
Perspective on the Use of DNA Repair Inhibitors as a Tool for Imaging and Radionuclide Therapy of Glioblastoma

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
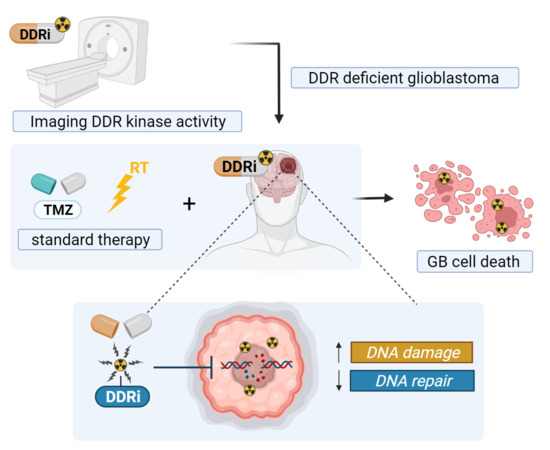
1. Introduction
2. Targeting DDR Pathways in GB
3. DDR (Radio)Pharmaceuticals
3.1. ATM/ATR Inhibitors
3.1.1. Targeting ATM/ATR as an Anti-GB Strategy
3.1.2. Current Status of ATM/ATR Targeted Therapy in GB
3.1.3. ATM/ATR Radiopharmaceuticals
3.2. CHK1/2 Inhibitors
3.2.1. Current Status of CHK1/2 Targeted Therapy in GB
3.2.2. CHK1/2 Radiopharmaceuticals
3.3. PARP Inhibitors
3.3.1. Current Status of PARP Targeted Therapy in GB
3.3.2. PARP Radiopharmaceuticals
3.4. DNA-PK Inhibitors
3.4.1. Current Status of DNA-PK Targeted Therapy in GB
3.4.2. DNA-PK Radiopharmaceuticals
4. Development of Other DDR Radiopharmaceuticals
5. Challenges and Risks of DDRi (Radio)Pharmaceuticals
6. Selection of New GB Radiopharmaceuticals Targeting the DDR
6.1. ATMi AZD1390
6.2. DNA-PKi Nedisertib (M3814)
6.3. CHK1i SAR-020106 and MK-8776 (SCH900776)
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caragher, S.P.; Sachdev, S.; Ahmed, A. Radiotherapy and Glioma Stem Cells: Searching for Chinks in Cellular Armor. Curr. Stem Cell Rep. 2017, 3, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Holland, E.C. Targeting brain cancer: Advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 2010, 10, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Ferri, A.; Stagni, V.; Barilà, D. Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 4910. [Google Scholar] [CrossRef]
- Li, L.-Y.; Guan, Y.-D.; Chen, X.-S.; Yang, J.-M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 2520. [Google Scholar] [CrossRef] [PubMed]
- Velic, D.; Couturier, A.M.; Ferreira, M.T.; Rodrigue, A.; Poirier, G.G.; Fleury, F.; Masson, J.Y. DNA Damage Signalling and Repair Inhibitors: The Long-Sought-After Achilles’ Heel of Cancer. Biomolecules 2015, 5, 3204–3259. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal. Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Lanz, M.C.; Dibitetto, D.; Smolka, M.B. DNA damage kinase signaling: Checkpoint and repair at 30 years. EMBO J. 2019, 38, e101801. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.S.; Gasol, A.; Slangen, P.L.G.; Cornelissen, F.M.G.; Lagerweij, T.; Veldman, H.; Dik, R.; van den Berg, J.; Slotman, B.J.; Würdinger, T.; et al. Identification of MEK162 as a Radiosensitizer for the Treatment of Glioblastoma. Mol. Cancer Ther. 2018, 17, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA Replication Stress for Cancer Therapy. Genes 2016, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, A.; Yung, W.K.A.; Majd, N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 22, 351. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Reuvers, T.G.A.; Kanaar, R.; Nonnekens, J. DNA Damage-Inducing Anticancer Therapies: From Global to Precision Damage. Cancers 2020, 12, 2098. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Shibata, A.; Jeggo, P.; Löbrich, M. The pendulum of the Ku-Ku clock. DNA Repair 2018, 71, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Squatrito, M.; Brennan, C.W.; Helmy, K.; Huse, J.T.; Petrini, J.H.; Holland, E.C. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell 2010, 18, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Pappula, A.L.; Rasheed, S.; Mirzaei, G.; Petreaca, R.C.; Bouley, R.A. A Genome-Wide Profiling of Glioma Patients with an IDH1 Mutation Using the Catalogue of Somatic Mutations in Cancer Database. Cancers 2021, 13, 4299. [Google Scholar] [CrossRef]
- Núñez, F.J.; Mendez, F.M.; Kadiyala, P.; Alghamri, M.S.; Savelieff, M.G.; Garcia-Fabiani, M.B.; Haase, S.; Koschmann, C.; Calinescu, A.-A.; Kamran, N.; et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 2019, 11, eaaq1427. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Benitez, J.A.; Li, J.; Miki, S.; Ponte de Albuquerque, C.; Galatro, T.; Orellana, L.; Zanca, C.; Reed, R.; Boyer, A.; et al. Inhibition of Nuclear PTEN Tyrosine Phosphorylation Enhances Glioma Radiation Sensitivity through Attenuated DNA Repair. Cancer Cell 2019, 35, 504–518. [Google Scholar] [CrossRef] [Green Version]
- Kaina, B.; Ochs, K.; Grösch, S.; Fritz, G.; Lips, J.; Tomicic, M.; Dunkern, T.; Christmann, M. BER, MGMT, and MMR in defense against alkylation-induced genotoxicity and apoptosis. Prog. Nucleic. Acid Res. Mol. Biol. 2001, 68, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/home (accessed on 7 December 2021).
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 2020, 78, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, M.A.; Rezende, F.; Titze-de-Almeida, R.; Cornelissen, B. BRCAness as a Biomarker of Susceptibility to PARP Inhibitors in Glioblastoma Multiforme. Biomolecules 2021, 11, 1188. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Cai, J.; Tan, Z.; Meng, X.; Li, R.; Li, Y.; Jiang, C. Mutant IDH1 Enhances Temozolomide Sensitivity via Regulation of the ATM/CHK2 Pathway in Glioma. Cancer Res. Treat. 2021, 53, 367–377. [Google Scholar] [CrossRef]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, C.B.; Noorbakhsh, S.I.; Sundaram, R.K.; Kalathil, A.N.; Ganesa, S.; Jia, L.; Breslin, H.; Burgenske, D.M.; Gilad, O.; Sarkaria, J.N.; et al. Temozolomide Sensitizes MGMT-Deficient Tumor Cells to ATR Inhibitors. Cancer Res. 2019, 79, 4331–4338. [Google Scholar] [CrossRef] [PubMed]
- Färkkilä, A.; Gulhan, D.C.; Casado, J.; Jacobson, C.A.; Nguyen, H.; Kochupurakkal, B.; Maliga, Z.; Yapp, C.; Chen, Y.A.; Schapiro, D.; et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat. Commun. 2020, 11, 1459. [Google Scholar] [CrossRef] [Green Version]
- Gulhan, D.C.; Lee, J.J.; Melloni, G.E.M.; Cortés-Ciriano, I.; Park, P.J. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef]
- Carlucci, G.; Carney, B.; Sadique, A.; Vansteene, A.; Tang, J.; Reiner, T. Evaluation of [(18)F]-ATRi as PET tracer for in vivo imaging of ATR in mouse models of brain cancer. Nucl. Med. Biol. 2017, 48, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.C.; Koustoulidou, S.; Cornelissen, B. Imaging the DNA damage response with PET and SPECT. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1065–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly, C.; Vidal, A.; Bonnemaire, C.; Kraeber-Bodéré, F.; Chérel, M.; Pallardy, A.; Rousseau, C.; Garcion, E.; Lacoeuille, F.; Hindré, F.; et al. Potential for Nuclear Medicine Therapy for Glioblastoma Treatment. Front Pharmacol. 2019, 10, 772. [Google Scholar] [CrossRef] [PubMed]
- Bolcaen, J.; Kleynhans, J.; Nair, S.; Verhoeven, J.; Goethals, I.; Sathekge, M.; Vandevoorde, C.; Ebenhan, T. A perspective on the radiopharmaceutical requirements for imaging and therapy of glioblastoma. Theranostics 2021, 11, 7911–7947. [Google Scholar] [CrossRef] [PubMed]
- Puttemans, J.; Lahoutte, T.; D’Huyvetter, M.; Devoogdt, N. Beyond the Barrier: Targeted Radionuclide Therapy in Brain Tumors and Metastases. Pharmaceutics 2019, 11, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ersahin, D.; Doddamane, I.; Cheng, D. Targeted radionuclide therapy. Cancers 2011, 3, 3838–3855. [Google Scholar] [CrossRef] [PubMed]
- Sgouros, G.; Roeske, J.C.; McDevitt, M.R.; Palm, S.; Allen, B.J.; Fisher, D.R.; Brill, A.B.; Song, H.; Howell, R.W.; Akabani, G.; et al. MIRD Pamphlet No. 22 (abridged): Radiobiology and dosimetry of alpha-particle emitters for targeted radionuclide therapy. J. Nucl. Med. 2010, 51, 311–328. [Google Scholar] [CrossRef] [Green Version]
