P.A.; di Blasio, L.; Puliafito, A.; Bouvard, C.; Sessa, R.; Tarone, G.; Sorokin, L.; Helley, D.; et al. Endothelial podosome rosettes regulate vascular branching in tumour angiogenesis. Nat. Cell Biol. 2014, 16, 931–938. [Google Scholar] [CrossRef]
- Varon, C.; Tatin, F.; Moreau, V.; van Obberghen-Schilling, E.; Fernandez-Sauze, S.; Reuzeau, E.; Kramer, I.; Génot, E. Transforming Growth Factor β Induces Rosettes of Podosomes in Primary Aortic Endothelial Cells. Mol. Cell. Biol. 2006, 26, 3582–3594. [Google Scholar] [CrossRef] [Green Version]
- Rottiers, P.; Saltel, F.; Daubon, T.; Chaigne-Delalande, B.; Tridon, V.; Billottet, C.; Reuzeau, E.; Génot, E. TGFβ-induced endothelial podosomes mediate basement membrane collagen degradation in arterial vessels. J. Cell Sci. 2009, 122, 4311–4318. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- De Smet, F.; Segura, I.; de Bock, K.; Hohensinner, P.J.; Carmeliet, P. Mechanisms of Vessel Branching. Arter. Thromb. Vasc. Biol. 2009, 29, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Aquila, G.; Kostina, A.; Sega, F.V.D.; Shlyakhto, E.; Kostareva, A.; Marracino, L.; Ferrari, R.; Rizzo, P.; Malaschicheva, A. The Notch pathway: A novel therapeutic target for cardiovascular diseases? Expert Opin. Ther. Targets 2019, 23, 695–710. [Google Scholar] [CrossRef]
- Phng, L.-K.; Gerhardt, H. Angiogenesis: A Team Effort Coordinated by Notch. Dev. Cell 2009, 16, 196–208. [Google Scholar] [CrossRef]
- Suchting, S.; Freitas, C.; le Noble, F.; Benedito, R.; Bréant, C.; Duarte, A.; Eichmann, A. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc. Natl. Acad. Sci. USA 2007, 104, 3225–3230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellström, M.; Phng, L.-K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.-K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.; Hirashima, M.; Benedito, R.; Trindade, A.; Diniz, P.; Bekman, E.; Costa, L.; Henrique, D.; Rossant, J. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004, 18, 2474–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gale, N.W.; Dominguez, M.G.; Noguera, I.; Pan, L.; Hughes, V.; Valenzuela, D.M.; Murphy, A.J.; Adams, N.C.; Lin, H.C.; Holash, J.; et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc. Natl. Acad. Sci. USA 2004, 101, 15949–15954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siekmann, A.F.; Lawson, N. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 2007, 445, 781–784. [Google Scholar] [CrossRef]
- Weinstein, B.M. Vessels and Nerves: Marching to the Same Tune. Cell 2005, 120, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Eichmann, A.; Makinen, T.; Alitalo, K. Neural guidance molecules regulate vascular remodeling and vessel navigation. Genes Dev. 2005, 19, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Tessier-Lavigne, M. Common mechanisms of nerve and blood vessel wiring. Nature 2005, 436, 193–200. [Google Scholar] [CrossRef]
- Tamagnone, L.; Mazzone, M. Semaphorin Signals on the Road of Endothelial Tip Cells. Dev. Cell 2011, 21, 189–190. [Google Scholar] [CrossRef] [Green Version]
