
Extracellular Vesicle-Mediated Mitochondrial Reprogramming in Cancer


Abstract
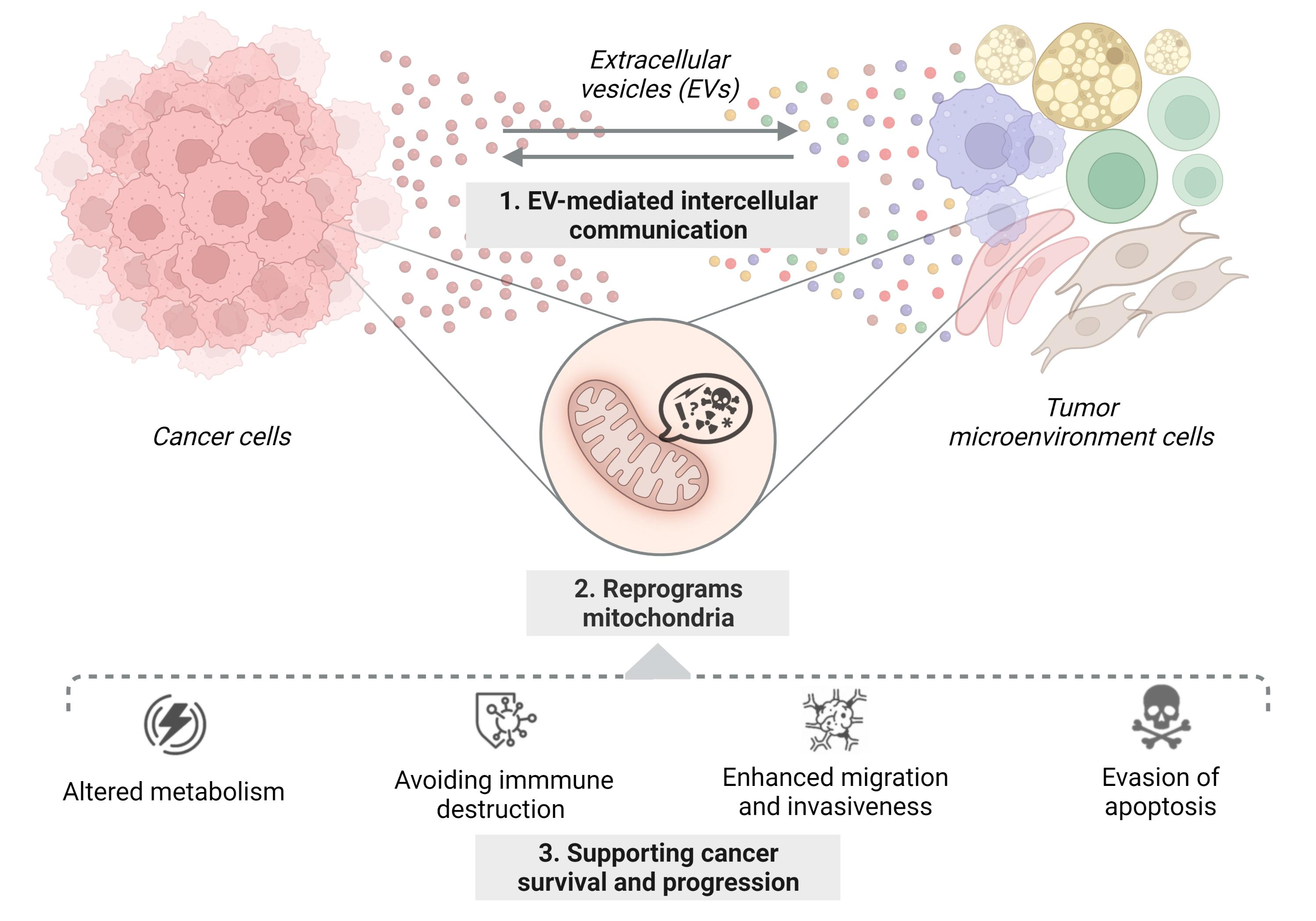
:Simple Summary
Abstract

1. Introduction
2. Extracellular Vesicle Release and Cargo in Cancer
2.1. Overview
2.2. Alterations in EV Biogenesis and Release
2.3. Alteration in EV Cargo
3. Mitochondria in Cancer
3.1. Mitochondrial Metabolism
3.2. Mitochondrial Dynamics
3.3. Mitochondrial-Mediated Apoptosis
4. EV Modulation of Mitochondrial Processes in Cancer
4.1. Dysregulating Cellular Energetics
4.1.1. Metabolic Coupling with Cancer-Associated Fibroblasts (CAFs)
4.1.2. Metabolic Coupling with Adipocytes
4.1.3. Metabolic Subjugation of Neighboring Cells
4.2. Resisting Cell Death
4.2.1. Modulation of the Bcl-2 Pathway
4.2.2. Chemotherapy Resistance
4.2.3. Enhancing Cancer Survival and Chemotherapy Resistance through EV Transfer of Functional Mitochondria and Mitochondrial Components
4.3. Avoiding Immune Destruction
4.3.1. Macrophage Mitochondrial Reprogramming in Cancer
4.3.2. EV-Related Immunosuppression of T Cells
4.4. Activating Invasion and Metastasis
4.4.1. Increased Motility and Migration
4.4.2. Intravasation (Trans-Endothelial Migration into Vessels)
5. Nanoparticle-Based Therapeutic Strategies Targeting Mitochondria in Cancer
5.1. Naturally Occurring EVs
5.2. Altered EVs
5.2.1. EVs Enriched with Biological Cargo
5.2.2. EVs Loaded with Exogenous Drugs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2019, 21, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bebelman, M.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Maisano, D.; Mimmi, S.; Dattilo, V.; Marino, F.; Gentile, M.; Vecchio, E.; Fiume, G.; Nisticò, N.; Aloisio, A.; de Santo, M.P.; et al. A novel phage display based platform for exosome diversity characterization. Nanoscale 2022, 14, 2998–3003. [Google Scholar] [CrossRef]
- Hurley, J.H.; Hanson, P.I. Membrane Budding and Scission by the Escrt Machinery: It’s All in the Neck. Mol. Cell Biol. 2010, 11, 556–566. [Google Scholar] [CrossRef]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.-H.; Kim, J.-H. A Comprehensive Review on Factors Influences Biogenesis, Functions, Therapeutic and Clinical Implications of Exosomes. Int. J. Nanomed. 2021, 16, 1281–1312. [Google Scholar] [CrossRef]
- Xavier, C.P.R.; Caires, H.R.; Barbosa, M.A.G.; Bergantim, R.; Guimarães, J.E.; Vasconcelos, M.H. The Role of Extracellular Vesicles in the Hallmarks of Cancer and Drug Resistance. Cells 2020, 9, 1141. [Google Scholar] [CrossRef]
- Jabalee, J.; Towle, R.; Garnis, C. The Role of Extracellular Vesicles in Cancer: Cargo, Function, and Therapeutic Implications. Cells 2018, 7, 93. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.-T.; Huang, C.-C.; You, H.-L.; Chou, F.-F.; Hu, C.-C.A.; Chao, F.-P.; Chen, C.-M.; Cheng, J.-T. Overexpression of tumor susceptibility gene TSG101 in human papillary thyroid carcinomas. Oncogene 2002, 21, 4830–4837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, K.B.; Stanton, M.J.; West, W.W.; Todd, G.L.; Wagner, K.-U. Tsg101 is upregulated in a subset of invasive human breast cancers and its targeted overexpression in transgenic mice reveals weak oncogenic properties for mammary cancer initiation. Oncogene 2007, 26, 5950–5959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoshima, M.; Tanaka, N.; Aoki, J.; Tanaka, Y.; Murata, K.; Kyuuma, M.; Kobayashi, H.; Ishii, N.; Yaegashi, N.; Sugamura, K. Inhibition of Tumor Growth and Metastasis by Depletion of Vesicular Sorting Protein Hrs: Its Regulatory Role on E-Cadherin and β-Catenin. Cancer Res. 2007, 67, 5162–5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roucourt, B.; Meeussen, S.; Bao, J.; Zimmermann, P.; David, G. Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 2015, 25, 412–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlodavsky, I.; Friedmann, Y.; Elkin, M.; Aingorn, H.; Atzmon, R.; Ishai-Michaeli, R.; Bitan, M.; Pappo, O.; Peretz, T.; Michal, I.; et al. Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nat. Med. 1999, 5, 793–802. [Google Scholar] [CrossRef]
- Kegelman, T.P.; Das, S.K.; Emdad, L.; Hu, B.; Menezes, M.E.; Bhoopathi, P.; Wang, X.-Y.; Pellecchia, M.; Sarkar, D.; Fisher, P.B. Targeting Tumor Invasion: The Roles of Mda-9/Syntenin. Expert Opin. Ther. Targets 2015, 19, 97–112. [Google Scholar] [CrossRef] [Green Version]
- McAndrews, K.M.; Kalluri, R. Mechanisms associated with biogenesis of exosomes in cancer. Mol. Cancer 2019, 18, 52. [Google Scholar] [CrossRef]
- Kucharzewska, P.; Belting, M. Emerging roles of extracellular vesicles in the adaptive response of tumour cells to microenvironmental stress. J. Extracell. Vesicles 2013, 2, 20304. [Google Scholar] [CrossRef]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Severino, P.E.; Goliwas, K.; Hough, K.; Van Vessem, D.; Wang, H.; Frost, A.R.; Ponnazhagen, S.; Berry, J.L.; Deshane, J.S. Abstract 1177: Mechanical strain induces phenotypic changes in breast cancer cells and promotes immunosuppression in the tumor microenvironment. Lab. Investig. 2019, 100, 1503–1516. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.; Yin, Y.; Jia, X.; Mao, L. Mechanism of cargo sorting into small extracellular vesicles. Bioengineered 2021, 12, 8186–8201. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Samuel, M.; Kumar, S.; Mathivanan, S. Ticket to a bubble ride: Cargo sorting into exosomes and extracellular vesicles. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 140203. [Google Scholar] [CrossRef] [PubMed]
- Ruan, K.; Fang, X.; Ouyang, G. MicroRNAs: Novel regulators in the hallmarks of human cancer. Cancer Lett. 2009, 285, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Macgregor-Das, A.M.; Das, S. A Microrna’s Journey to the Center of the Mitochondria. Am. J. Physiol. -Heart Circ. Physiol. 2018, 315, H206–H215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragomir, M.P.; Knutsen, E.; Calin, G. SnapShot: Unconventional miRNA Functions. Cell 2018, 174. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.L.; Adams, A.; King, K.E.; Dunn, W.; Christenson, L.K.; Hung, W.-T.; Weinman, S.A. The RNA binding protein FMR1 controls selective exosomal miRNA cargo loading during inflammation. J. Cell Biol. 2020, 219, e201912074. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Gutierrez-Vazquez, C.; Sanchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, L.; Giurato, G.; Cicchini, C.; Montaldo, C.; Mancone, C.; Tarallo, R.; Battistelli, C.; Alonzi, T.; Weisz, A.; Tripodi, M. The RNA-Binding Protein SYNCRIP Is a Component of the Hepatocyte Exosomal Machinery Controlling MicroRNA Sorting. Cell Rep. 2016, 17, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Fabbiano, F.; Corsi, J.; Gurrieri, E.; Trevisan, C.; Notarangelo, M.; D’Agostino, V.G. Rna Packaging into Extracellular Vesicles: An Orchestra of Rna-Binding Proteins? J. Extracell. Vesicles 2020, 10, e12043. [Google Scholar] [CrossRef]
- Krishn, S.R.; Salem, I.; Quaglia, F.; Naranjo, N.M.; Agarwal, E.; Liu, Q.; Sarker, S.; Kopenhaver, J.; McCue, P.A.; Weinreb, P.H.; et al. The Avβ6 Integrin in Cancer Cell-Derived Small Extracellular Vesicles Enhances Angiogenesis. J. Extracell. Vesicles 2020, 9, 1763594. [Google Scholar] [CrossRef]