- Zalutsky, M.R.; Reardon, D.A.; Akabani, G.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; McLendon, R.E.; Wong, T.Z.; Bigner, D.D. Clinical experience with alpha-particle emitting 211At: Treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J. Nucl. Med. 2008, 49, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Królicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Pawlak, D.; Kuliński, R.; Rola, R.; Merlo, A.; Morgenstern, A. Dose escalation study of targeted alpha therapy with [(225)Ac]Ac-DOTA-substance P in recurrence glioblastoma-safety and efficacy. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3595–3605. [Google Scholar] [CrossRef]
- Reilly, S.W.; Makvandi, M.; Xu, K.; Mach, R.H. Rapid Cu-Catalyzed [(211)At]Astatination and [(125)I]Iodination of Boronic Esters at Room Temperature. Org. Lett. 2018, 20, 1752–1755. [Google Scholar] [CrossRef]
- Makvandi, M.; Lee, H.; Puentes, L.N.; Reilly, S.W.; Rathi, K.S.; Weng, C.C.; Chan, H.S.; Hou, C.; Raman, P.; Martinez, D.; et al. Targeting PARP-1 with Alpha-Particles Is Potently Cytotoxic to Human Neuroblastoma in Preclinical Models. Mol. Cancer Ther. 2019, 18, 1195–1204. [Google Scholar] [CrossRef] [Green Version]
- Pirovano, G.; Wilson, T.C.; Reiner, T. Auger: The future of precision medicine. Nucl. Med. Biol. 2021, 96, 50–53. [Google Scholar] [CrossRef]
- Carlucci, G.; Carney, B.; Brand, C.; Kossatz, S.; Irwin, C.P.; Carlin, S.D.; Keliher, E.J.; Weber, W.; Reiner, T. Dual-Modality Optical/PET Imaging of PARP1 in Glioblastoma. Mol. Imaging Biol. 2015, 17, 848–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carney, B.; Carlucci, G.; Salinas, B.; Di Gialleonardo, V.; Kossatz, S.; Vansteene, A.; Longo, V.A.; Bolaender, A.; Chiosis, G.; Keshari, K.R.; et al. Non-invasive PET Imaging of PARP1 Expression in Glioblastoma Models. Mol. Imaging Biol. 2016, 18, 386–392. [Google Scholar] [CrossRef] [Green Version]
- Zmuda, F.; Blair, A.; Liuzzi, M.C.; Malviya, G.; Chalmers, A.J.; Lewis, D.; Sutherland, A.; Pimlott, S.L. An (18)F-Labeled Poly(ADP-ribose) Polymerase Positron Emission Tomography Imaging Agent. J. Med. Chem. 2018, 61, 4103–4114. [Google Scholar] [CrossRef] [PubMed]
- Zmuda, F.; Malviya, G.; Blair, A.; Boyd, M.; Chalmers, A.J.; Sutherland, A.; Pimlott, S.L. Synthesis and Evaluation of a Radioiodinated Tracer with Specificity for Poly(ADP-ribose) Polymerase-1 (PARP-1) in Vivo. J. Med. Chem. 2015, 58, 8683–8693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jannetti, S.A.; Carlucci, G.; Carney, B.; Kossatz, S.; Shenker, L.; Carter, L.M.; Salinas, B.; Brand, C.; Sadique, A.; Donabedian, P.L.; et al. PARP-1-Targeted Radiotherapy in Mouse Models of Glioblastoma. J. Nucl. Med. 2018, 59, 1225–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirovano, G.; Jannetti, S.A.; Carter, L.M.; Sadique, A.; Kossatz, S.; Guru, N.; Demétrio De Souza França, P.; Maeda, M.; Zeglis, B.M.; Lewis, J.S.; et al. Targeted Brain Tumor Radiotherapy Using an Auger Emitter. Clin. Cancer Res. 2020, 26, 2871–2881. [Google Scholar] [CrossRef] [Green Version]
- Salinas, B.; Irwin, C.P.; Kossatz, S.; Bolaender, A.; Chiosis, G.; Pillarsetty, N.; Weber, W.A.; Reiner, T. Radioiodinated PARP1 tracers for glioblastoma imaging. EJNMMI Res. 2015, 5, 123. [Google Scholar] [CrossRef] [Green Version]
- Jucaite, A.; Stenkrona, P.; Cselényi, Z.; De Vita, S.; Buil-Bruna, N.; Varnäs, K.; Savage, A.; Varrone, A.; Johnström, P.; Schou, M.; et al. Brain exposure of the ATM inhibitor AZD1390 in humans-a positron emission tomography study. Neuro Oncol. 2021, 23, 687–696. [Google Scholar] [CrossRef]
- Wickremsinhe, E.R.; Hynes, S.M.; Payne, C.D.; Guo, Y.; Cassidy, K.C. Disposition of [(14)C]LY2606368 following intravenous administration in patients with advanced and/or metastatic solid tumours. Xenobiotica 2020, 50, 793–804. [Google Scholar] [CrossRef]
- Schöder, H.; França, P.D.S.; Nakajima, R.; Burnazi, E.; Roberts, S.; Brand, C.; Grkovski, M.; Mauguen, A.; Dunphy, M.P.; Ghossein, R.A.; et al. Safety and Feasibility of PARP1/2 Imaging with (18)F-PARPi in Patients with Head and Neck Cancer. Clin. Cancer Res. 2020, 26, 3110–3116. [Google Scholar] [CrossRef] [Green Version]
- Guibbal, F.; Hopkins, S.L.; Pacelli, A.; Isenegger, P.G.; Mosley, M.; Torres, J.B.; Dias, G.M.; Mahaut, D.; Hueting, R.; Gouverneur, V.; et al. [(18)F]AZD2461, an Insight on Difference in PARP Binding Profiles for DNA Damage Response PET Imaging. Mol. Imaging Biol. 2020, 22, 1226–1234. [Google Scholar] [CrossRef]
- Reilly, S.W.; Puentes, L.N.; Schmitz, A.; Hsieh, C.J.; Weng, C.C.; Hou, C.; Li, S.; Kuo, Y.M.; Padakanti, P.; Lee, H.; et al. Synthesis and evaluation of an AZD2461 [(18)F]PET probe in non-human primates reveals the PARP-1 inhibitor to be non-blood-brain barrier penetrant. Bioorg. Chem. 2019, 83, 242–249. [Google Scholar] [CrossRef]
- Huang, T.; Hu, P.; Banizs, A.B.; He, J. Initial evaluation of Cu-64 labeled PARPi-DOTA PET imaging in mice with mesothelioma. Bioorg. Med. Chem. Lett. 2017, 27, 3472–3476. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Chen, H.; Mpoy, C.; Afrin, S.; Rogers, B.E.; Garbow, J.R.; Katzenellenbogen, J.A.; Xu, J. Radiosynthesis and Evaluation of Talazoparib and Its Derivatives as PARP-1-Targeting Agents. Biomedicines 2021, 9, 565. [Google Scholar] [CrossRef] [PubMed]
- Sander Effron, S.; Makvandi, M.; Lin, L.; Xu, K.; Li, S.; Lee, H.; Hou, C.; Pryma, D.A.; Koch, C.; Mach, R.H. PARP-1 Expression Quantified by [(18)F]FluorThanatrace: A Biomarker of Response to PARP Inhibition Adjuvant to Radiation Therapy. Cancer Biother. Radiopharm 2017, 32, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmonds, C.E.; Makvandi, M.; Lieberman, B.P.; Xu, K.; Zeng, C.; Li, S.; Hou, C.; Lee, H.; Greenberg, R.A.; Mankoff, D.A.; et al. [(18)F]FluorThanatrace uptake as a marker of PARP1 expression and activity in breast cancer. Am J. Nucl. Med. Mol. Imaging 2016, 6, 94–101. [Google Scholar] [PubMed]
- Lee, H.; Riad, A.; Martorano, P.; Mansfield, A.; Samanta, M.; Batra, V.; Mach, R.H.; Maris, J.M.; Pryma, D.A.; Makvandi, M. PARP-1-Targeted Auger Emitters Display High-LET Cytotoxic Properties In Vitro but Show Limited Therapeutic Utility in Solid Tumor Models of Human Neuroblastoma. J. Nucl. Med. 2020, 61, 850–856. [Google Scholar] [CrossRef]
- Liao, M.; Watkins, S.; Nash, E.; Isaacson, J.; Etter, J.; Beltman, J.; Fan, R.; Shen, L.; Mutlib, A.; Kemeny, V.; et al. Evaluation of absorption, distribution, metabolism, and excretion of [(14)C]-rucaparib, a poly(ADP-ribose) polymerase inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 765–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makvandi, M.; Xu, K.; Lieberman, B.P.; Anderson, R.C.; Effron, S.S.; Winters, H.D.; Zeng, C.; McDonald, E.S.; Pryma, D.A.; Greenberg, R.A.; et al. A Radiotracer Strategy to Quantify PARP-1 Expression In Vivo Provides a Biomarker That Can Enable Patient Selection for PARP Inhibitor Therapy. Cancer Res. 2016, 76, 4516–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.C.; Makvandi, M.; Xu, K.; Lieberman, B.P.; Zeng, C.; Pryma, D.A.; Mach, R.H. Iodinated benzimidazole PARP radiotracer for evaluating PARP1/2 expression in vitro and in vivo. Nucl. Med. Biol. 2016, 43, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Riss, P.J.; Soskic, V.; Schrattenholz, A.; Roesch, F. Synthesis and radiosynthesis of N5-[18F]fluoroethyl-pirenzepine and its metabolite N5-[18F]fluoroethyl-LS 75. J. Label. Compd. Radiopharm 2009, 52, 576–579. [Google Scholar] [CrossRef]