- Mukouyama, Y.-S.; Gerber, H.-P.; Ferrara, N.; Gu, C.; Anderson, D.J. Peripheral nerve-derived VEGF promotes arterial differentiation via neuropilin 1-mediated positive feedback. Development 2005, 132, 941–952. [Google Scholar] [CrossRef] [Green Version]
- Mukouyama, Y.-S.; Shin, D.; Britsch, S.; Taniguchi, M.; Anderson, D.J. Sensory Nerves Determine the Pattern of Arterial Differentiation and Blood Vessel Branching in the Skin. Cell 2002, 109, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Tamagnone, L. Emerging Role of Semaphorins as Major Regulatory Signals and Potential Therapeutic Targets in Cancer. Cancer Cell 2012, 22, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.; Bando, Y.; Sato, K. Involvement of Netrins and Their Receptors in Neuronal Migration in the Cerebral Cortex. Front. Cell Dev. Biol. 2020, 8, 590009. [Google Scholar] [CrossRef]
- Dai, C.; Gong, Q.; Cheng, Y.; Su, G. Regulatory mechanisms of Robo4 and their effects on angiogenesis. Biosci. Rep. 2019, 39, BSR20190513. [Google Scholar] [CrossRef] [Green Version]
- Filippini, A.; D’Amore, A.; D’Alessio, A. Calcium Mobilization in Endothelial Cell Functions. Int. J. Mol. Sci. 2019, 20, 4525. [Google Scholar] [CrossRef] [Green Version]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2–dependent Ca 2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef] [Green Version]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [Green Version]
- Leung, S.W.S.; Shi, Y. The glycolytic process in endothelial cells and its implications. Acta Pharmacol. Sin. 2021, 43, 251–259. [Google Scholar] [CrossRef]
- Li, X.; Kumar, A.; Carmeliet, P. Metabolic Pathways Fueling the Endothelial Cell Drive. Annu. Rev. Physiol. 2019, 81, 483–503. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Locasale, J.W.; Cantley, L.C. Metabolic Flux and the Regulation of Mammalian Cell Growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Yetkin-Arik, B.; Vogels, I.M.C.; Neyazi, N.; Van Duinen, V.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. Endothelial tip cells in vitro are less glycolytic and have a more flexible response to metabolic stress than non-tip cells. Sci. Rep. 2019, 9, 10414. [Google Scholar] [CrossRef] [Green Version]
- Dryden, N.H.; Sperone, A.; Martin-Almedina, S.; Hannah, R.L.; Birdsey, G.M.; Khan, S.T.; Layhadi, J.A.; Mason, J.C.; Haskard, D.O.; Göttgens, B.; et al. The Transcription Factor Erg Controls Endothelial Cell Quiescence by Repressing Activity of Nuclear Factor (NF)-κB p65. J. Biol. Chem. 2012, 287, 12331–12342. [Google Scholar] [CrossRef] [Green Version]
- Andrade, J.; Shi, C.; Costa, A.S.H.; Choi, J.; Kim, J.; Doddaballapur, A.; Sugino, T.; Ong, Y.T.; Castro, M.; Zimmermann, B.; et al. Control of endothelial quiescence by FOXO-regulated metabolites. Nat. Cell Biol. 2021, 23, 413–423. [Google Scholar] [CrossRef]