- Mansour, M.A. Ubiquitination: Friend and Foe in Cancer. Int. J. Biochem. Cell Biol. 2018, 101, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Möller, A.; Lobb, R.J. The evolving translational potential of small extracellular vesicles in cancer. Nat. Cancer 2020, 20, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Tiepolo, T.; Angelin, A.; Sabatelli, P.; Maraldi, N.M.; Basso, E.; Forte, M.A.; Bernardi, P.; Bonaldo, P. Genetic ablation of cyclophilin D rescues mitochondrial defects and prevents muscle apoptosis in collagen VI myopathic mice. Hum. Mol. Genet. 2009, 18, 2024–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, E.; Riva, A.; Moreno, C.; Odena, G.; Mudan, S.; Manyakin, N.; Miquel, R.; Degré, D.; Trepo, E.; Sancho-Bru, P.; et al. Perturbations in Mitochondrial Dynamics Are Closely Involved in the Progression of Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2020, 44, 856–865. [Google Scholar] [CrossRef]
- Palma, E.; Ma, X.; Riva, A.; Iansante, V.; Dhawan, A.; Wang, S.; Ni, H.-M.; Sesaki, H.; Williams, R.; Ding, W.-X.; et al. Dynamin-1–Like Protein Inhibition Drives Megamitochondria Formation as an Adaptive Response in Alcohol-Induced Hepatotoxicity. Am. J. Pathol. 2018, 189, 580–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, W.-X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.G.; Wikman, H.; Pantel, K.; Haigis, M.C.; De Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.F.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Chen, J.; Yang, L.; Liu, J.; Zhang, X.; Zhang, Y.; Tu, Q.; Yin, D.; Lin, D.; Wong, P.P.; et al. Extracellular vesicle-packaged HIF-1α-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat. Cell Biol. 2019, 21, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.S.; Kang, C.W.; Kang, S.; Jang, Y.; Chae, Y.C.; Kim, B.G.; Cho, N.H. ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene 2020, 39, 664–676. [Google Scholar] [CrossRef]
- Fonseca, P.; Vardaki, I.; Occhionero, A.; Panaretakis, T. Chapter Five—Metabolic and Signaling Functions of Cancer Cell-Derived Extracellular Vesicles. In International Review of Cell and Molecular Biology; Kwang, W.J., Lorenzo, G., Eds.; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef]
- Deep, G.; Schlaepfer, I.R. Aberrant Lipid Metabolism Promotes Prostate Cancer: Role in Cell Survival under Hypoxia and Extracellular Vesicles Biogenesis. Int. J. Mol. Sci. 2016, 17, 1061. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhu, N.; Yan, T.; Shi, Y.-N.; Chen, J.; Zhang, C.-J.; Xie, X.-J.; Liao, D.-F.; Qin, L. The crosstalk: Exosomes and lipid metabolism. Cell Commun. Signal. 2020, 18, 1–12. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Rider, M.A.; Bundy, J.L.; Liu, X.; Singh, R.K.; Meckes, D.G., Jr. Proteomic profiling of NCI-60 extracellular vesicles uncovers common protein cargo and cancer type-specific biomarkers. Oncotarget 2016, 7, 86999–87015. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Peng, P.; Chen, S.; Li, L.; Zhang, M.; Cao, D.; Yang, J.; Li, H.; Gui, T.; Li, X.; et al. Characterization and proteomic analysis of ovarian cancer-derived exosomes. J. Proteom. 2013, 80, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Park, J.O.; Choi, D.-Y.; Choi, D.-S.; Kim, H.J.; Kang, J.W.; Jung, J.H.; Lee, J.H.; Kim, J.; Freeman, M.R.; Lee, K.Y.; et al. Identification and characterization of proteins isolated from microvesicles derived from human lung cancer pleural effusions. Proteomics 2013, 13, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Clement, E.; Lazar, I.; Attané, C.; Carrié, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef]
- Puhka, M.; Takatalo, M.; Nordberg, M.-E.; Valkonen, S.; Nandania, J.; Aatonen, M.; Yliperttula, M.; Laitinen, S.; Velagapudi, V.; Mirtti, T.; et al. Metabolomic Profiling of Extracellular Vesicles and Alternative Normalization Methods Reveal Enriched Metabolites and Strategies to Study Prostate Cancer-Related Changes. Theranostics 2017, 7, 3824–3841. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.-L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Villar, V.H.; Merhi, F.; Djavaheri-Mergny, M.; Durán, R.V. Glutaminolysis and Autophagy in Cancer. Autophagy 2015, 11, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to Clinic: Glutamine Metabolism to Cancer Therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef]
- Ko, Y.-H.; Lin, Z.; Flomenberg, N.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P.; Martinez-Outschoorn, U.E. Glutamine Fuels a Vicious Cycle of Autophagy in the Tumor Stroma and Oxidative Mitochondrial Metabolism in Epithelial Cancer Cells: Implications for Preventing Chemotherapy Resistance. Cancer Biol. Ther. 2011, 12, 1085–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Kroeger, B.; Marie, P.P.; Bridges, E.M.; Mason, J.D.; McCormick, K.; E Zois, C.; Sheldon, H.; Alham, N.K.; Johnson, E.; et al. Glutamine deprivation alters the origin and function of cancer cell exosomes. EMBO J. 2020, 39, e103009. [Google Scholar] [CrossRef] [PubMed]
- Simula, L.; Nazio, F.; Campello, S. The mitochondrial dynamics in cancer and immune-surveillance. Semin. Cancer Biol. 2017, 47, 29–42. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, T.-E.; Chen, M.; Xu, D.; Zhu, Y.; Hu, B.-Y.; Lin, Z.-F.; Pan, J.-J.; Wang, X.; Wu, C.; et al. MFN1-dependent alteration of mitochondrial dynamics drives hepatocellular carcinoma metastasis by glucose metabolic reprogramming. Br. J. Cancer 2019, 122, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Kashatus, D.F. The regulation of tumor cell physiology by mitochondrial dynamics. Biochem. Biophys. Res. Commun. 2018, 500, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, L.; Jia, R. The role of mitochondrial dynamics in human cancers. Am. J. Cancer Res. 2020, 10, 1278–1293. [Google Scholar] [PubMed]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Kannan, A.; Wells, R.B.; Sivakumar, S.; Komatsu, S.; Singh, K.P.; Samten, B.; Philley, J.V.; Sauter, E.R.; Ikebe, M.; Idell, S.; et al. Mitochondrial Reprogramming Regulates Breast Cancer Progression. Clin. Cancer Res. 2016, 22, 3348–3360. [Google Scholar] [CrossRef] [Green Version]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of Mitochondrial Fission Prevents Cell Cycle Progression in Lung Cancer. FASEB J. 2012, 26, 2175–2186. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Wu, Q.; Horbinski, C.M.; Flavahan, W.A.; Yang, K.; Zhou, W.; Dombrowski, S.M.; Huang, Z.; Fang, X.; Shi, Y.; et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat. Neurosci. 2015, 18, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Chen, G.; Li, X.; Wu, X.; Chang, Z.; Xu, J.; Zhu, Y.; Yin, P.; Liang, X.; Dong, L. Mfn2 Suppresses Cancer Progression through Inhibition of Mtorc2/Akt Signaling. Sci. Rep. 2017, 7, 41718. [Google Scholar] [CrossRef]
- Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Behera, B.P.; Mishra, S.R.; Bhutia, S.K. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin. Cancer Biol. 2019, 66, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Ferro, F.; Servais, S.; Besson, P.; Roger, S.; Dumas, J.-F.; Brisson, L. Autophagy and Mitophagy in Cancer Metabolic Remodelling. Semin. Cell Dev. Biol. 2020, 98, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Suárez, H.; Andreu, Z.; Mazzeo, C.; Toribio, V.; Pérez-Rivera, A.E.; López-Martín, S.; García-Silva, S.; Hurtado, B.; Morato, E.; Peláez, L.; et al. CD9 inhibition reveals a functional connection of extracellular vesicle secretion with mitophagy in melanoma cells. J. Extracell. Vesicles 2021, 10, e12082. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.; Sazanov, L. Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [Green Version]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Colina-Tenorio, L.; Horten, P.; Pfanner, N.; Rampelt, H. Shaping the mitochondrial inner membrane in health and disease. J. Intern. Med. 2020, 287, 645–664. [Google Scholar] [CrossRef]