- Gao, M.; Wang, M.; Miller, K.D.; Zheng, Q.H. Simple synthesis of carbon-11-labeled chromen-4-one derivatives as new potential PET agents for imaging of DNA-dependent protein kinase (DNA-PK) in cancer. Appl. Radiat Isot. 2012, 70, 1558–1563. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.; Palmer, D.; Fitzgerald, R.; Andreu-Vieyra, C.; Zhang, H.; Tang, Z.; Su, D.; Sahasranaman, S. Human Mass Balance and Metabolite Profiling of [(14) C]-Pamiparib, a Poly (ADP-Ribose) Polymerase Inhibitor, in Patients With Advanced Cancer. Clin. Pharmacol. Drug Dev. 2021, 10, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 2018, 4, eaat1719. [Google Scholar] [CrossRef] [Green Version]
- Young, R.J.; Demétrio De Souza França, P.; Pirovano, G.; Piotrowski, A.F.; Nicklin, P.J.; Riedl, C.C.; Schwartz, J.; Bale, T.A.; Donabedian, P.L.; Kossatz, S.; et al. Preclinical and first-in-human-brain-cancer applications of [(18)F]poly (ADP-ribose) polymerase inhibitor PET/MR. Neurooncol. Adv. 2020, 2, vdaa119. [Google Scholar] [CrossRef]
- Donabedian, P.L.; Kossatz, S.; Engelbach, J.A.; Jannetti, S.A.; Carney, B.; Young, R.J.; Weber, W.A.; Garbow, J.R.; Reiner, T. Discriminating radiation injury from recurrent tumor with [(18)F]PARPi and amino acid PET in mouse models. EJNMMI Res. 2018, 8, 59. [Google Scholar] [CrossRef]
- Reiner, T.; Keliher, E.J.; Earley, S.; Marinelli, B.; Weissleder, R. Synthesis and in vivo imaging of a 18F-labeled PARP1 inhibitor using a chemically orthogonal scavenger-assisted high-performance method. Angew. Chem. Int. Ed. 2011, 50, 1922–1925. [Google Scholar] [CrossRef]
- Laird, J.; Lok, B.H.; Carney, B.; Kossatz, S.; de Stanchina, E.; Reiner, T.; Poirier, J.T.; Rudin, C.M. Positron-Emission Tomographic Imaging of a Fluorine 18-Radiolabeled Poly(ADP-Ribose) Polymerase 1 Inhibitor Monitors the Therapeutic Efficacy of Talazoparib in SCLC Patient-Derived Xenografts. J. Thorac. Oncol. 2019, 14, 1743–1752. [Google Scholar] [CrossRef]
- Carney, B.; Kossatz, S.; Lok, B.H.; Schneeberger, V.; Gangangari, K.K.; Pillarsetty, N.V.K.; Weber, W.A.; Rudin, C.M.; Poirier, J.T.; Reiner, T. Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nat. Commun. 2018, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Demétrio de Souza França, P.; Roberts, S.; Kossatz, S.; Guru, N.; Mason, C.; Zanoni, D.K.; Abrahão, M.; Schöder, H.; Ganly, I.; Patel, S.G.; et al. Fluorine-18 labeled poly (ADP-ribose) polymerase1 inhibitor as a potential alternative to 2-deoxy-2-[(18)F]fluoro-d-glucose positron emission tomography in oral cancer imaging. Nucl. Med. Biol. 2020, 84, 80–87. [Google Scholar] [CrossRef]
- Tang, J.; Salloum, D.; Carney, B.; Brand, C.; Kossatz, S.; Sadique, A.; Lewis, J.S.; Weber, W.A.; Wendel, H.G.; Reiner, T. Targeted PET imaging strategy to differentiate malignant from inflamed lymph nodes in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2017, 114, e7441–e7449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.C.; Xavier, M.A.; Knight, J.; Verhoog, S.; Torres, J.B.; Mosley, M.; Hopkins, S.L.; Wallington, S.; Allen, P.D.; Kersemans, V.; et al. PET Imaging of PARP Expression Using (18)F-Olaparib. J. Nucl. Med. 2019, 60, 504–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, T.L.; Friis, S.D.; Audrain, H.; Nordeman, P.; Antoni, G.; Skrydstrup, T. Efficient 11C-carbonylation of isolated aryl palladium complexes for PET: Application to challenging radiopharmaceutical synthesis. J. Am Chem. Soc. 2015, 137, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Makvandi, M.; Pantel, A.; Schwartz, L.; Schubert, E.; Xu, K.; Hsieh, C.J.; Hou, C.; Kim, H.; Weng, C.C.; Winters, H.; et al. A PET imaging agent for evaluating PARP-1 expression in ovarian cancer. J. Clin. Investig. 2018, 128, 2116–2126. [Google Scholar] [CrossRef]
- Michel, L.S.; Dyroff, S.; Brooks, F.J.; Spayd, K.J.; Lim, S.; Engle, J.T.; Phillips, S.; Tan, B.; Wang-Gillam, A.; Bognar, C.; et al. PET of Poly (ADP-Ribose) Polymerase Activity in Cancer: Preclinical Assessment and First In-Human Studies. Radiology 2017, 282, 453–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Xu, J.; Mpoy, C.; Chu, W.; Kim, S.H.; Li, H.; Rogers, B.E.; Katzenellenbogen, J.A. Preliminary evaluation of a novel (18)F-labeled PARP-1 ligand for PET imaging of PARP-1 expression in prostate cancer. Nucl. Med. Biol. 2018, 66, 26–31. [Google Scholar] [CrossRef]
- Zhou, D.; Chu, W.; Xu, J.; Jones, L.A.; Peng, X.; Li, S.; Chen, D.L.; Mach, R.H. Synthesis, [18F] radiolabeling, and evaluation of poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors for in vivo imaging of PARP-1 using positron emission tomography. Bioorg. Med. Chem. 2014, 22, 1700–1707. [Google Scholar] [CrossRef] [Green Version]
- Riad, A.; Gitto, S.B.; Lee, H.; Winters, H.D.; Martorano, P.M.; Hsieh, C.J.; Xu, K.; Omran, D.K.; Powell, D.J., Jr.; Mach, R.H.; et al. PARP Theranostic Auger Emitters Are Cytotoxic in BRCA Mutant Ovarian Cancer and Viable Tumors from Ovarian Cancer Patients Enable Ex-Vivo Screening of Tumor Response. Molecules 2020, 25, 6029. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Halladay, J.S.; Siu, M.; Chen, Y.; Hop, C.E.; Khojasteh, S.C.; Ma, S. Novel Mechanism of Decyanation of GDC-0425 by Cytochrome P450. Drug Metab. Dispos. 2017, 45, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Wickremsinhe, E.R.; Hynes, S.M.; Palmieri, M.D.; Mitchell, M.I.; Abraham, T.L.; Rehmel, J.F.; Chana, E.; Jost, L.M.; Cassidy, K.C. Disposition and metabolism of LY2603618, a Chk-1 inhibitor following intravenous administration in patients with advanced and/or metastatic solid tumors. Xenobiotica 2014, 44, 827–841. [Google Scholar] [CrossRef]
- Wickstroem, K.; Hagemann, U.B.; Cruciani, V.; Wengner, A.M.; Kristian, A.; Ellingsen, C.; Siemeister, G.; Bjerke, R.M.; Karlsson, J.; Ryan, O.B.; et al. Synergistic Effect of a Mesothelin-Targeted (227)Th Conjugate in Combination with DNA Damage Response Inhibitors in Ovarian Cancer Xenograft Models. J. Nucl. Med. 2019, 60, 1293–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helt, C.E.; Cliby, W.A.; Keng, P.C.; Bambara, R.A.; O’Reilly, M.A. Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage. J. Biol. Chem. 2005, 280, 1186–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Bennetzen, M.V.; Larsen, D.H.; Bunkenborg, J.; Bartek, J.; Lukas, J.; Andersen, J.S. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell. Proteom. 2010, 9, 1314–1323. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.M.; Harnden, D.G.; Arlett, C.F.; Harcourt, S.A.; Lehmann, A.R.; Stevens, S.; Bridges, B.A. Ataxia telangiectasia: A human mutation with abnormal radiation sensitivity. Nature 1975, 258, 427–429. [Google Scholar] [CrossRef]