- Sun, J.-X.; Dou, G.-R.; Yang, Z.-Y.; Liang, L.; Duan, J.-L.; Ruan, B.; Li, M.-H.; Chang, T.-F.; Xu, X.-Y.; Chen, J.-J.; et al. Notch activation promotes endothelial quiescence by repressing MYC expression via miR-218. Mol. Ther. Nucleic Acids 2021, 25, 554–566. [Google Scholar] [CrossRef]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Doddaballapur, A.; Michalik, K.M.; Manavski, Y.; Lucas, T.; Houtkooper, R.H.; You, X.; Chen, W.; Zeiher, A.M.; Potente, M.; Dimmeler, S.; et al. Laminar Shear Stress Inhibits Endothelial Cell Metabolism via KLF2-Mediated Repression of PFKFB3. Arter. Thromb. Vasc. Biol. 2015, 35, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Rohlenova, K.; Goveia, J.; García-Caballero, M.; Subramanian, A.; Kalucka, J.; Treps, L.; Falkenberg, K.D.; de Rooij, L.P.; Zheng, Y.; Lin, L.; et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab. 2020, 31, 862.e14–877.e14. [Google Scholar] [CrossRef]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef] [PubMed]
- West, J.B. Ibn al-Nafis, the pulmonary circulation, and the Islamic Golden Age. J. Appl. Physiol. 2008, 105, 1877–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racker, E. From Pasteur to Mitchell: A hundred years of bioenergetics. Fed. Proc. 1980, 39, 210–215. [Google Scholar] [PubMed]
- Krebs, H.A. The History of the Tricarboxylic Acid Cycle. Perspect. Biol. Med. 1970, 14, 154–170. [Google Scholar] [CrossRef]
- Barnett, J.A. A history of research on yeasts 2: Louis Pasteur and his contemporaries, 1850–1880. Yeast 2000, 16, 755–771. [Google Scholar] [CrossRef]
- Heidenreich, R.; Röcken, M.; Ghoreschi, K. Angiogenesis drives psoriasis pathogenesis. Int. J. Exp. Pathol. 2009, 90, 232–248. [Google Scholar] [CrossRef]
- Ma, H.H.; Liutkevičienė, R. Age-Related Macular Degeneration: What Do We Know So Far? Acta Medica Litu. 2021, 28, 36–47. [Google Scholar] [CrossRef]
- Droho, S.; Cuda, C.M.; Perlman, H.; Lavine, J.A. Macrophage-derived interleukin-6 is necessary and sufficient for choroidal angiogenesis. Sci. Rep. 2021, 11, 18084. [Google Scholar] [CrossRef]
- Cheng, R.; Ma, J.-X. Angiogenesis in diabetes and obesity. Rev. Endocr. Metab. Disord. 2015, 16, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Okonkwo, U.A.; Chen, L.; Ma, D.; Haywood, V.A.; Barakat, M.; Urao, N.; DiPietro, L.A. Compromised angiogenesis and vascular Integrity in impaired diabetic wound healing. PLoS ONE 2020, 15, e0231962. [Google Scholar] [CrossRef]
- Khurana, R.; Simons, M.; Martin, J.F.; Zachary, I.C. Role of Angiogenesis in Cardiovascular Disease. Circulation 2005, 112, 1813–1824. [Google Scholar] [CrossRef]
- Zachary, I.; Morgan, R.D. Therapeutic angiogenesis for cardiovascular disease: Biological context, challenges, prospects. Heart 2011, 97, 181–189. [Google Scholar] [CrossRef]
- Seto, S.-W.; Chang, D.; Jenkins, A.; Bensoussan, A.; Kiat, H. Angiogenesis in Ischemic Stroke and Angiogenic Effects of Chinese Herbal Medicine. J. Clin. Med. 2016, 5, 56. [Google Scholar] [CrossRef]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Ntellas, P.; Mavroeidis, L.; Gkoura, S.; Gazouli, I.; Amylidi, A.-L.; Papadaki, A.; Zarkavelis, G.; Mauri, D.; Karpathiou, G.; Kolettas, E.; et al. Old Player-New Tricks: Non Angiogenic Effects of the VEGF/VEGFR Pathway in Cancer. Cancers 2020, 12, 3145. [Google Scholar] [CrossRef]