- Eramo, M.; Lisnyak, V.; Formosa, L.; Ryan, M.T. The ‘mitochondrial contact site and cristae organising system’ (MICOS) in health and human disease. J. Biochem. 2019, 167, 243–255. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and Beyond—Mitochondrial Performance in Apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.Y. Apoptosis in Cancer: From Pathogenesis to Treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic Reprogramming of Cancer-Associated Fibroblasts by IDH3α Downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [Green Version]
- Albino, D.; Falcione, M.; Uboldi, V.; Temilola, D.O.; Sandrini, G.; Merulla, J.; Civenni, G.; Kokanovic, A.; Stürchler, A.; Shinde, D.; et al. Circulating extracellular vesicles release oncogenic miR-424 in experimental models and patients with aggressive prostate cancer. Commun. Biol. 2021, 4, 119. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Greening, D.W.; Chen, M.; Xu, R.; Ji, H.; Simpson, R.J. Exosomes Derived from Human Primary and Metastatic Colorectal Cancer Cells Contribute to Functional Heterogeneity of Activated Fibroblasts by Reprogramming Their Proteome. Proteomics 2019, 19, 1800148. [Google Scholar] [CrossRef] [PubMed]
- La Shu, S.; Yang, Y.; Allen, C.L.; Maguire, O.; Minderman, H.; Sen, A.; Ciesielski, M.J.; Collins, K.A.; Bush, P.J.; Singh, P.; et al. Metabolic reprogramming of stromal fibroblasts by melanoma exosome microRNA favours a pre-metastatic microenvironment. Sci. Rep. 2018, 8, 12905. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Xu, G.; Chang, Z.; Zhu, L.; Yao, J. Mir-210 Transferred by Lung Cancer Cell-Derived Exosomes May Act as Proangiogenic Factor in Cancer-Associated Fibroblasts by Modulating Jak2/Stat3 Pathway. Clin. Sci. 2020, 134, 807–825. [Google Scholar] [CrossRef]
- Puissegur, M.-P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2010, 18, 465–478. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhou, Z.; Xu, S.; Liao, C.; Chen, X.; Li, B.; Peng, J.; Li, D.; Yang, L. Extracellular vesicle packaged LMP1-activated fibroblasts promote tumor progression via autophagy and stroma-tumor metabolism coupling. Cancer Lett. 2020, 478, 93–106. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Z.; Liu, W.; Li, X. SNHG3 Functions as miRNA Sponge to Promote Breast Cancer Cells Growth Through the Metabolic Reprogramming. Appl. Biochem. Biotechnol. 2020, 191, 1084–1099. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; Lucas, F.A.S.; et al. Author response: Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2015. [Google Scholar] [CrossRef]
- Achreja, A.; Zhao, H.; Yang, L.; Yun, T.H.; Marini, J.; Nagrath, D. Exo-MFA—A 13C metabolic flux analysis framework to dissect tumor microenvironment-secreted exosome contributions towards cancer cell metabolism. Metab. Eng. 2017, 43, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Ham, I.-H.; Lee, D.; Hur, H. Cancer-Associated Fibroblast-Induced Resistance to Chemotherapy and Radiotherapy in Gastrointestinal Cancers. Cancers 2021, 13, 1172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jiang, H.; Li, X.; Chen, X.; Huang, Y. Mir-92a Regulates Brown Adipocytes Differentiation, Mitochondrial Oxidative Respiration, and Heat Generation by Targeting Smad7. J. Cell. Biochem. 2020, 121, 3825–3836. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Sun, S.; Li, Z.; Yang, Q.; Li, B.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Yang, C.; et al. Tumour-originated exosomal miR-155 triggers cancer-associated cachexia to promote tumour progression. Mol. Cancer 2018, 17, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Li, J.; Li, Z.; Sun, S.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Zhang, Y.; Sun, S.; et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J. Exp. Clin. Cancer Res. 2019, 38, 223. [Google Scholar] [CrossRef] [Green Version]
- Lazar, I.; Clement, E.; Dauvillier, S.; Milhas, D.; Ducoux-Petit, M.; LeGonidec, S.; Moro, C.; Soldan, V.; Dalle, S.; Balor, S.; et al. Adipocyte Exosomes Promote Melanoma Aggressiveness through Fatty Acid Oxidation: A Novel Mechanism Linking Obesity and Cancer. Cancer Res. 2016, 76, 4051–4057. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tan, J.; Ou, S.; Chen, J.; Chen, L. Adipose-derived exosomes deliver miR-23a/b to regulate tumor growth in hepatocellular cancer by targeting the VHL/HIF axis. J. Physiol. Biochem. 2019, 75, 391–401. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Yogev, O.; Henderson, S.; Hayes, M.J.; Marelli, S.S.; Ofir-Birin, Y.; Regev-Rudzki, N.; Herrero, J.; Enver, T. Herpesviruses shape tumour microenvironment through exosomal transfer of viral microRNAs. PLoS Pathog. 2017, 13, e1006524. [Google Scholar] [CrossRef]
- Matsumoto, A.; Takahashi, Y.; Nishikawa, M.; Sano, K.; Morishita, M.; Charoenviriyakul, C.; Saji, H.; Takakura, Y. Accelerated growth of B16 BL 6 tumor in mice through efficient uptake of their own exosomes by B16 BL 6 cells. Cancer Sci. 2017, 108, 1803–1810. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wu, X.-H.; Wang, D.; Luo, C.-L.; Chen, L.-X. Bladder cancer cell-derived exosomes inhibit tumor cell apoptosis and induce cell proliferation in vitro. Mol. Med. Rep. 2013, 8, 1272–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Ming, X.; Xu, J.; Xiao, Y. Circ_0009910 Shuttled by Exosomes Regulates Proliferation, Cell Cycle and Apoptosis of Acute Myeloid Leukemia Cells by Regulating Mir-5195-3p/Grb10 Axis. Hematol. Oncol. 2021, 39, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.-L.; Hu, G.-W.; Zhang, B.; Kuang, W.; Chen, Y.; Wu, L.; Xu, G.-H. Glioma cells enhance angiogenesis and inhibit endothelial cell apoptosis through the release of exosomes that contain long non-coding RNA CCAT2. Oncol. Rep. 2017, 38, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Deng, M.; Yuan, H.; Liu, S.; Hu, Z.; Xiao, H. Exosome-transmitted LINC00461 promotes multiple myeloma cell proliferation and suppresses apoptosis by modulating microRNA/BCL-2 expression. Cytotherapy 2019, 21, 96–106. [Google Scholar] [CrossRef]
- Dong, H.; Wang, W.; Chen, R.; Zhang, Y.; Zou, K.; Ye, M.; He, X.; Zhang, F.; Han, J. Exosome-mediated transfer of lncRNA-SNHG14 promotes trastuzumab chemoresistance in breast cancer. Int. J. Oncol. 2018, 53, 1013–1026. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Ren, M.; Li, Y.; Fu, Y.; Deng, M.; Li, C. Exosome-mediated transfer of lncRNA PART1 induces gefitinib resistance in esophageal squamous cell carcinoma via functioning as a competing endogenous RNA. J. Exp. Clin. Cancer Res. 2018, 37, 171. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, M.; Hu, C. Exosomal transfer of miR-214 mediates gefitinib resistance in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2018, 507, 457–464. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, Z.; Li, X.; Liu, J.-L.; Tian, L.; Chen, J.-Q. Exosome-mediated transfer of CLIC1 contributes to the vincristine-resistance in gastric cancer. Mol. Cell. Biochem. 2019, 462, 97–105. [Google Scholar] [CrossRef]