- Barnieh, F.M.; Loadman, P.M.; Falconer, R.A. Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr. Res. Pharmacol. Drug Dis. 2021, 2, 100017. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.U.; Carruthers, R.; Gilmour, L.; Yildirim, S.; Watts, C.; Chalmers, A.J. Selective Inhibition of Parallel DNA Damage Response Pathways Optimizes Radiosensitization of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 4416–4428. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Reinhardt, H.C.; Bartkova, J.; Tommiska, J.; Blomqvist, C.; Nevanlinna, H.; Bartek, J.; Yaffe, M.B.; Hemann, M.T. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009, 23, 1895–1909. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Frosina, G.; Marubbi, D.; Marcello, D.; Vecchio, D.; Daga, A. The efficacy and toxicity of ATM inhibition in glioblastoma initiating cells-driven tumor models. Crit. Rev. Oncol. Hematol. 2019, 138, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Adams, B.R.; Wignarajah, S.; Beckta, J.M.; O’Connor, M.J.; Valerie, K. Dynamic inhibition of ATM kinase provides a strategy for glioblastoma multiforme radiosensitization and growth control. Cell Cycle 2012, 11, 1167–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.C.; Quek, H.; Offenhäuser, C.; Fazry, S.; Boyd, A.; Lavin, M.; Roberts, T.; Day, B. ATM inhibition prevents interleukin-6 from contributing to the proliferation of glioblastoma cells after ionizing radiation. J. Neurooncol. 2018, 138, 509–518. [Google Scholar] [CrossRef]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2020, 207, 107450. [Google Scholar] [CrossRef]
- Gorecki, L.; Andrs, M.; Rezacova, M.; Korabecny, J. Discovery of ATR kinase inhibitor berzosertib (VX-970, M6620): Clinical candidate for cancer therapy. Pharmacol. Ther. 2020, 210, 107518. [Google Scholar] [CrossRef]
- Yap, T.A.; Tolcher, A.W.; Plummer, E.R.; Becker, A.; Fleuranceau-Morel, P.; Goddemeier, T.; Locatelli, G.; Gounaris, I.; Bono, J.S.D. A first-in-human phase I study of ATR inhibitor M1774 in patients with solid tumors. J. Clin. Oncol. 2021, 39, TPS3153. [Google Scholar] [CrossRef]
- Nandakumar, P.; Mansouri, A.; Das, S. The Role of ATRX in Glioma Biology. Front. Oncol. 2017, 7, 236. [Google Scholar] [CrossRef]
- Garbarino, J.; Eckroate, J.; Sundaram, R.K.; Jensen, R.B.; Bindra, R.S. Loss of ATRX confers DNA repair defects and PARP inhibitor sensitivity. Transl. Oncol. 2021, 14, 101147. [Google Scholar] [CrossRef]
- Karlin, J.; Allen, J.; Ahmad, S.F.; Hughes, G.; Sheridan, V.; Odedra, R.; Farrington, P.; Cadogan, E.B.; Riches, L.C.; Garcia-Trinidad, A.; et al. Orally Bioavailable and Blood-Brain Barrier-Penetrating ATM Inhibitor (AZ32) Radiosensitizes Intracranial Gliomas in Mice. Mol. Cancer Ther. 2018, 17, 1637–1647. [Google Scholar] [CrossRef] [Green Version]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.N.; Rooney, C.; Griffin, N.; Roudier, M.; Young, L.A.; Garcia-Trinidad, A.; Hughes, G.D.; Whiteaker, J.R.; Wilson, Z.; Odedra, R.; et al. pRAD50: A novel and clinically applicable pharmacodynamic biomarker of both ATM and ATR inhibition identified using mass spectrometry and immunohistochemistry. Br. J. Cancer 2018, 119, 1233–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainey, M.D.; Charlton, M.E.; Stanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadkarni, A.; Shrivastav, M.; Mladek, A.C.; Schwingler, P.M.; Grogan, P.T.; Chen, J.; Sarkaria, J.N. ATM inhibitor KU-55933 increases the TMZ responsiveness of only inherently TMZ sensitive GBM cells. J. Neurooncol. 2012, 110, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Li, Z.; Yang, L.; Shen, L.; Wang, Y. A potential new role of ATM inhibitor in radiotherapy: Suppressing ionizing Radiation-Activated EGFR. Int. J. Radiat. Biol. 2020, 96, 461–468. [Google Scholar] [CrossRef]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2015, 9, 192–203. [Google Scholar] [CrossRef]
- Vecchio, D.; Daga, A.; Carra, E.; Marubbi, D.; Baio, G.; Neumaier, C.E.; Vagge, S.; Corvò, R.; Pia Brisigotti, M.; Louis Ravetti, J.; et al. Predictability, efficacy and safety of radiosensitization of glioblastoma-initiating cells by the ATM inhibitor KU-60019. Int. J. Cancer 2014, 135, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [Green Version]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer Ther. 2020, 19, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, J.P.Y.; Ma, H.T.; Poon, R.Y.C. Synergism between ATM and PARP1 Inhibition Involves DNA Damage and Abrogating the G(2) DNA Damage Checkpoint. Mol. Cancer Ther. 2020, 19, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; de Miguel Luken, M.J.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination with Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Bang, Y.J.; Carter, L.; Azaro, A.; Krebs, M.; Im, S.-A.; Chen, Y.; Buil-Bruna, N.; Li, Y.; Eaton, D.; et al. Abstract A094: Phase I modular study of AZD0156, a first-in-class oral selective inhibitor of ataxia telangiectasia mutated protein kinase (ATM), in combination with olaparib (AToM Study, Module 1). Mol. Cancer Ther. 2018, 17, A094. [Google Scholar] [CrossRef]
- Batey, M.A.; Zhao, Y.; Kyle, S.; Richardson, C.; Slade, A.; Martin, N.M.; Lau, A.; Newell, D.R.; Curtin, N.J. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer Ther. 2013, 12, 959–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, L.; Li, B.; Wu, Z.; Wu, Y.; Wang, Y.; Jin, F.; Li, D.; Ma, H.; Wang, D. Silencing of ataxia-telangiectasia mutated by siRNA enhances the in vitro and in vivo radiosensitivity of glioma. Oncol. Rep. 2016, 35, 3303–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eich, M.; Roos, W.P.; Nikolova, T.; Kaina, B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Mol. Cancer Ther. 2013, 12, 2529–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, C.; Kurian, K.M.; Williams, K.; Watts, C.; Jackson, A.; Carruthers, R.; Strathdee, K.; Cruickshank, G.; Dunn, L.; Erridge, S.; et al. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: Results of the phase I OPARATIC trial. Neuro Oncol. 2020, 22, 1840–1850. [Google Scholar] [CrossRef] [PubMed]
- Sim, H.W.; McDonald, K.L.; Lwin, Z.; Barnes, E.H.; Rosenthal, M.; Foote, M.C.; Koh, E.S.; Back, M.; Wheeler, H.; Sulman, E.P.; et al. A randomized phase II trial of veliparib, radiotherapy and temozolomide in patients with unmethylated MGMT glioblastoma: The VERTU study. Neuro Oncol. 2021, 32, 1736–1749. [Google Scholar] [CrossRef]
- Robins, H.I.; Zhang, P.; Gilbert, M.R.; Chakravarti, A.; de Groot, J.F.; Grimm, S.A.; Wang, F.; Lieberman, F.S.; Krauze, A.; Trotti, A.M.; et al. A randomized phase I/II study of ABT-888 in combination with temozolomide in recurrent temozolomide resistant glioblastoma: An NRG oncology RTOG group study. J. Neurooncol. 2016, 126, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurzrock, R.; Galanis, E.; Johnson, D.R.; Kansra, V.; Wilcoxen, K.; Mcclure, T.; Martell, R.E.; Agarwal, S. A phase I study of niraparib in combination with temozolomide (TMZ) in patients with advanced cancer. J. Clin. Oncol. 2014, 32, 2092. [Google Scholar] [CrossRef]
- Mei, L.; Zhang, J.; He, K. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: Where we stand. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; El-Khouiery, A.; Soria, J.C.; Lopez, J.; Berges, A.; Cheung, S.Y.A.; Irurzun-Arana, I.; Goldwin, A.; et al. Ceralasertib (AZD6738), an oral ATR kinase inhibitor, in combination with carboplatin in patients with advanced solid tumors: A Phase I study. Clin. Cancer Res. 2021, 27, 5213–5224. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Redon, C.E.; Sciuto, L.; Padiernos, E.; Ji, J.; Lee, M.J.; Yuno, A.; Lee, S.; Zhang, Y.; Tran, L.; et al. Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Milton, S.; Murphy, C.E.; et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [CrossRef] [Green Version]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [Green Version]