- Solimando, A.G.; De Summa, S.; Vacca, A.; Ribatti, D. Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling. Cancers 2020, 12, 3380. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.S.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Correction: Corrigendum: Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 526, 144. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.-M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous Deficiency of PHD2 Restores Tumor Oxygenation and Inhibits Metastasis via Endothelial Normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Girdhar, K.; Powis, A.; Raisingani, A.; Chrudinová, M.; Huang, R.; Tran, T.; Sevgi, K.; Dogru, Y.D.; Altindis, E. Viruses and Metabolism: The Effects of Viral Infections and Viral Insulins on Host Metabolism. Annu. Rev. Virol. 2021, 8, 373–391. [Google Scholar] [CrossRef]
- Sumbria, D.; Berber, E.; Mathayan, M.; Rouse, B.T. Virus Infections and Host Metabolism—Can We Manage the Interactions? Front. Immunol. 2020, 11, 594963. [Google Scholar] [CrossRef]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the Cellular Metabolome during Human Cytomegalovirus Infection. PLOS Pathog. 2006, 2, e132. [Google Scholar] [CrossRef] [Green Version]
- Thai, M.; Graham, N.A.; Braas, D.; Nehil, M.; Komisopoulou, E.; Kurdistani, S.; McCormick, F.; Graeber, T.; Christofk, H.R. Adenovirus E4ORF1-Induced MYC Activation Promotes Host Cell Anabolic Glucose Metabolism and Virus Replication. Cell Metab. 2014, 19, 694–701. [Google Scholar] [CrossRef] [Green Version]
- Ripoli, M.; D’Aprile, A.; Quarato, G.; Sarasin-Filipowicz, M.; Gouttenoire, J.; Scrima, R.; Cela, O.; Boffoli, D.; Heim, M.H.; Moradpour, D.; et al. Hepatitis C Virus-Linked Mitochondrial Dysfunction Promotes Hypoxia-Inducible Factor 1α-Mediated Glycolytic Adaptation. J. Virol. 2010, 84, 647–660. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Van Gorp, E.C.M.; Suharti, C.; Cate, H.T.; Dolmans, W.M.V.; van der Meer, J.W.M.; Cate, J.W.T.; Brandjes, D.P.M. Review: Infectious Diseases and Coagulation Disorders. J. Infect. Dis. 1999, 180, 176–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, H.M. Infection of Endothelial Cells by Common Human Viruses. Clin. Infect. Dis. 1989, 11 (Suppl. 4), S700–S704. [Google Scholar] [CrossRef]
- Singh, S.; Singh, P.K.; Suhail, H.; Arumugaswami, V.; Pellett, P.E.; Giri, S.; Kumar, A. AMP-Activated Protein Kinase Restricts Zika Virus Replication in Endothelial Cells by Potentiating Innate Antiviral Responses and Inhibiting Glycolysis. J. Immunol. 2020, 204, 1810–1824. [Google Scholar] [CrossRef]
- Delgado, T.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Global Metabolic Profiling of Infection by an Oncogenic Virus: KSHV Induces and Requires Lipogenesis for Survival of Latent Infection. PLOS Pathog. 2012, 8, e1002866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, T.; Carroll, P.A.; Punjabi, A.S.; Margineantu, D.; Hockenbery, D.M.; Lagunoff, M. Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10696–10701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Han, K.; Blair, R.; Kenst, K.; Qin, Z.; Upcin, B.; Wörsdörfer, P.; Midkiff, C.C.; Mudd, J.; Belyaeva, E.; et al. SARS-CoV-2 Infects Endothelial Cells In Vivo and In Vitro. Front. Cell. Infect. Microbiol. 