- Hu, W.; Xu, Z.; Zhu, S.; Sun, W.; Wang, X.; Tan, C.; Zhang, Y.; Zhang, G.; Xu, Y.; Tang, J. Small extracellular vesicle-mediated Hsp70 intercellular delivery enhances breast cancer adriamycin resistance. Free Radic. Biol. Med. 2021, 164, 85–95. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Zhang, L.-L.; Bi, Q.-C.; Gan, L.-J.; Wei, M.-J.; Hong, T.; Tan, R.-J.; Lan, X.-M.; Liu, L.-H.; Han, X.-J.; et al. Exosomal connexin 43 regulates the resistance of glioma cells to temozolomide. Oncol. Rep. 2021, 45, 44. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Q.; Liu, J.-T.; Fan, L.-L.; Liu, Y.; Cheng, L.; Wang, F.; Yu, H.-Q.; Gao, J.; Wei, W.; Wang, H.; et al. Exosomes derived from gefitinib-treated EGFR-mutant lung cancer cells alter cisplatin sensitivity via up-regulating autophagy. Oncotarget 2016, 7, 24585–24595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Lu, Y.; Li, A.; Song, H.; Zhang, W.; Gilbert, M.; Yang, C. CBMT-46, Microenvironment-derived mitochondria prime glioma chemoresistance by augmenting Nad+ Metabolism and parp-dependent DNA repair. Neuro Oncol. 2018, 20, vi42. [Google Scholar] [CrossRef] [Green Version]
- Salaud, C.; Alvarez-Arenas, A.; Geraldo, F.; Belmonte-Beitia, J.; Calvo, G.F.; Gratas, C.; Pecqueur, C.; Garnier, D.; Pérez-Garcià, V.; Vallette, F.M.; et al. Mitochondria transfer from tumor-activated stromal cells (TASC) to primary Glioblastoma cells. Biochem. Biophys. Res. Commun. 2020, 533, 139–147. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [Green Version]
- Seo, W.; Gao, Y.; He, Y.; Sun, J.; Xu, H.; Feng, D.; Park, S.H.; Cho, Y.-E.; Guillot, A.; Ren, T.; et al. ALDH2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles. J. Hepatol. 2019, 71, 1000–1011. [Google Scholar] [CrossRef]
- Pritchard, A.; Tousif, S.; Wang, Y.; Hough, K.; Khan, S.; Strenkowski, J.; Chacko, B.K.; Darley-Usmar, V.M.; Deshane, J.S. Lung Tumor Cell-Derived Exosomes Promote M2 Macrophage Polarization. Cells 2020, 9, 1303. [Google Scholar] [CrossRef]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-Induced Tumor Exosomes Promote M2-Like Macrophage Polarization of Infiltrating Myeloid Cells and Microrna-Mediated Metabolic Shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef]
- Bland, C.L.; Byrne-Hoffman, C.N.; Fernandez, A.; Rellick, S.L.; Deng, W.; Klinke, D.J. Exosomes derived from B16F0 melanoma cells alter the transcriptome of cytotoxic T cells that impacts mitochondrial respiration. FEBS J. 2018, 285, 1033–1050. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yang, Y.; Wang, W.; Zhang, Y.; Chen, Z.; Hao, C.; Zhang, J. Melanoma-released exosomes directly activate the mitochondrial apoptotic pathway of CD4+ T cells through their microRNA cargo. Exp. Cell Res. 2018, 371, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wu, X.; Wang, D.; Luo, C.; Chen, L. Renal Carcinoma Cell-Derived Exosomes Induce Human Immortalized Line of Jurkat T Lymphocyte Apoptosis in vitro. Urol. Int. 2013, 91, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Czystowska-Kuźmicz, M.; Han, J.; Szczepanski, M.J.; Szajnik, M.; Quadrini, K.; Brandwein, H.; Hadden, J.W.; Signorelli, K.; Whiteside, T.L. IRX-2, a novel immunotherapeutic, protects human T cells from tumor-induced cell death. Cell Death Differ. 2009, 16, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, M.H.; Borin, T.F.; Ara, R.; Piranlioglu, R.; Achyut, B.R.; Korkaya, H.; Liu, Y.; Arbab, A.S. Critical immunosuppressive effect of MDSC-derived exosomes in the tumor microenvironment. Oncol. Rep. 2021, 45, 1171–1181. [Google Scholar] [CrossRef]
- Hildeman, D.A.; Mitchell, T.; Kappler, J.; Marrack, P. T cell apoptosis and reactive oxygen species. J. Clin. Investig. 2003, 111, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, I.; Ghosh, J.C.; Kossenkov, A.V.; Mulugu, S.; Krishn, S.R.; Vaira, V.; Qin, J.; Plow, E.F.; Languino, L.R.; Altieri, D.C. Small Extracellular Vesicle Regulation of Mitochondrial Dynamics Reprograms a Hypoxic Tumor Microenvironment. Dev. Cell 2020, 55, 163–177.e6. [Google Scholar] [CrossRef]
- Wu, C.-H.; Silvers, C.R.; Messing, E.M.; Lee, Y.-F. Bladder cancer extracellular vesicles drive tumorigenesis by inducing the unfolded protein response in endoplasmic reticulum of nonmalignant cells. J. Biol. Chem. 2019, 294, 3207–3218. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Zhou, W.; Rong, Y.; Kuang, T.; Xu, X.; Wu, W.; Wang, D.; Lou, W. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2019, 383, 111543. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, L.; Gao, Y.-F.; Fan, Z.-M.; Cai, X.-Y.; Liu, M.-Y.; Guo, X.-R.; Gao, C.-L.; Xia, Z.-K. MicroRNA-106b induces mitochondrial dysfunction and insulin resistance in C2C12 myotubes by targeting mitofusin-2. Mol. Cell. Endocrinol. 2013, 381, 230–240. [Google Scholar] [CrossRef]
- Kim, J.; Lee, C.; Kim, I.; Ro, J.; Kim, J.; Min, Y.; Park, J.; Sunkara, V.; Park, Y.-S.; Michael, I.; et al. Three-Dimensional Human Liver-Chip Emulating Premetastatic Niche Formation by Breast Cancer-Derived Extracellular Vesicles. ACS Nano 2020, 14, 14971–14988. [Google Scholar] [CrossRef]
- Hua, W.; Dijke, P.T.; Kostidis, S.; Giera, M.; Hornsveld, M. TGFβ-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell. Mol. Life Sci. 2019, 77, 2103–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lötvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood–brain barrier. Nat. Commun. 2015, 6, 6716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusic, M.; Prokisch, H. Ncrnas: New Players in Mitochondrial Health and Disease? Front. Genet. 2020, 11, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Gu, H.; Lan, Z.; Lei, Q.; Wang, W.; Ruan, J.; Yu, M.; Lin, J.; Cui, Q. Downregulation of Cyclooxygenase-1 Stimulates Mitochondrial Apoptosis through the Nf-Κb Signaling Pathway in Colorectal Cancer Cells. Oncol. Rep. 2019, 41, 559–569. [Google Scholar] [CrossRef] [Green Version]
- Gentric, G.; Mechta-Grigoriou, F. Tumor Cells and Cancer-Associated Fibroblasts: An Updated Metabolic Perspective. Cancers 2021, 13, 399. [Google Scholar] [CrossRef]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2019, 146, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. BioMed Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Bera, S. CAF cellular glycolysis: Linking cancer cells with the microenvironment. Tumor Biol. 2016, 37, 8503–8514. [Google Scholar] [CrossRef]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef]
- Barquissau, V.; Beuzelin, D.; Pisani, D.F.; Beranger, G.E.; Mairal, A.; Montagner, A.; Roussel, B.; Tavernier, G.; Marques, M.-A.; Moro, C.; et al. White-to-brite conversion in human adipocytes promotes metabolic reprogramming towards fatty acid anabolic and catabolic pathways. Mol. Metab. 2016, 5, 352–365. [Google Scholar] [CrossRef]
- Sah, R.P.; Sharma, A.; Nagpal, S.; Patlolla, S.H.; Sharma, A.; Kandlakunta, H.; Anani, V.; Angom, R.S.; Kamboj, A.K.; Ahmed, N.; et al. Phases of Metabolic and Soft Tissue Changes in Months Preceding a Diagnosis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2019, 156, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K.A.; Ni Chonghaile, T.; Letai, A. Mitochondria: Gatekeepers of response to chemotherapy. Trends Cell Biol. 2013, 23, 612–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, R.L.; Jiang, X.P.; Head, J.F. Mitochondria organelle transplantation: Introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res. Treat. 2012, 136, 347–354. [Google Scholar] [CrossRef]