- Talele, S.; Mohammad, A.; Kim, M.; Burgenske, D.; Tuma, A.C.M.; Sarkaria, J.N.; Elmquist, W.F. Abstract 4858: CNS delivery of VX-970: A selective ATR inhibitor for radiosensitization in GBM. Cancer Res. 2019, 79, 4858. [Google Scholar] [CrossRef]
- Chen, E.M.; Quijano, A.R.; Seo, Y.E.; Jackson, C.; Josowitz, A.D.; Noorbakhsh, S.; Merlettini, A.; Sundaram, R.K.; Focarete, M.L.; Jiang, Z.; et al. Biodegradable PEG-poly(ω-pentadecalactone-co-p-dioxanone) nanoparticles for enhanced and sustained drug delivery to treat brain tumors. Biomaterials 2018, 178, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Wakimoto, H.; Martuza, R.L.; Rabkin, S.D. Abstract 1122: ATR inhibitors synergize with PARP inhibitors in killing glioblastoma stem cells and treating glioblastoma. Cancer Res. 2017, 77, 1122. [Google Scholar] [CrossRef]
- Liang, J.; Zhao, H.; Diplas, B.H.; Liu, S.; Liu, J.; Wang, D.; Lu, Y.; Zhu, Q.; Wu, J.; Wang, W.; et al. Genome-Wide CRISPR-Cas9 Screen Reveals Selective Vulnerability of ATRX-Mutant Cancers to WEE1 Inhibition. Cancer Res. 2020, 80, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 Kinase in Cancer. Trends. Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef]
- Vendetti, F.P.; Karukonda, P.; Clump, D.A.; Teo, T.; Lalonde, R.; Nugent, K.; Ballew, M.; Kiesel, B.F.; Beumer, J.H.; Sarkar, S.N.; et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J. Clin. Investig. 2018, 128, 3926–3940. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Nissink, J.W.M.; McGuire, T.; Turner, P.; Guichard, S.; Yates, J.W.T.; Lau, A.; Blades, K.; Heathcote, D.; Odedra, R.; et al. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J. Med. Chem. 2018, 61, 9889–9907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fròsina, G.; Profumo, A.; Marubbi, D.; Marcello, D.; Ravetti, J.L.; Daga, A. ATR kinase inhibitors NVP-BEZ235 and AZD6738 effectively penetrate the brain after systemic administration. Radiat. Oncol. 2018, 13, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Xie, G.; Zhou, G.; Cheng, Y.; Zhang, G.; Yao, G.; Chen, Y.; Li, Y.; Zhao, G. NVP-BEZ235, a novel dual PI3K-mTOR inhibitor displays anti-glioma activity and reduces chemoresistance to temozolomide in human glioma cells. Cancer Lett. 2015, 367, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Gil del Alcazar, C.R.; Hardebeck, M.C.; Mukherjee, B.; Tomimatsu, N.; Gao, X.; Yan, J.; Xie, X.J.; Bachoo, R.; Li, L.; Habib, A.A.; et al. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clin. Cancer Res. 2014, 20, 1235–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.J.; Long, L.M.; Yang, N.; Zhang, Q.Q.; Ji, W.J.; Zhao, J.H.; Qin, Z.H.; Wang, Z.; Chen, G.; Liang, Z.Q. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol. Sin. 2013, 34, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerniglia, G.J.; Karar, J.; Tyagi, S.; Christofidou-Solomidou, M.; Rengan, R.; Koumenis, C.; Maity, A. Inhibition of autophagy as a strategy to augment radiosensitization by the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Mol. Pharmacol. 2012, 82, 1230–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuger, S.; Graus, D.; Brendtke, R.; Günther, N.; Katzer, A.; Lutyj, P.; Polat, B.; Chatterjee, M.; Sukhorukov, V.L.; Flentje, M.; et al. Radiosensitization of Glioblastoma Cell Lines by the Dual PI3K and mTOR Inhibitor NVP-BEZ235 Depends on Drug-Irradiation Schedule. Transl. Oncol. 2013, 6, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netland, I.A.; Førde, H.E.; Sleire, L.; Leiss, L.; Rahman, M.A.; Skeie, B.S.; Gjerde, C.H.; Enger, P.; Goplen, D. Dactolisib (NVP-BEZ235) toxicity in murine brain tumour models. BMC Cancer 2016, 16, 657. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, B.; Tomimatsu, N.; Amancherla, K.; Camacho, C.V.; Pichamoorthy, N.; Burma, S. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia 2012, 14, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise-Draper, T.M.; Moorthy, G.; Salkeni, M.A.; Karim, N.A.; Thomas, H.E.; Mercer, C.A.; Beg, M.S.; O’Gara, S.; Olowokure, O.; Fathallah, H.; et al. A Phase Ib Study of the Dual PI3K/mTOR Inhibitor Dactolisib (BEZ235) Combined with Everolimus in Patients with Advanced Solid Malignancies. Target Oncol. 2017, 12, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Fazio, N.; Buzzoni, R.; Baudin, E.; Antonuzzo, L.; Hubner, R.A.; Lahner, H.; De Herder, W.W.; Raderer, M.; Teulé, A.; Capdevila, J.; et al. A Phase II Study of BEZ235 in Patients with Everolimus-resistant, Advanced Pancreatic Neuroendocrine Tumours. Anticancer Res. 2016, 36, 713–719. [Google Scholar]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Busino, L.; Chiesa, M.; Draetta, G.F.; Donzelli, M. Cdc25A phosphatase: Combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene 2004, 23, 2050–2056. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, Y.; Xie, T.; Luo, L.; Xu, C.; Gao, Q.; Shen, L.; Wan, F.; Lei, T.; Ye, F. Radiation-induced G2/M arrest rarely occurred in glioblastoma stem-like cells. Int. J. Radiat. Biol. 2018, 94, 394–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.P.; Jones, A.M.; Collins, I. Structure-based design, discovery and development of checkpoint kinase inhibitors as potential anticancer therapies. Expert Opin. Drug Discov. 2013, 8, 621–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daud, A.I.; Ashworth, M.T.; Strosberg, J.; Goldman, J.W.; Mendelson, D.; Springett, G.; Venook, A.P.; Loechner, S.; Rosen, L.S.; Shanahan, F.; et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 1060–1066. [Google Scholar] [CrossRef]
- Sausville, E.A.; Arbuck, S.G.; Messmann, R.; Headlee, D.; Bauer, K.S.; Lush, R.M.; Murgo, A.; Figg, W.D.; Lahusen, T.; Jaken, S.; et al. Phase I trial of 72-h continuous infusion UCN-01 in patients with refractory neoplasms. J. Clin. Oncol. 2001, 19, 2319–2333. [Google Scholar] [CrossRef]
- Laquente, B.; Lopez-Martin, J.; Richards, D.; Illerhaus, G.; Chang, D.Z.; Kim, G.; Stella, P.; Richel, D.; Szcylik, C.; Cascinu, S.; et al. A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC Cancer 2017, 17, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scagliotti, G.; Kang, J.H.; Smith, D.; Rosenberg, R.; Park, K.; Kim, S.W.; Su, W.C.; Boyd, T.E.; Richards, D.A.; Novello, S.; et al. Phase II evaluation of LY2603618, a first-generation CHK1 inhibitor, in combination with pemetrexed in patients with advanced or metastatic non-small cell lung cancer. Investig. New Drugs 2016, 34, 625–635. [Google Scholar] [CrossRef]
- Wehler, T.; Thomas, M.; Schumann, C.; Bosch-Barrera, J.; Viñolas Segarra, N.; Dickgreber, N.J.; Dalhoff, K.; Sebastian, M.; Corral Jaime, J.; Alonso, M.; et al. A randomized, phase 2 evaluation of the CHK1 inhibitor, LY2603618, administered in combination with pemetrexed and cisplatin in patients with advanced nonsquamous non-small cell lung cancer. Lung Cancer 2017, 108, 212–216. [Google Scholar] [CrossRef]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.S.; Moore, K.; Patel, M.; Grant, S.C.; Burris, H.A., 3rd; William, W.N., Jr.; Jones, S.; Meric-Bernstam, F.; Infante, J.; Golden, L.; et al. Evaluation of Prexasertib, a Checkpoint Kinase 1 Inhibitor, in a Phase Ib Study of Patients with Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 3263–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet. Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Karzai, F.H.; Soltani, S.N.; Zimmer, A.; Green, J.E.; Lee, M.J.; Trepel, J.B.; Yuno, A.; Lipkowitz, S.; Nair, J.; et al. A Phase II Single Arm Pilot Study of the CHK1 Inhibitor Prexasertib (LY2606368) in BRCA Wild-Type, Advanced Triple-Negative Breast Cancer. Oncologist 2020, 25, 1013. [Google Scholar] [CrossRef]