2021, 11, 701278. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.M.; Macarak, E.J.; MacGregor, R.R.; Wolfe, J.; Kefalides, N.A. Virus Infection of Endothelial Cells. J. Infect. Dis. 1981, 143, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Mubarik, S.; Liu, X.; Eshak, E.S.; Liu, K.; Liu, Q.; Wang, F.; Shi, F.; Wen, H.; Bai, J.; Yu, C.; et al. The Association of Hypertension with the Severity of and Mortality From the COVID-19 in the Early Stage of the Epidemic in Wuhan, China: A Multicenter Retrospective Cohort Study. Front. Med. 2021, 8, 623608. [Google Scholar] [CrossRef] [PubMed]
- Dan, S.; Pant, M.; Upadhyay, S.K. The Case Fatality Rate in COVID-19 Patients with Cardiovascular Disease: Global Health Challenge and Paradigm in the Current Pandemic. Curr. Pharmacol. Rep. 2020, 6, 315–324. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Clementi, N.; Scagnolari, C.; D’Amore, A.; Palombi, F.; Criscuolo, E.; Frasca, F.; Pierangeli, A.; Mancini, N.; Antonelli, G.; Clementi, M.; et al. Naringenin is a powerful inhibitor of SARS-CoV-2 infection in vitro. Pharmacol. Res. 2021, 163, 105255. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Structure of Detergent-Resistant Membrane Domains: Does Phase Separation Occur in Biological Membranes? Biochem. Biophys. Res. Commun. 1997, 240, 1–7. [Google Scholar] [CrossRef]
- Moldovan, N.I.; Heltianu, C.; Simionescu, N.; Simionescu, M. Ultrastructural Evidence of Differential Solubility in Triton X-100 of Endothelial Vesicles and Plasma Membrane. Exp. Cell Res. 1995, 219, 309–313. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Palade, G.E. Fine structure of blood capillaries. J. Appl. Phys. 1953, 24, 1424. [Google Scholar] [CrossRef] [Green Version]
- Yamada, E. The fine structure of the gall bladder epithelium of the mouse. J. Biophys. Biochem. Cytol. 1955, 1, 445–458. [Google Scholar] [CrossRef] [Green Version]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.-S.; Glenney, J.R.; Anderson, R.G. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Scherer, P.E.; Okamoto, T.; Chun, M.; Nishimoto, I.; Lodish, H.F.; Lisanti, M.P. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc. Natl. Acad. Sci. USA 1996, 93, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Song, K.S.; Scherer, P.E.; Tang, Z.; Okamoto, T.; Li, S.; Chafel, M.; Chu, C.; Kohtz, D.S.; Lisanti, M.P. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J. Biol. Chem. 1996, 271, 15160–15165. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, T.; Schlegel, A.; Scherer, P.E.; Lisanti, M.P. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J. Biol. Chem. 1998, 273, 5419–5422. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, O.; Tillu, V.; Ariotti, N.; Parton, R.G.; Collins, B.M. Cavin family proteins and the assembly of caveolae. J. Cell Sci. 2015, 128, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.M.; Bastiani, M.; Luetterforst, R.; Kirkham, M.; Kirkham, A.; Nixon, S.J.; Walser, P.; Abankwa, D.; Oorschot, V.M.; Martin, S.; et al. PTRF-Cavin, a Conserved Cytoplasmic Protein Required for Caveola Formation and Function. Cell 2008, 132, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Parton, R.G.; McMahon, K.-A.; Wu, Y. Caveolae: Formation, dynamics, and function. Curr. Opin. Cell Biol. 2020, 65, 8–16. [Google Scholar] [CrossRef]