- Sun, C.; Wang, Z.; Liu, Y.; Liu, Y.; Li, H.; Di, C.; Wu, Z.; Gan, L.; Zhang, H. Carbon ion beams induce hepatoma cell death by NADPH oxidase-mediated mitochondrial damage. J. Cell. Physiol. 2013, 229, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Kreger, B.T.; Johansen, E.R.; Cerione, R.A.; Antonyak, M.A. The Enrichment of Survivin in Exosomes from Breast Cancer Cells Treated with Paclitaxel Promotes Cell Survival and Chemoresistance. Cancers 2016, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappellesso, R.; Tinazzi, A.; Giurici, T.; Simonato, F.; Guzzardo, V.; Ventura, L.; Crescenzi, M.; Chiarelli, S.; Fassina, A. Programmed cell death 4 and microRNA 21 inverse expression is maintained in cells and exosomes from ovarian serous carcinoma effusions. Cancer Cytopathol. 2014, 122, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Hmadcha, A.; Martin-Montalvo, A.; Gauthier, B.; Soria, B.; Capilla-Gonzalez, V. Therapeutic Potential of Mesenchymal Stem Cells for Cancer Therapy. Front. Bioeng. Biotechnol. 2020, 8, 43. [Google Scholar] [CrossRef]
- Wang, H.; Huang, H.; Wang, L.; Liu, Y.; Wang, M.; Zhao, S.; Lu, G.; Kang, X. Cancer-associated fibroblasts secreted miR-103a-3p suppresses apoptosis and promotes cisplatin resistance in non-small cell lung cancer. Aging 2021, 13, 14456–14468. [Google Scholar] [CrossRef]
- Cai, J.; Wang, J.; Huang, Y.; Wu, H.-X.; Xia, T.; Xiao, J.; Chen, X.; Li, H.; Qiu, Y.; Wang, Y.; et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis. 2016, 7, e2459. [Google Scholar] [CrossRef]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MohammadAlipour, A.; Dumbali, S.P.; Wenzel, P.L. Mitochondrial Transfer and Regulators of Mesenchymal Stromal Cell Function and Therapeutic Efficacy. Front. Cell Dev. Biol. 2020, 8, 603292. [Google Scholar] [CrossRef] [PubMed]
- Hough, K.; Trevor, J.L.; Strenkowski, J.G.; Wang, Y.; Chacko, B.K.; Tousif, S.; Chanda, D.; Steele, C.; Antony, V.B.; Dokland, T.; et al. Exosomal transfer of mitochondria from airway myeloid-derived regulatory cells to T cells. Redox Biol. 2018, 18, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.-F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 2017, 6, e22187. [Google Scholar] [CrossRef] [Green Version]
- Griessinger, E.; Moschoi, R.; Biondani, G.; Peyron, J.-F. Mitochondrial Transfer in the Leukemia Microenvironment. Trends Cancer 2017, 3, 828–839. [Google Scholar] [CrossRef]
- Li, Y.; Guo, X.; Guo, S.; Wang, Y.; Chen, L.; Liu, Y.; Jia, M.; An, J.; Tao, K.; Xing, J. Next generation sequencing-based analysis of mitochondrial DNA characteristics in plasma extracellular vesicles of patients with hepatocellular carcinoma. Oncol. Lett. 2020, 20, 2820–2828. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Gahan, P. Exosomes in Immune Regulation. Non-Coding RNA 2021, 7, 4. [Google Scholar] [CrossRef]
- Jang, S.C.; Crescitelli, R.; Cvjetkovic, A.; Belgrano, V.; Bagge, R.O.; Sundfeldt, K.; Ochiya, T.; Kalluri, R.; Lötvall, J. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J. Extracell. Vesicles 2019, 8, 1635420. [Google Scholar] [CrossRef] [Green Version]
- Marconi, S.; Santamaria, S.; Bartolucci, M.; Stigliani, S.; Aiello, C.; Gagliani, M.; Bellese, G.; Petretto, A.; Cortese, K.; Castagnola, P. Trastuzumab Modulates the Protein Cargo of Extracellular Vesicles Released by ERBB2+ Breast Cancer Cells. Membranes 2021, 11, 199. [Google Scholar] [CrossRef]
- Ruivo, C.F.; Adem, B.; Silva, M.; Melo, S.A. The Biology of Cancer Exosomes: Insights and New Perspectives. Cancer Res. 2017, 77, 6480–6488. [Google Scholar] [CrossRef] [Green Version]
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural Transm. 2010, 117, 1. [Google Scholar] [CrossRef] [PubMed]
- Keserű, J.S.; Soltész, B.; Lukács, J.; Márton, É.; Szilágyi-Bónizs, M.; Penyige, A.; Póka, R.; Nagy, B. Detection of cell-free, exosomal and whole blood mitochondrial DNA copy number in plasma or whole blood of patients with serous epithelial ovarian cancer. J. Biotechnol. 2019, 298, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.S.; Baty, J.W.; Dong, L.-F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells without Mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef]
- Bates, J.P.; Derakhshandeh, R.; Jones, L.; Webb, T.J. Mechanisms of Immune Evasion in Breast Cancer. BMC Cancer 2018, 18, 556. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Investig. 2016, 126, 1216–1223. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Cai, S.; Li, M.; Salma, K.I.; Zhou, X.; Han, F.; Chen, J.; Huyan, T. Tumor-Derived Extracellular Vesicles: Their Role in Immune Cells and Immunotherapy. Int. J. Nanomed. 2021, 16, 5395–5409. [Google Scholar] [CrossRef]
- Sheehan, C.; D’Souza-Schorey, C. Tumor-derived extracellular vesicles: Molecular parcels that enable regulation of the immune response in cancer. J. Cell Sci. 2019, 132, jcs235085. [Google Scholar] [CrossRef] [Green Version]
- She, Z.; Xie, M.; Hun, M.; Abdirahman, A.S.; Li, C.; Wu, F.; Luo, S.; Wan, W.; Wen, C.; Tian, J. Immunoregulatory Effects of Mitochondria Transferred by Extracellular Vesicles. Front. Immunol. 2021, 11, 628576. [Google Scholar] [CrossRef]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.G.; Huang, L.; VanderVen, B.C. Immunometabolism at the Interface between Macrophages and Pathogens. Immunology 2019, 19, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, S.; Pucci, M.; Alessandro, R.; Fontana, S. Extracellular Vesicles and Tumor-Immune Escape: Biological Functions and Clinical Perspectives. Int. J. Mol. Sci. 2020, 21, 2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graner, M.W.; Schnell, S.; Olin, M.R. Tumor-Derived Exosomes, Micrornas, and Cancer Immune Suppression. Semin. Immunopathol. 2018, 40, 505–515. [Google Scholar] [CrossRef]
- Syed, S.N.; Frank, A.-C.; Raue, R.; Brüne, B. Microrna-a Tumor Trojan Horse for Tumor-Associated Macrophages. Cells 2019, 8, 1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.; Kim, M.; Kim, Y.; Jung, H.S.; Jeoung, D. Exosomal MicroRNAs as Mediators of Cellular Interactions Between Cancer Cells and Macrophages. Front. Immunol. 2020, 11, 1167. [Google Scholar] [CrossRef]
- Yi, M.; Xu, L.; Jiao, Y.; Luo, S.; Li, A.; Wu, K. The role of cancer-derived microRNAs in cancer immune escape. J. Hematol. Oncol. 2020, 13, 25. [Google Scholar] [CrossRef] [Green Version]
- Garaude, J.; Acín-Pérez, R.; Martínez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villán, E.; Hervás-Stubbs, S.; Pelegrín, P.; Sander, L.E.; Enríquez, J.A.; et al. Mitochondrial Respiratory-Chain Adaptations in Macrophages Contribute to Antibacterial Host Defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef] [Green Version]
- Zaini, R.G.; Al-Rehaili, A.A. The Therapeutic Strategies of Regulatory T Cells in Malignancies and Stem Cell Transplantations. J. Oncol. 2019, 2019, 5981054. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.D.; Dorronsoro, A.; Booker, C.N. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J. Clin. Investig. 2016, 126, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.N.; Cheng, L.-C.; Kuo, C.-L.; Lo, Y.K.; Chou, H.-Y.; Chen, C.-H.; Wang, Y.-H.; Chuang, T.-H.; Cheng, S.-J.; Lee, A.Y.-L. Mitochondrial Lon-induced mtDNA leakage contributes to PD-L1–mediated immunoescape via STING-IFN signaling and extracellular vesicles. J. Immunother. Cancer 2020, 8, e001372. [Google Scholar] [CrossRef] [PubMed]