- Do, K.; Kochupurakkal, B.S.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 inhibitor prexasertib, and the PARP inhibitor olaparib, in high-grade serous ovarian cancer and other solid tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Hollebecque, A.; Postel-Vinay, S.; Bauer, T.M.; Blackwood, E.M.; Evangelista, M.; Mahrus, S.; Peale, F.V.; Lu, X.; Sahasranaman, S.; et al. Phase I Study of GDC-0425, a Checkpoint Kinase 1 Inhibitor, in Combination with Gemcitabine in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2017, 23, 2423–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, A.; Infante, J.R.; Shapiro, G.I.; Moore, K.N.; LoRusso, P.M.; Hamilton, E.; Cousin, S.; Toulmonde, M.; Postel-Vinay, S.; Tolaney, S.; et al. Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors. Ann. Oncol. 2018, 29, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Krishnamurthy Nemani, V.; Du, G.; Montano, R.; Song, R.; Gimi, B.; Swartz, H.M.; Eastman, A.; Khan, N. Monitoring oxygen levels in orthotopic human glioma xenograft following carbogen inhalation and chemotherapy by implantable resonator-based oximetry. Int. J. Cancer 2015, 136, 1688–1696. [Google Scholar] [CrossRef] [Green Version]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. Abrogation of the Chk1-mediated G(2) checkpoint pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res. 2001, 61, 5843–5849. [Google Scholar] [PubMed]
- Signore, M.; Pelacchi, F.; di Martino, S.; Runci, D.; Biffoni, M.; Giannetti, S.; Morgante, L.; De Majo, M.; Petricoin, E.F.; Stancato, L.; et al. Combined PDK1 and CHK1 inhibition is required to kill glioblastoma stem-like cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1223. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Dai, Y.; Grant, S.; Dent, P. Enhancing CHK1 inhibitor lethality in glioblastoma. Cancer Biol. Ther. 2012, 13, 379–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, T.M.; Galbán, S.; Li, F.; Heist, K.A.; Galbán, C.J.; Lawrence, T.S.; Holland, E.C.; Thomae, T.L.; Chenevert, T.L.; Rehemtulla, A.; et al. DW-MRI as a Predictive Biomarker of Radiosensitization of GBM through Targeted Inhibition of Checkpoint Kinases. Transl. Oncol. 2013, 6, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patties, I.; Kallendrusch, S.; Böhme, L.; Kendzia, E.; Oppermann, H.; Gaunitz, F.; Kortmann, R.D.; Glasow, A. The Chk1 inhibitor SAR-020106 sensitizes human glioblastoma cells to irradiation, to temozolomide, and to decitabine treatment. J. Exp. Clin. Cancer Res. 2019, 38, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, A.; Thoms, J.A.I.; Stringer, B.W.; Chung, S.A.; Ensbey, K.S.; Jue, T.R.; Jahan, Z.; Subramanian, S.; Anande, G.; Shen, H.; et al. Constitutive CHK1 Expression Drives a pSTAT3-CIP2A Circuit that Promotes Glioblastoma Cell Survival and Growth. Mol. Cancer Res. 2020, 18, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Lai, G.; Wan, F.; Xiao, Z.; Zeng, L.; Wang, X.; Ye, F.; Lei, T. Knockdown of checkpoint kinase 1 is associated with the increased radiosensitivity of glioblastoma stem-like cells. Tohoku J. Exp. Med. 2012, 226, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobson, A.G.; Lountos, G.T.; Lorenzi, P.L.; Llamas, J.; Connelly, J.; Cerna, D.; Tropea, J.E.; Onda, A.; Zoppoli, G.; Kondapaka, S.; et al. Cellular inhibition of checkpoint kinase 2 (Chk2) and potentiation of camptothecins and radiation by the novel Chk2 inhibitor PV1019 [7-nitro-1H-indole-2-carboxylic acid {4-[1-(guanidinohydrazone)-ethyl]-phenyl}-amide]. J. Pharmacol. Exp. Ther. 2009, 331, 816–826. [Google Scholar] [CrossRef] [Green Version]
- Vanan, I.; Dong, Z.; Tosti, E.; Warshaw, G.; Symons, M.; Ruggieri, R. Role of a DNA damage checkpoint pathway in ionizing radiation-induced glioblastoma cell migration and invasion. Cell Mol. Neurobiol. 2012, 32, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yan, Z.; Painter, C.L.; Zhang, Q.; Chen, E.; Arango, M.E.; Kuszpit, K.; Zasadny, K.; Hallin, M.; Hallin, J.; et al. PF-00477736 mediates checkpoint kinase 1 signaling pathway and potentiates docetaxel-induced efficacy in xenografts. Clin. Cancer Res. 2009, 15, 4630–4640. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.Y.; Tan, K.V.; Cornelissen, B. PARP Inhibitors in Cancer Diagnosis and Therapy. Clin. Cancer Res. 2021, 27, 1585–1594. [Google Scholar] [CrossRef]
- Gupta, S.K.; Smith, E.J.; Mladek, A.C.; Tian, S.; Decker, P.A.; Kizilbash, S.H.; Kitange, G.J.; Sarkaria, J.N. PARP Inhibitors for Sensitization of Alkylation Chemotherapy in Glioblastoma: Impact of Blood-Brain Barrier and Molecular Heterogeneity. Front Oncol. 2018, 8, 670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, J.F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Rabkin, S.D. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10, 2910. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, F.; Nagashima, H.; Ning, J.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Sandhu, S.K.; Carden, C.P.; de Bono, J.S. Poly(ADP-ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J. Clin. 2011, 61, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.T.; Muzik, H.; Turhan, A.G.; Zamò, A.; O’Connor, M.J.; Bebb, D.G.; Lees-Miller, S.P. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol. Cancer Ther. 2010, 9, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Majuelos-Melguizo, J.; Rodríguez, M.I.; López-Jiménez, L.; Rodríguez-Vargas, J.M.; Martí Martín-Consuegra, J.M.; Serrano-Sáenz, S.; Gavard, J.; de Almodóvar, J.M.; Oliver, F.J. PARP targeting counteracts gliomagenesis through induction of mitotic catastrophe and aggravation of deficiency in homologous recombination in PTEN-mutant glioma. Oncotarget 2015, 6, 4790–4803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.; de Gooijer, M.C.; Roig, E.M.; Buil, L.C.; Christner, S.M.; Beumer, J.H.; Würdinger, T.; Beijnen, J.H.; van Tellingen, O. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin. Cancer Res. 2014, 20, 2703–2713. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Guo, Y.; Liu, Y.; Wang, H.; Gong, W.; Wang, X.; Gao, Y.; Yu, F.; Su, D.; Wang, F.; et al. Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia 2020, 22, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Lester, A.; Rapkins, R.; Nixdorf, S.; Khasraw, M.; McDonald, K. Combining PARP inhibitors with radiation therapy for the treatment of glioblastoma: Is PTEN predictive of response? Clin. Transl. Oncol. 2017, 19, 273–278. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carruthers, R.; Chalmers, A.J. The potential of PARP inhibitors in neuro-oncology. CNS Oncol. 2012, 1, 85–97. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Li, X.; Gao, F.; de Groot, J.F.; Koul, D.; Yung, W.K.A. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro Oncol. 2021, 23, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, A.; Im, S.A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Färkkilä, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Anderson, V.E.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; Antoni, L.; Caldwell, J.J.; Aherne, W.; Pearl, L.H.; Oliver, A.W.; Collins, I.; et al. CCT241533 is a potent and selective inhibitor of CHK2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res. 2011, 71, 463–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Vandenberg, C.J.; Lieschke, E.; Di Rago, L.; Scott, C.L.; Majewski, I.J. CHK2 Inhibition Provides a Strategy to Suppress Hematologic Toxicity from PARP Inhibitors. Mol. Cancer Res. 2021, 19, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Wang, J.; Mishail, D.; Wang, C.Y. Recent advancements in PARP inhibitors-based targeted cancer therapy. Precis. Clin. Med. 2020, 3, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Ghorai, A.; Mahaddalkar, T.; Thorat, R.; Dutt, S. Sustained inhibition of PARP-1 activity delays glioblastoma recurrence by enhancing radiation-induced senescence. Cancer Lett. 2020, 490, 44–53. [Google Scholar] [CrossRef]