- Parton, R.G.; Tillu, V.; Collins, B. Caveolae. Curr. Biol. 2018, 28, R402–R405. [Google Scholar] [CrossRef] [Green Version]
- Parton, R.G. Caveolae: Structure, Function, and Relationship to Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 111–136. [Google Scholar] [CrossRef]
- Bourseau-Guilmain, E.; Menard, J.A.; Lindqvist, E.; Indira Chandran, V.; Christianson, H.C.; Cerezo Magaña, M.; Lidfeldt, J.; Marko-Varga, G.; Welinder, C.; Belting, M. Hypoxia regulates global membrane protein endocytosis through caveolin-1 in cancer cells. Nat. Commun. 2016, 7, 11371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ramírez, C.M.; Aryal, B.; Madrigal-Matute, J.; Liu, X.; Diaz, A.; Torrecilla-Parra, M.; Suárez, Y.; Cuervo, A.M.; Sessa, W.C.; et al. Cav-1 (Caveolin-1) Deficiency Increases Autophagy in the Endothelium and Attenuates Vascular Inflammation and Atherosclerosis. Arter. Thromb. Vasc. Biol. 2020, 40, 1510–1522. [Google Scholar] [CrossRef]
- Morais, C.; Ebrahem, Q.; Anand-Apte, B.; Parat, M.-O. Altered Angiogenesis in Caveolin-1 Gene–Deficient Mice Is Restored by Ablation of Endothelial Nitric Oxide Synthase. Am. J. Pathol. 2012, 180, 1702–1714. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Bergaya, S.; Murata, T.; Alp, I.F.; Bauer, M.P.; Lin, M.I.; Drab, M.; Kurzchalia, T.V.; Stan, R.; Sessa, W.C. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J. Clin. Investig. 2006, 116, 1284–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razani, B.; Engelman, J.A.; Wang, X.B.; Schubert, W.; Zhang, X.L.; Marks, C.B.; Macaluso, F.; Russell, R.G.; Li, M.; Pestell, R.G.; et al. Caveolin-1 Null Mice Are Viable but Show Evidence of Hyperproliferative and Vascular Abnormalities. J. Biol. Chem. 2001, 276, 38121–38138. [Google Scholar] [CrossRef] [PubMed]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of Caveolae, Vascular Dysfunction, and Pulmonary Defects in Caveolin-1 Gene-Disrupted Mice. Science 2001, 293, 2449–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippini, A.; D’Alessio, A. Caveolae and Lipid Rafts in Endothelium: Valuable Organelles for Multiple Functions. Biomolecules 2020, 10, 1218. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, C.M.; Zhang, X.; Bandyopadhyay, C.; Rotllan, N.; Sugiyama, M.G.; Aryal, B.; Liu, X.; He, S.; Kraehling, J.R.; Ulrich, V.; et al. Caveolin-1 Regulates Atherogenesis by Attenuating Low-Density Lipoprotein Transcytosis and Vascular Inflammation Independently of Endothelial Nitric Oxide Synthase Activation. Circulation 2019, 140, 225–239. [Google Scholar] [CrossRef]