- Simula, L.; Antonucci, Y.; Scarpelli, G.; Cancila, V.; Colamatteo, A.; Manni, S.; De Angelis, B.; Quintarelli, C.; Procaccini, C.; Matarese, G.; et al. Pd-1-Induced T Cell Exhaustion Is Controlled by a Drp1-Dependent Mechanism. Mol. Oncol. 2022, 16, 188–205. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Wu, Y.; Wan, M.; You, Y.; Zhai, Z.; Song, Z. Metformin Increases Sensitivity of Melanoma Cells to Cisplatin by Blocking Exosomal-Mediated miR-34a Secretion. J. Oncol. 2021, 2021, 5525231. [Google Scholar] [CrossRef]
- Bukeirat, M.; Sarkar, S.N.; Hu, H.; Quintana, D.D.; Simpkins, J.W.; Ren, X. Mir-34a Regulates Blood-Brain Barrier Permeability and Mitochondrial Function by Targeting Cytochrome C. J. Cereb. Blood Flow Metab 2016, 36, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Claridge, B.; Lozano, J.; Poh, Q.H.; Greening, D.W. Development of Extracellular Vesicle Therapeutics: Challenges, Considerations, and Opportunities. Front. Cell Dev. Biol. 2021, 9, 734720. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sawada, K.; Miyamoto, M.; Shimizu, A.; Yamamoto, M.; Kinose, Y.; Nakamura, K.; Kawano, M.; Kodama, M.; Hashimoto, K.; et al. Exploring the potential of engineered exosomes as delivery systems for tumor-suppressor microRNA replacement therapy in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 527, 153–161. [Google Scholar] [CrossRef]
- Zhang, K.; Shao, C.-X.; Zhu, J.-D.; Lv, X.-L.; Tu, C.-Y.; Jiang, C.; Shang, M.-J. Exosomes function as nanoparticles to transfer miR-199a-3p to reverse chemoresistance to cisplatin in hepatocellular carcinoma. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Vakhshiteh, F.; Rahmani, S.; Ostad, S.N.; Madjd, Z.; Dinarvand, R.; Atyabi, F. Exosomes derived from miR-34a-overexpressing mesenchymal stem cells inhibit in vitro tumor growth: A new approach for drug delivery. Life Sci. 2020, 266, 118871. [Google Scholar] [CrossRef]
- Tomasetti, M.; Nocchi, L.; Staffolani, S.; Manzella, N.; Amati, M.; Goodwin, J.; Kluckova, K.; Nguyen, M.; Strafella, E.; Bajzikova, M.; et al. MicroRNA-126 Suppresses Mesothelioma Malignancy by Targeting IRS1 and Interfering with the Mitochondrial Function. Antioxid. Redox Signal. 2014, 21, 2109–2125. [Google Scholar] [CrossRef] [Green Version]
- Monaco, F.; Gaetani, S.; Alessandrini, F.; Tagliabracci, A.; Bracci, M.; Valentino, M.; Neuzil, J.; Amati, M.; Bovenzi, M.; Tomasetti, M.; et al. Exosomal transfer of miR-126 promotes the anti-tumour response in malignant mesothelioma: Role of miR-126 in cancer-stroma communication. Cancer Lett. 2019, 463, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Tao, H.; Xu, H.; Li, C.; Qiao, G.; Guo, M.; Cao, S.; Liu, M.; Lin, X. Exosomes-Coated miR-34a Displays Potent Antitumor Activity in Pancreatic Cancer Both in vitro and in vivo. Drug Des. Dev. Ther. 2020, 14, 3495–3507. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Wu, X.H.; Fan, Y.R.; Tan, B.; Quan, Z.; Luo, C.L. Exosome-Derived Microrna-29c Induces Apoptosis of Biu-87 Cells by Down Regulating Bcl-2 and Mcl-1. Asian Pac. J. Cancer Prev. 2014, 15, 3471–3476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleki, N.; MirHakimi, S.; Babashah, S.; Sayadi, A.; Parnian, G.; HadiZadeh, M. Use of cellular exosomes as a new carrier in breast cancer gene therapy. Klin. Onkol. 2021, 34, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Xu, H.; Zuo, L.; Li, C.; Qiao, G.; Guo, M.; Zheng, L.; Leitgeb, M.; Lin, X. Exosomes-coated bcl-2 siRNA inhibits the growth of digestive system tumors both in vitro and in vivo. Int. J. Biol. Macromol. 2020, 161, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liao, C.; Liang, S.; Ye, B.-C. A Novel Peptide-Equipped Exosomes Platform for Delivery of Antisense Oligonucleotides. ACS Appl. Mater. Interfaces 2021, 13, 10760–10767. [Google Scholar] [CrossRef] [PubMed]
- Lobos-González, L.; Bustos, R.; Campos, A.; Silva, V.; Silva, V.; Jeldes, E.; Salomon, C.; Varas-Godoy, M.; Cáceres-Verschae, A.; Duran, E.; et al. Exosomes released upon mitochondrial ASncmtRNA knockdown reduce tumorigenic properties of malignant breast cancer cells. Sci. Rep. 2020, 10, 343. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Liu, Z.; Li, Y.; Huang, Q.; Xia, L.; Li, K. Delivery of manganese carbonyl to the tumor microenvironment using Tumor-Derived exosomes for cancer gas therapy and low dose radiotherapy. Biomaterials 2021, 274, 120894. [Google Scholar] [CrossRef]
- Srivastava, A.; Amreddy, N.; Babu, A.; Panneerselvam, J.; Mehta, M.; Muralidharan, R.; Chen, A.; Zhao, Y.D.; Razaq, M.; Riedinger, N.; et al. Nanosomes carrying doxorubicin exhibit potent anticancer activity against human lung cancer cells. Sci. Rep. 2016, 6, 38541. [Google Scholar] [CrossRef]
- Hosseini, H.M.; Fooladi, A.A.I.; Soleimanirad, J.; Nourani, M.R.; Davaran, S.; Mahdavi, M. Staphylococcal entorotoxin B anchored exosome induces apoptosis in negative esterogen receptor breast cancer cells. Tumor Biol. 2014, 35, 3699–3707. [Google Scholar] [CrossRef]
- Hosseini, H.M.; Fooladi, A.A.I.; Soleimanirad, J.; Nourani, M.R.; Mahdavi, M. Exosome/staphylococcal enterotoxin B, an anti tumor compound against pancreatic cancer. J. BUON 2014, 19, 440–448. [Google Scholar]
- Han, D.; Wang, K.; Zhang, T.; Gao, G.-C.; Xu, H. Natural killer cell-derived exosome-entrapped paclitaxel can enhance its anti-tumor effect. Eur. Rev. Med Pharmacol. Sci. 2020, 24, 5703–5713. [Google Scholar] [PubMed]
- Phinney, D.G.; Pittenger, M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Stem Cells 2017, 35, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Cheng, Y.; Gao, H.; Zhuang, J.; Zhang, W.; Bian, Q.; Wang, F.; Du, Y.; Li, Z.; Kong, D.; et al. In Vivo Tracking of Mesenchymal Stem Cell-Derived Extracellular Vesicles Improving Mitochondrial Function in Renal Ischemia–Reperfusion Injury. ACS Nano 2020, 14, 4014–4026. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, S.; Wang, C.; Wang, Y.; Wan, M.; Liu, F.; Gong, M.; Yuan, Y.; Chen, Y.; Cheng, J.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Attenuate Mitochondrial Damage and Inflammation by Stabilizing Mitochondrial DNA. ACS Nano 2021, 15, 1519–1538. [Google Scholar] [CrossRef] [PubMed]
- Collino, F.; Lopes, J.A.; Tapparo, M.; Tortelote, G.G.; Kasai-Brunswick, T.H.; Lopes, G.M.; Almeida, D.B.; Skovronova, R.; Wendt, C.H.C.; De Miranda, K.R.; et al. Extracellular Vesicles Derived from Induced Pluripotent Stem Cells Promote Renoprotection in Acute Kidney Injury Model. Cells 2020, 9, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, X.; Zhu, W.; Zhang, Y.; Hong, Y.; Liang, X.; Fan, B.; Zhao, H.; He, H.; Zhang, F. Exosomes from mesenchymal stem cells overexpressing MIF enhance myocardial repair. J. Cell. Physiol. 2020, 235, 8010–8022. [Google Scholar] [CrossRef]
- Lee, M.; Liu, T.; Im, W.; Kim, M. Exosomes from Adipose-Derived Stem Cells Ameliorate Phenotype of Huntington’s Disease in Vitro Model. Eur. J. Neurosci. 2016, 44, 2114–2119. [Google Scholar] [CrossRef]