- Venere, M.; Hamerlik, P.; Wu, Q.; Rasmussen, R.D.; Song, L.A.; Vasanji, A.; Tenley, N.; Flavahan, W.A.; Hjelmeland, A.B.; Bartek, J.; et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014, 21, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Lesueur, P.; Lequesne, J.; Grellard, J.M.; Dugué, A.; Coquan, E.; Brachet, P.E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, A.J.; Gutierrez-Quintana, R.; Walker, D.J.; Williams, K.; Forster, D.; Jackson, M.R.; Derby, S.; Stobo, J.; Sweeting, L.; Kelly, C.; et al. Abstract IA-006: Enhancing the therapeutic ratio for glioblastoma by combining radiation therapy with PARP inhibitors. Clin. Cancer Res. 2021, 27, IA-006. [Google Scholar] [CrossRef]
- Barazzuol, L.; Jena, R.; Burnet, N.G.; Meira, L.B.; Jeynes, J.C.; Kirkby, K.J.; Kirkby, N.F. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat. Oncol. 2013, 8, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dungey, F.A.; Löser, D.A.; Chalmers, A.J. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-Ribose) polymerase: Mechanisms and therapeutic potential. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.L.; Kwon, H.C.; Burgan, W.E.; Carter, D.; Beam, K.; Weizheng, X.; Zhang, J.; Slusher, B.S.; Chakravarti, A.; Tofilon, P.J.; et al. In vitro and in vivo radiosensitization of glioblastoma cells by the poly (ADP-ribose) polymerase inhibitor E7016. Clin. Cancer Res. 2009, 15, 607–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, M.J.; Mulligan, E.A.; Grogan, P.T.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Curtin, N.J.; Lou, Z.; Decker, P.A.; Wu, W.; et al. Effective sensitization of temozolomide by ABT-888 is lost with development of temozolomide resistance in glioblastoma xenograft lines. Mol. Cancer Ther. 2009, 8, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Lemasson, B.; Wang, H.; Galbán, S.; Li, Y.; Zhu, Y.; Heist, K.A.; Tsein, C.; Chenevert, T.L.; Rehemtulla, A.; Galbán, C.J.; et al. Evaluation of Concurrent Radiation, Temozolomide and ABT-888 Treatment Followed by Maintenance Therapy with Temozolomide and ABT-888 in a Genetically Engineered Glioblastoma Mouse Model. Neoplasia 2016, 18, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Jue, T.R.; Nozue, K.; Lester, A.J.; Joshi, S.; Schroder, L.B.; Whittaker, S.P.; Nixdorf, S.; Rapkins, R.W.; Khasraw, M.; McDonald, K.L. Veliparib in combination with radiotherapy for the treatment of MGMT unmethylated glioblastoma. J. Transl. Med. 2017, 15, 61. [Google Scholar] [CrossRef] [Green Version]
- Albert, J.M.; Cao, C.; Kim, K.W.; Willey, C.D.; Geng, L.; Xiao, D.; Wang, H.; Sandler, A.; Johnson, D.H.; Colevas, A.D.; et al. Inhibition of poly(ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin. Cancer Res. 2007, 13, 3033–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajesh, M.; Mukhopadhyay, P.; Godlewski, G.; Bátkai, S.; Haskó, G.; Liaudet, L.; Pacher, P. Poly(ADP-ribose)polymerase inhibition decreases angiogenesis. Biochem. Biophys Res. Commun. 2006, 350, 1056–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, A.L.; Ricks, C.B.; Bohm, A.K.; Lun, X.; Maxwell, L.; Safdar, S.; Bukhari, S.; Gerber, A.; Sayeed, W.; Bering, E.A.; et al. ABT-888 restores sensitivity in temozolomide resistant glioma cells and xenografts. PLoS ONE 2018, 13, e0202860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, A.L.; Meode, M.; Tan, M.; Maxwell, L.; Bering, E.A.; Pedersen, H.; Willms, J.; Liao, J.; Black, S.; Cairncross, J.G.; et al. PARP inhibition suppresses the emergence of temozolomide resistance in a model system. J. Neurooncol. 2020, 148, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Mladek, A.C.; Carlson, B.L.; Boakye-Agyeman, F.; Bakken, K.K.; Kizilbash, S.H.; Schroeder, M.A.; Reid, J.; Sarkaria, J.N. Discordant in vitro and in vivo chemopotentiating effects of the PARP inhibitor veliparib in temozolomide-sensitive versus -resistant glioblastoma multiforme xenografts. Clin. Cancer Res. 2014, 20, 3730–3741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinberg, L.; Supko, J.G.; Mikkelsen, T.; Blakeley, J.O.N.; Stevens, G.; Ye, X.; Desideri, S.; Ryu, S.; Desai, B.; Giranda, V.L.; et al. Phase I adult brain tumor consortium (ABTC) trial of ABT-888 (veliparib), temozolomide (TMZ), and radiotherapy (RT) for newly diagnosed glioblastoma multiforme (GBM) including pharmacokinetic (PK) data. J. Clin. Oncol. 2013, 31, 2065. [Google Scholar] [CrossRef]
- Zhang, S.; Peng, X.; Li, X.; Liu, H.; Zhao, B.; Elkabets, M.; Liu, Y.; Wang, W.; Wang, R.; Zhong, Y.; et al. BKM120 sensitizes glioblastoma to the PARP inhibitor rucaparib by suppressing homologous recombination repair. Cell Death Dis. 2021, 12, 546. [Google Scholar] [CrossRef] [PubMed]
- Choi, P.J.; Cooper, E.; Schweder, P.; Mee, E.; Turner, C.; Faull, R.; Denny, W.A.; Dragunow, M.; Park, T.I.; Jose, J. PARP inhibitor cyanine dye conjugate with enhanced cytotoxic and antiproliferative activity in patient derived glioblastoma cell lines. Bioorg. Med. Chem. Lett. 2020, 30, 127252. [Google Scholar] [CrossRef] [PubMed]
- Parrish, K.E.; Cen, L.; Murray, J.; Calligaris, D.; Kizilbash, S.; Mittapalli, R.K.; Carlson, B.L.; Schroeder, M.A.; Sludden, J.; Boddy, A.V.; et al. Efficacy of PARP Inhibitor Rucaparib in Orthotopic Glioblastoma Xenografts Is Limited by Ineffective Drug Penetration into the Central Nervous System. Mol. Cancer Ther. 2015, 14, 2735–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, R.; Jones, C.; Middleton, M.; Wilson, R.; Evans, J.; Olsen, A.; Curtin, N.; Boddy, A.; McHugh, P.; Newell, D.; et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 7917–7923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesueur, P.; Chevalier, F.; El-Habr, E.A.; Junier, M.P.; Chneiweiss, H.; Castera, L.; Müller, E.; Stefan, D.; Saintigny, Y. Radiosensitization Effect of Talazoparib, a Parp Inhibitor, on Glioblastoma Stem Cells Exposed to Low and High Linear Energy Transfer Radiation. Sci. Rep. 2018, 8, 3664. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Yuan, B.; Chen, H.D.; Xu, L.; Tian, Y.N.; Zhang, A.; He, J.X.; Miao, Z.H. Acquired resistance of phosphatase and tensin homolog-deficient cells to poly(ADP-ribose) polymerase inhibitor and Ara-C mediated by 53BP1 loss and SAMHD1 overexpression. Cancer Sci. 2018, 109, 821–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Gao, F.; Zheng, S.; Zhang, C.; Martinez-Ledesma, E.; Ezhilarasan, R.; Ding, J.; Li, X.; Feng, N.; Multani, A.; et al. EGFR Amplification Induces Increased DNA Damage Response and Renders Selective Sensitivity to Talazoparib (PARP Inhibitor) in Glioblastoma. Clin. Cancer Res. 2020, 26, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Kizilbash, S.H.; Gupta, S.K.; Chang, K.; Kawashima, R.; Parrish, K.E.; Carlson, B.L.; Bakken, K.K.; Mladek, A.C.; Schroeder, M.A.; Decker, P.A.; et al. Restricted Delivery of Talazoparib Across the Blood-Brain Barrier Limits the Sensitizing Effects of PARP Inhibition on Temozolomide Therapy in Glioblastoma. Mol. Cancer Ther. 2017, 16, 2735–2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambade, M.J.; Van Swearingen, A.E.D.; McClure, M.B.; Deal, A.M.; Santos, C.; Sun, K.; Wang, J.; Mikule, K.; Anders, C.K. Efficacy and pharmacodynamics of niraparib in BRCA-mutant and wild-type intracranial triple-negative breast cancer murine models. Neuro-Oncol. Adv. 2019, 1, vdz005. [Google Scholar] [CrossRef] [PubMed]