- Filippini, A.; Sica, G.; D’Alessio, A. The caveolar membrane system in endothelium: From cell signaling to vascular pathology. J. Cell. Biochem. 2018, 119, 5060–5071. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.P.; Mendoza-Topaz, C.; Howard, G.; Chadwick, J.; Shvets, E.; Cowburn, A.; Dunmore, B.J.; Crosby, A.; Morrell, N.; Nichols, B.J. Caveolae protect endothelial cells from membrane rupture during increased cardiac output. J. Cell Biol. 2015, 211, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Madaro, L.; Antonangeli, F.; Favia, A.; Esposito, B.; Biamonte, F.; Bouché, M.; Ziparo, E.; Sica, G.; Filippini, A.; D’Alessio, A. Knock down of caveolin-1 affects morphological and functional hallmarks of human endothelial cells. J. Cell. Biochem. 2013, 114, 1843–1851. [Google Scholar] [CrossRef]
- Sowa, G. Caveolae, caveolins, cavins, and endothelial cell function: New insights. Front. Physiol. 2012, 2, 120. [Google Scholar] [CrossRef] [Green Version]
- D’Alessio, A.; Esposito, B.; Giampietri, C.; Ziparo, E.; Pober, J.S.; Filippini, A. Plasma membrane micro domains regulate TACE-dependent TNFR1 shedding in human endothelial cells. J. Cell Mol. Med. 2011, 16, 626–635. [Google Scholar] [CrossRef]
- D’Alessio, A.; Kluger, M.S.; Li, J.H.; Al-Lamki, R.; Bradley, J.; Pober, J.S. Targeting of Tumor Necrosis Factor Receptor 1 to Low Density Plasma Membrane Domains in Human Endothelial Cells. J. Biol. Chem. 2010, 285, 23868–23879. [Google Scholar] [CrossRef] [Green Version]
- D’Alessio, A.; Al-Lamki, R.S.; Bradley, J.R.; Pober, J.S. Caveolae Participate in Tumor Necrosis Factor Receptor 1 Signaling and Internalization in a Human Endothelial Cell Line. Am. J. Pathol. 2005, 166, 1273–1282. [Google Scholar] [CrossRef] [Green Version]
- Gratton, J.-P.; Bernatchez, P.; Sessa, W. Caveolae and Caveolins in the Cardiovascular System. Circ. Res. 2004, 94, 1408–1417. [Google Scholar] [CrossRef]
- Razani, B.; Combs, T.P.; Wang, X.B.; Frank, P.G.; Park, D.S.; Russell, R.G.; Li, M.; Tang, B.; Jelicks, L.A.; Scherer, P.E.; et al. Caveolin-1-deficient Mice Are Lean, Resistant to Diet-induced Obesity, and Show Hypertriglyceridemia with Adipocyte Abnormalities. J. Biol. Chem. 2002, 277, 8635–8647. [Google Scholar] [CrossRef] [Green Version]
- Kuo, A.; Lee, M.Y.; Sessa, W.C. Lipid Droplet Biogenesis and Function in the Endothelium. Circ. Res. 2017, 120, 1289–1297. [Google Scholar] [CrossRef]
- Kuo, A.; Lee, M.Y.; Yang, K.; Gross, R.W.; Sessa, W.C. Caveolin-1 regulates lipid droplet metabolism in endothelial cells via autocrine prostacyclin–stimulated, cAMP-mediated lipolysis. J. Biol. Chem. 2018, 293, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Ha, T.-K.; Her, N.-G.; Lee, M.-G.; Ryu, B.-K.; Lee, J.-H.; Han, J.; Jeong, S.-I.; Kang, M.-J.; Kim, N.-H.; Kim, H.-J.; et al. Caveolin-1 Increases Aerobic Glycolysis in Colorectal Cancers by Stimulating HMGA1-Mediated GLUT3 Transcription. Cancer Res. 2012, 72, 4097–4109. [Google Scholar] [CrossRef] [Green Version]
- Ouporov, I.V.; Knull, H.R.; Thomasson, K.A. Brownian Dynamics Simulations of Interactions between Aldolase and G- or F-Actin. Biophys. J. 1999, 76, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Pagliaro, L. Aldolase exists in both the fluid and solid phases of cytoplasm. J. Cell Biol. 1988, 107, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, J.D.; Madden, D.P.; Steck, T.L. Association of phosphofructokinase and aldolase with the membrane of the intact erythrocyte. J. Biol. Chem. 1984, 259, 9374–9378. [Google Scholar] [CrossRef]