- Silva, J.D.; Su, Y.; Calfee, C.S.; Delucchi, K.L.; Weiss, D.; McAuley, D.F.; O’Kane, C.; Krasnodembskaya, A.D. Mesenchymal stromal cell extracellular vesicles rescue mitochondrial dysfunction and improve barrier integrity in clinically relevant models of ARDS. Eur. Respir. J. 2020, 58, 2002978. [Google Scholar] [CrossRef]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef]
- Lee, M.; Ban, J.-J.; Kim, K.Y.; Jeon, G.S.; Im, W.; Sung, J.-J.; Kim, M. Adipose-derived stem cell exosomes alleviate pathology of amyotrophic lateral sclerosis in vitro. Biochem. Biophys. Res. Commun. 2016, 479, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Bonafede, R.; Brandi, J.; Manfredi, M.; Scambi, I.; Schiaffino, L.; Merigo, F.; Turano, E.; Bonetti, B.; Marengo, E.; Cecconi, D.; et al. The Anti-Apoptotic Effect of ASC-Exosomes in an In Vitro ALS Model and Their Proteomic Analysis. Cells 2019, 8, 1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabria, E.; Scambi, I.; Bonafede, R.; Schiaffino, L.; Peroni, D.; Potrich, V.; Capelli, C.; Schena, F.; Mariotti, R. ASCs-Exosomes Recover Coupling Efficiency and Mitochondrial Membrane Potential in an in vitro Model of ALS. Front. Neurosci. 2019, 13, 1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, S.; Collino, F.; Deregibus, M.C.; Grange, C.; Tetta, C.; Camussi, G. Microvesicles Derived from Human Bone Marrow Mesenchymal Stem Cells Inhibit Tumor Growth. Stem Cells Dev. 2013, 22, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Reza, A.M.M.T.; Choi, Y.-J.; Yasuda, H.; Kim, J.-H. Human Adipose Mesenchymal Stem Cell-Derived Exosomal-Mirnas Are Critical Factors for Inducing Anti-Proliferation Signalling to A2780 and Skov-3 Ovarian Cancer Cells. Sci. Rep. 2016, 6, 38498. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Ma, Y.; Chen, X.; Ji, X.; Gao, J.; Zhang, L.; Ye, K.; Qiao, F.; Dai, Y.; Wang, H.; et al. Microvesicles released from human embryonic stem cell derived-mesenchymal stem cells inhibit proliferation of leukemia cells. Oncol. Rep. 2017, 38, 1013–1020. [Google Scholar] [CrossRef] [Green Version]
- Ardeshirylajimi, A.; Ashkezari, M.D.; Rezaie, Z.; Seifati, S.M. Antitumoral potential of microvesicles extracted from human adipose-derived mesenchymal stem cells on human breast cancer cells. J. Cancer Res. Ther. 2019, 15, 1114–1119. [Google Scholar] [CrossRef]
- Dibavar, M.A.; Soleimani, M.; Atashi, A.; Rassaei, N.; Amiri, S. The effect of simultaneous administration of arsenic trioxide and microvesicles derived from human bone marrow mesenchymal stem cells on cell proliferation and apoptosis of acute myeloid leukemia cell line. Artif. Cells Nanomed. Biotechnol. 2018, 46, S138–S146. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Song, B.; Wei, Y.; Chen, F.; Chi, Y.; Fan, H.; Liu, N.; Li, Z.; Han, Z.; Ma, F. Exosomes from mesenchymal stromal cells enhance imatinib-induced apoptosis in human leukemia cells via activation of caspase signaling pathway. Cytotherapy 2018, 20, 181–188. [Google Scholar] [CrossRef]
- Loussouarn, C.; Pers, Y.-M.; Bony, C.; Jorgensen, C.; Noël, D. Mesenchymal Stromal Cell-Derived Extracellular Vesicles Regulate the Mitochondrial Metabolism via Transfer of miRNAs. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial Degeneration Precedes the Development of Muscle Atrophy in Progression of Cancer Cachexia in Tumour-Bearing Mice. J. Cachexia Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Shuler, K.T.; Wilson, B.E.; Muñoz, E.R.; Mitchell, A.D.; Selsby, J.T.; Hudson, M.B. Muscle Stem Cell-Derived Extracellular Vesicles Reverse Hydrogen Peroxide-Induced Mitochondrial Dysfunction in Mouse Myotubes. Cells 2020, 9, 2544. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial Spare Respiratory Capacity: Mechanisms, Regulation, and Significance in Non-Transformed and Cancer Cells. FASEB J. 2020, 34, 13106–13124. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Gopal, S.K.; Xu, R.; Simpson, R.J.; Chen, W. Exosomes and Their Roles in Immune Regulation and Cancer. Semin. Cell Dev. Biol. 2015, 40, 72–81. [Google Scholar] [CrossRef]
- Wu, C.-H.; Li, J.; Li, L.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C.; Jong, A.Y. Extracellular vesicles derived from natural killer cells use multiple cytotoxic proteins and killing mechanisms to target cancer cells. J. Extracell. Vesicles 2019, 8, 1588538. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, N.J.; Sedelies, K.A.; Browne, K.A.; Wowk, M.E.; Newbold, A.; Sutton, V.R.; Clarke, C.J.P.; Oliaro, J.; Lindemann, R.K.; Bird, P.; et al. A Central Role for Bid in Granzyme B-induced Apoptosis. J. Biol. Chem. 2005, 280, 4476–4482. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lee, S.-A.; Gu, N.-Y.; Jeong, S.Y.; Byeon, J.S.; Jeong, D.-U.; Ouh, I.-O.; Lee, Y.-H.; Hyun, B.-H. Canine Natural Killer Cell-Derived Exosomes Exhibit Antitumor Activity in a Mouse Model of Canine Mammary Tumor. BioMed Res. Int. 2021, 2021, 6690704. [Google Scholar] [CrossRef]
- Ristorcelli, E.; Beraud, E.; Verrando, P.; Villard, C.; Lafitte, D.; Sbarra, V.; Lombardo, D.; Verine, A. Human tumor nanoparticles induce apoptosis of pancreatic cancer cells. FASEB J. 2008, 22, 3358–3369. [Google Scholar] [CrossRef]
- Liu, Y.D.; Zhuang, X.P.; Cai, D.L.; Cao, C.; Gu, Q.S.; Ni Liu, X.; Bin Zheng, B.; Guan, B.J.; Yu, L.; Li, J.K.; et al. Let-7a regulates EV secretion and mitochondrial oxidative phosphorylation by targeting SNAP23 in colorectal cancer. J. Exp. Clin. Cancer Res. 2021, 40, 31. [Google Scholar] [CrossRef]
- Philley, J.V.; Kannan, A.; Qin, W.; Sauter, E.R.; Ikebe, M.; Hertweck, K.L.; Troyer, D.A.; Semmes, O.J.; Dasgupta, S. Complex-I Alteration and Enhanced Mitochondrial Fusion Are Associated With Prostate Cancer Progression. J. Cell. Physiol. 2015, 231, 1364–1374. [Google Scholar] [CrossRef] [Green Version]
- Behzadi, E.; Mahmoodzadeh Hosseini, H.; Imani Fooladi, A.A. The inhibitory impacts of Lactobacillus rhamnosus GG-derived extracellular vesicles on the growth of hepatic cancer cells. Microb. Pathog. 2017, 110, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Reynolds, C.P. Bcl-2 Inhibitors: Targeting Mitochondrial Apoptotic Pathways in Cancer Therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EV Donor Cell | EV Recipient Cell | EV Cargo | ncRNA Target | Mitochondrial Effect | References |
|---|---|---|---|---|---|
| Dysregulation of cellular energetics | |||||
| Metabolic coupling with CAFs | |||||
| Prostate cancer cells | Fibroblasts | miR-424 | n.i. | IDH3a↓, NDUFA4L2(CI)↑→OXPHOS↓ | [84,85] |
| CRC cells | Fibroblasts | n.i. | n.i. | ATP5H(CV)↑, IDH2↑, ECH1↑, TUFM↓, ALSDH2↓ | [86] |
| Melanoma cells | Fibroblasts | miR-155, miR-210 | n.i. | OXPHOS↓ | [87] |
| Lung cancer cells | Fibroblasts | miR-210 | NDUFA4 (CI), SDHD (CII) | SDHD↓ → OXPHOS↓ | [88,89] |
| Nasopharyngeal carcinoma cells | Fibroblasts | LMP1 | NA | OXPHOS↓ | [90] |
| CAFs | Breast cancer cells | lncRNA SNHG3 | n.i. | OXPHOS↓ | [91] |
| CAFs | Prostate cancer cells | miR-22, let7a, miR-125b and metabolites (amino acids, lipids, TCA intermediates) | n.i. | CYTB(CIII)↓, COXI (CIV) ↓→ OXPHOS↓ | [92] |
| CAFs | Pancreatic cancer cells | miRNA, TCA metabolites | n.i. | TCA↑ | [93] |
| CAFs | CRC cells | miR-92a | n.i. | OXPHOS↓ | [94,95] |
| Metabolic coupling with adipocytes | |||||
| Breast cancer cells | Adipocytes | miR-155 | PPARγ | UCP1↑ | [96] |
| Breast cancer cells | Adipocytes | miR-144, miR-126 | n.i. | UCP1↑, mito matrix density↑ | [97] |