- Raikar, L.S.; Vallejo, J.; Lloyd, P.G.; Hardin, C.D. Overexpression of caveolin-1 results in increased plasma membrane targeting of glycolytic enzymes: The structural basis for a membrane associated metabolic compartment. J. Cell. Biochem. 2006, 98, 861–871. [Google Scholar] [CrossRef]
- Shiroto, T.; Romero, N.; Sugiyama, T.; Sartoretto, J.L.; Kalwa, H.; Yan, Z.; Shimokawa, H.; Michel, T. Caveolin-1 Is a Critical Determinant of Autophagy, Metabolic Switching, and Oxidative Stress in Vascular Endothelium. PLoS ONE 2014, 9, e87871. [Google Scholar] [CrossRef] [Green Version]




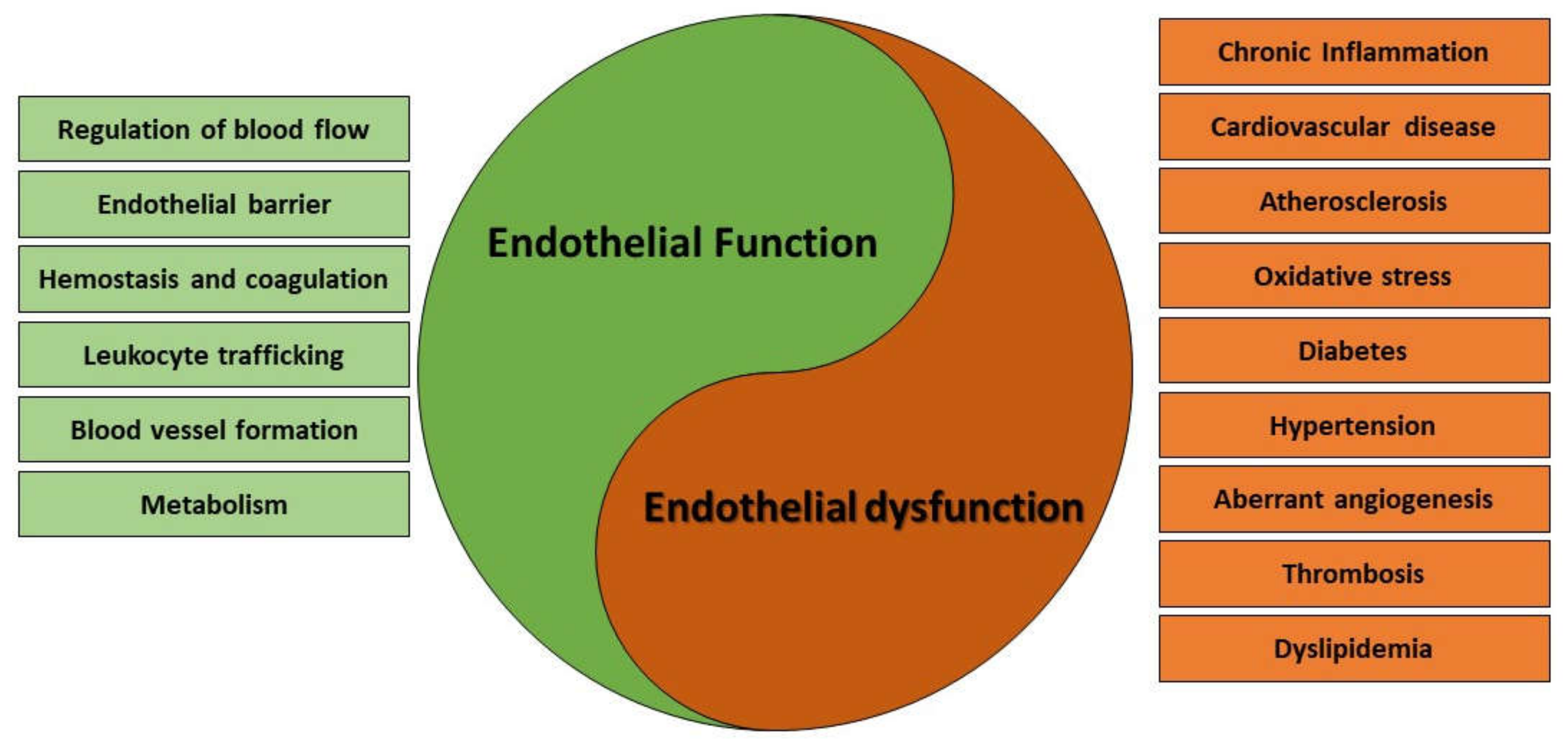{kind=link}
{kind=link}
{kind=link}
| Name | Mechanism of Action and Target | Clinical Indications |
|---|---|---|
| Axitinib (Inlyta) | Inhibit receptor tyrosine kinases VEGFR-1, VEGFR-2, and VEGFR-3 | Advanced renal cell carcinoma. |
| Bevacizumab (Avastin) | Prevents the interaction of VEGF to VEGFR1/Flt-1 and VEGFR2/KDR on the surface of ECs | Cervical cancer, Colorectal cancer, Glioblastoma, Hepatocellular carcinoma, Non-squamous non-small cell lung cancer, Renal cell carcinoma |
| Cabozantinib (Cometric) | Inhibits the tyrosine kinase activity of VEGFR-1, VEGFR-2, and other receptor tyrosine kinases. | Thyroid cancers, Hepatocellular carcinoma, Renal cell carcinoma |
| Everolimus (Afinitor) | Inhibits antigenic and interleukin (IL-2 and IL-15) stimulated activation and proliferation of T and B lymphocytes. | Astrocytoma, breast cancer, pancreatic cancer, gastrointestinal cancer, lung cancer, renal cell carcinoma |
| Pazopanib (Votrient) | Multi-tyrosine kinase inhibitor. | Renal Cell Carcinoma, Soft Tissue Sarcoma. |
| Ramucirumab (Cyramza) | VEGFR2 antagonist | Gastric Cancer, Non-Small Cell Lung Cancer, Colorectal Cancer, Hepatocellular Carcinoma |
| Regorafenib (Stivarga) | Surface and intracellular kinase inhibitor | Colorectal Cancer, Gastrointestinal Stromal Tumors, Hepatocellular Carcinoma |
| Sorafenib (Nexavar) | Surface and intracellular kinase inhibitor | Hepatocellular Carcinoma, Renal Cell Carcinoma, Thyroid Carcinoma |
| Sunitinib (Sutent) | Multiple receptor tyrosine kinases inhibitor (PDGFR, VEGFR1, VEGFR2, VEGFR3, etc) | Gastrointestinal Stromal Tumor, Advanced Renal Cell Carcinoma, Advanced Pancreatic Neuroendocrine Tumors |
| Vandetanib (Caprelsa) | Tyrosine kinase inhibitor | Medullary thyroid cancer |
| Ziv-Aflibercept (Zaltrap) | VEGF-A and VEGF-B inhibitor | Colorectal cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippini, A.; Tamagnone, L.; D’Alessio, A. Endothelial Cell Metabolism in Vascular Functions. Cancers 2022, 14, 1929. https://doi.org/10.3390/cancers14081929
Filippini A, Tamagnone L, D’Alessio A. Endothelial Cell Metabolism in Vascular Functions. Cancers. 2022; 14(8):1929. https://doi.org/10.3390/cancers14081929
Chicago/Turabian StyleFilippini, Antonio, Luca Tamagnone, and Alessio D’Alessio. 2022. "Endothelial Cell Metabolism in Vascular Functions" Cancers 14, no. 8: 1929. https://doi.org/10.3390/cancers14081929