| Adipocytes | Melanoma, prostate tumor cells | FAO proteins (ECHA, HCDH), TCA proteins, OXPHOS proteins | NA | Mito number and density↑, FAO↑ | [98] |
| Adipocytes | Melanoma cells | FAO enzymes (HCDH, ECHA, HCD2), OXPHOS subunits (NDUA6 and NDUAS2 (CI) and ATPG (CV)), mitochondrial ADP/ATP transporters (ADT1, ADT2 and ADT3), fatty acids | NA | FAO↑, mito activity↑, mito intracellular trafficking↑ | [55] |
| Adipocytes | HCC cells | miR-23a/b | Mitochondrial glutaminase | Glutamine metabolism regulation | [99,100] |
| Metabolic subjugation of neighboring cells | |||||
| Kaposi’s sarcoma-associated herpes virus cells | Uninfected cells | miRNA | n.i. | Mito biogenesis↓, mito respiration↓ | [101] |
| Resistance to cell death | |||||
| Modulation of the Bcl-2 pathway | |||||
| Melanoma cells | Melanoma cells | n.i. | n.i. | Bax↓, Bcl-2↑ | [102] |
| Bladder cancer cells | Bladder cancer cells | n.i. | n.i. | Bax↓, Bcl-2↑ | [103] |
| Acute myeloid leukemia cells | Acute myeloid leukemia cells | circ_0009910 | miR-5195–3p | Bax↓, Bcl-2↑ | [104] |
| Glioma cells | Endothelial cells | lncRNA-CCAT2 | n.i. | Bax↓, Bcl-2↑ | [105] |
| MSCs | Multiple myeloma cells | lncRNA LINC00461 | miR-15a/16 | Bcl-2↑ | [106] |
| Chemotherapy resistance | |||||
| Trastuzamab-resistant breast cancer cells | Trastuzamab-sensitive breast cancer cells | lncRNA snhg14 | n.i. | Bax↓, Bcl-2↑ | [107] |
| Gefitinib-resistant esophageal squamous cell carcinoma cells | Esophageal squamous cell carcinoma cells | lncRNA PART1 | miR-129 | Bax↓, Bcl-2↑ | [108] |
| Gefitinib-resistant non-small cell lung cancer cells | Gefitinib-sensitive non-small cell lung cancer cells | miR-214 | n.i. | Bax↓, Bcl-2↑ | [109] |
| Vincristine-resistant gastric cancer cells | Vincristine-sensitive gastric cancer cells | CLIC1 | NA | Bcl-2↑ | [110] |
| Doxorubicin-resistant breast cancer cells | Doxorubicin-sensitive breast cancer cells | Hsp70 | NA | mito damage↑→ OXPHOS↓ | [111] |
| Temozolomide-resistant glioblastoma cells | Temozolomide-sensitive glioblastoma cells | Connexin 43 | NA | Bax↓, Bcl-2↑ | [112] |
| Gefitinib-treated NSCLC cells | Untreated NSCLC cells | n.i. | n.i. | Bax↓, Bcl-2↑ | [113] |
| CAFs | CRC cells | miR-92a-3p | n.i. | Bax↓ | [114] |
| Enhancing survival and chemoresistance through transfer of mitochondrial components | |||||
| Astrocytes | Glioma cells | mitochondria | NA | Mito metabolism↑ | [115] |
| Tumor-activated stromal cells | Glioblastoma cells | mitochondria | NA | OXPHOS↑ | [116] |
| CAFs | Breast cancer cells | mtDNA | NA | OXPHOS↑ | [117] |
| Aldh2-deficient hepatocytes | HCC cells | Oxidized mtDNA | NA | Bcl-2↑ | [118] |
| Escape from immune destruction | |||||
| Macrophage mitochondrial reprogramming in cancer | |||||
| Lung tumor cells | M0 macrophages | n.i. | n.i. | CI↓→OXPHOS ↓ | [119] |
| Hypoxia-induced TEVs | Infiltrating macrophages | let-7a | n.i. | OXPHOS↑ | [120] |
| Immunosuppression of T cells | |||||
| Melanoma cells | Tumor infiltrating cytotoxic CD8+ T lymphocytes | n.i. | n.i. | OXPHOS↑ | [121] |
| Melanoma cells | CD4+ T helper cells | miR-690 | n.i. | Bcl-2↓, MCL-1↓, Bcl-xl↓ | [122] |
| Renal carcinoma cells | Jurkat T Lymphocytes | FasL | NA | Bax↑, Bcl-2↓ | [123] |
| HNSCC cells | CD8+ T cells | FasL | NA | Release of CytC, mito membrane potential↓, Bcl-2↓, Bcl-XL↓, Bax↑, Bim↑ | [124] |
| MDSCs | CD8+ T cells | immuno-modulatory cytokines | NA | Mito ROS↑ → Bcl-2↓ | [125,126] |
| Activation of invasion and metastasis | |||||
| Increased motility and migration | |||||
| Hypoxic breast cancer cells | Epithelial cells | n.i. | n.i. | Drp1 phosphorylation, Mfn-1↑, Mfn-2↑, mito intracellular movement↑ | [127] |
| Bladder cancer cells | Urothelial cells | n.i. | n.i. | Mito size↓ | [128] |
| Adipocytes | Melanoma cells | Fission regulators (FIS1, OPA1), FA, FAO enzymes, OXPHOS proteins | NA | fission↑, mito size↓, mito intracellular movement↑ | [55] |
| CAFs | Pancreatic cancer cells | miR-106b | n.i. | Mfn-2↓ | [129,130] |
| Intravasation | |||||
| Breast cancer cells | Liver sinusoidal endothelial cells | n.i. | n.i. | Mito disfunction | [131,132] |
| Brain metastatic cancer cells | Endothelial cells | miR-181c | COX1 | Bax↑, Bcl-2↓ | [133,134,135] |
| EV Source | Target Cancer | Enrichment Method | (Bio)molecule | Target | Effect on Recipient Cell Mitochondria | Study Type | References |
|---|---|---|---|---|---|---|---|
| EVs enriched in biological cargo | |||||||
| MSCs | Breast | Cell transfection with lentivector | miR-34a | Bcl-2 | Bcl-2↓ | In vitro | [190] |
| HUVECs | Malignant mesothelioma | Cell transfection with transfection reagent | miR-126 | IRS1 | Affected mito metabolism, mito respiration↓ | In vitro | [191,192] |
| Human embryonic kidney (HEK293) cells | Pancreatic | EV transfection via ultrasound | miR-34 | Bcl-2 | Bcl-2↓ | In vitro and in vivo | [193] |
| Human bladder cancer (BIU-87) cells | Bladder | Cell transfection via viral vector (adenovirus) | miR-29c | n.i. | Bcl-2↓, MCL-1↓ | In vitro | [194] |
| Breast cancer (MDA-MB-231 class) cells | Breast | Cell transfection | miR-205 | n.i. | Bcl-2↓ | In vitro | [195] |
| Bovine milk | Pancreatic and colorectal | EV transfection via ultrasound | siRNA | Bcl-2 | Bcl-2↓ | In vitro and in vivo | [196] |
| Acute myeloid leukemia (HL-60 and MOLM-13) cells | Acute myeloid leukemia | Cell transfection with lipofectamine | siRNA | circ_0009910 | Bcl-2↓, Bax↑ | In vitro | [104] |
| Hepatocellular carcinoma (HepG2) cells | Liver | EV transfection via coincubation and oscillation | ASO-G3139 | Bcl-2 | Bcl-2↓ | In vitro | [197] |
| Breast cancer (MDA-MB-231) cells | Breast | Cell transfection with lipofectamine | ASO-1537S | ASncmtRNA | ASncmtRNA↓ | In vitro and in vivo | [198] |
| EVs loaded with exogenous drugs | |||||||
| Tumor cells | Metastatic breast | EV transfection with electroporation | Manganese carbonyl (MnCO) | NA | CO generation which induces mito toxicity | In vitro and in vivo | [199] |
| Non-small cell lung cancer (H1299) cells | Lung | EV coincubation with gold nanoparticles conjugated with doxorubicin | Doxorubicin | NA | Mito damage (perturbation of mito membrane potential), apoptosis via mito pathway↑ | In vitro | [200] |
| Epithelial-like breast cancer (MDA MB-231) cell | Estrogen receptor negative breast | Protein anchorage | Staphylococcal enterotoxin B (SEB) | NA | Bax↑, Bak↑, Bcl-2↓ | In vitro | [201] |
| Epithelial-like pancreatic cancer (MIA Paca-2) | Pancreatic | Protein anchorage | Staphylococcal enterotoxin B (SEB) | NA | Bax↑, Bak↑ | In vitro | [202] |
| Natural Killer (NK-92) | Breast | EV transfection via electroporation | Paclitaxel | NA | Bax/Bcl-2↑ | In vitro | [203] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carles-Fontana, R.; Heaton, N.; Palma, E.; Khorsandi, S.E. Extracellular Vesicle-Mediated Mitochondrial Reprogramming in Cancer. Cancers 2022, 14, 1865. https://doi.org/10.3390/cancers14081865
Carles-Fontana R, Heaton N, Palma E, Khorsandi SE. Extracellular Vesicle-Mediated Mitochondrial Reprogramming in Cancer. Cancers. 2022; 14(8):1865. https://doi.org/10.3390/cancers14081865
Chicago/Turabian StyleCarles-Fontana, Roger, Nigel Heaton, Elena Palma, and Shirin E. Khorsandi. 2022. "Extracellular Vesicle-Mediated Mitochondrial Reprogramming in Cancer" Cancers 14, no. 8: 1865. https://doi.org/10.3390/cancers14081865