- Miknyoczki, S.; Chang, H.; Grobelny, J.; Pritchard, S.; Worrell, C.; McGann, N.; Ator, M.; Husten, J.; Deibold, J.; Hudkins, R.; et al. The selective poly(ADP-ribose) polymerase-1(2) inhibitor, CEP-8983, increases the sensitivity of chemoresistant tumor cells to temozolomide and irinotecan but does not potentiate myelotoxicity. Mol. Cancer Ther. 2007, 6, 2290–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, A. Pamiparib: First Approval. Drugs 2021, 81, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Plummer, R.; Stephens, P.; Aissat-Daudigny, L.; Cambois, A.; Moachon, G.; Brown, P.D.; Campone, M. Phase 1 dose-escalation study of the PARP inhibitor CEP-9722 as monotherapy or in combination with temozolomide in patients with solid tumors. Cancer Chemothe.r Pharmacol. 2014, 74, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankaranarayanan, R.A.; Kossatz, S.; Weber, W.; Beheshti, M.; Morgenroth, A.; Mottaghy, F.M. Advancements in PARP1 Targeted Nuclear Imaging and Theranostic Probes. J. Clin. Med. 2020, 9, 2130. [Google Scholar] [CrossRef] [PubMed]
- Puentes, L.N.; Makvandi, M.; Mach, R.H. Molecular Imaging: PARP-1 and Beyond. J. Nucl. Med. 2021, 62, 765–770. [Google Scholar] [CrossRef]
- Thisgaard, H.; Halle, B.; Aaberg-Jessen, C.; Olsen, B.B.; Therkelsen, A.S.; Dam, J.H.; Langkjær, N.; Munthe, S.; Någren, K.; Høilund-Carlsen, P.F.; et al. Highly Effective Auger-Electron Therapy in an Orthotopic Glioblastoma Xenograft Model using Convection-Enhanced Delivery. Theranostics 2016, 6, 2278–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.C.; Jannetti, S.A.; Guru, N.; Pillarsetty, N.; Reiner, T.; Pirovano, G. Improved radiosynthesis of (123)I-MAPi, an auger theranostic agent. Int. J. Radiat. Biol. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Damia, G. Targeting DNA-PK in cancer. Mutat. Res. 2020, 821, 111692. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Huang, Z.; Zhai, K.; Huang, Q.; Tao, W.; Kim, L.; Wu, Q.; Almasan, A.; Yu, J.S.; Li, X.; et al. Inhibiting DNA-PK induces glioma stem cell differentiation and sensitizes glioblastoma to radiation in mice. Sci. Transl. Med. 2021, 13, eabc7275. [Google Scholar] [CrossRef] [PubMed]
- Kase, M.; Vardja, M.; Lipping, A.; Asser, T.; Jaal, J. Impact of PARP-1 and DNA-PK expression on survival in patients with glioblastoma multiforme. Radiother. Oncol. 2011, 101, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, H.; Liu, T.; Huang, M.; Butter, P.P.; Li, C.; Zhang, L.; Kao, G.D.; Gong, Y.; Maity, A.; et al. Temporal DNA-PK activation drives genomic instability and therapy resistance in glioma stem cells. JCI Insight 2018, 3, e98096. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Guo, Y.; Liu, X.; Czauderna, F.; Carr, M.I.; Zenke, F.T.; Blaukat, A.; Vassilev, L.T. Therapeutic Implications of p53 Status on Cancer Cell Fate Following Exposure to Ionizing Radiation and the DNA-PK Inhibitor M3814. Mol. Cancer Res. 2019, 17, 2457–2468. [Google Scholar] [CrossRef] [Green Version]
- Pospisilova, M.; Seifrtova, M.; Rezacova, M. Small molecule inhibitors of DNA-PK for tumor sensitization to anticancer therapy. J. Physiol. Pharmacol. 2017, 68, 337–344. [Google Scholar] [PubMed]
- Medová, M.; Medo, M.; Hovhannisyan, L.; Muñoz-Maldonado, C.; Aebersold, D.M.; Zimmer, Y. DNA-PK in human malignant disorders: Mechanisms and implications for pharmacological interventions. Pharmacol. Ther. 2020, 215, 107617. [Google Scholar] [CrossRef] [PubMed]
- Zenke, F.T.; Zimmermann, A.; Sirrenberg, C.; Dahmen, H.; Kirkin, V.; Pehl, U.; Grombacher, T.; Wilm, C.; Fuchss, T.; Amendt, C.; et al. Pharmacologic Inhibitor of DNA-PK, M3814, Potentiates Radiotherapy and Regresses Human Tumors in Mouse Models. Mol. Cancer Ther. 2020, 19, 1091–1101. [Google Scholar] [CrossRef] [Green Version]
- van Bussel, M.T.J.; Awada, A.; de Jonge, M.J.A.; Mau-Sørensen, M.; Nielsen, D.; Schöffski, P.; Verheul, H.M.W.; Sarholz, B.; Berghoff, K.; El Bawab, S.; et al. A first-in-man phase 1 study of the DNA-dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br. J. Cancer 2021, 124, 728–735. [Google Scholar] [CrossRef]
- Timme, C.R.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and as Orthotopic Xenografts. Mol. Cancer Ther. 2018, 17, 1207–1216. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.J.; Misenko, S.M.; Thandoni, A.; Schiff, D.; Jhawar, S.R.; Bunting, S.F.; Haffty, B.G. VX-984 is a selective inhibitor of non-homologous end joining, with possible preferential activity in transformed cells. Oncotarget 2018, 9, 25833–25841. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Karmokar, A.; Farrington, P.M.; James, N.H.; Ramos-Montoya, A.; Bickerton, S.J.; Hughes, G.D.; Illidge, T.M.; Cadogan, E.B.; Davies, B.R.; et al. Inhibition of DNA-PK with AZD7648 Sensitizes Tumor Cells to Radiotherapy and Induces Type I IFN-Dependent Durable Tumor Control. Clin. Cancer Res. 2021, 27, 4353–4366. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Sapinoso, L.M.; Tran, T.; Gaffney, B.; Wong, L.; Sankar, S.; Raymon, H.K.; Mortensen, D.S.; Xu, S. CC-115, a dual inhibitor of mTOR kinase and DNA-PK, blocks DNA damage repair pathways and selectively inhibits ATM-deficient cell growth in vitro. Oncotarget 2017, 8, 74688–74702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munster, P.; Mita, M.; Mahipal, A.; Nemunaitis, J.; Massard, C.; Mikkelsen, T.; Cruz, C.; Paz-Ares, L.; Hidalgo, M.; Rathkopf, D.; et al. First-In-Human Phase I Study Of A Dual mTOR Kinase And DNA-PK Inhibitor (CC-115) In Advanced Malignancy. Cancer Manag. Res. 2019, 11, 10463–10476. [Google Scholar] [CrossRef] [Green Version]
- Alexander, B.M.; Trippa, L.; Gaffey, S.; Arrillaga-Romany, I.C.; Lee, E.Q.; Rinne, M.L.; Ahluwalia, M.S.; Colman, H.; Fell, G.; Galanis, E.; et al. Individualized Screening Trial of Innovative Glioblastoma Therapy (INSIGhT): A Bayesian Adaptive Platform Trial to Develop Precision Medicines for Patients With Glioblastoma. JCO Precis. Oncol. 2019, 3, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Rasco, D.W.; Papadopoulos, K.P.; Pourdehnad, M.; Gandhi, A.K.; Hagner, P.R.; Li, Y.; Wei, X.; Chopra, R.; Hege, K.; DiMartino, J.; et al. A First-in-Human Study of Novel Cereblon Modulator Avadomide (CC-122) in Advanced Malignancies. Clin. Cancer Res. 2019, 25, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendell, J.C.; Varghese, A.M.; Hyman, D.M.; Bauer, T.M.; Pant, S.; Callies, S.; Lin, J.; Martinez, R.; Wickremsinhe, E.; Fink, A.; et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/mTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 3253–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein, M.M.; Hyman, D.M.; Caird, I.; Won, H.; Soldan, K.; Seier, K.; Iasonos, A.; Tew, W.P.; O’Cearbhaill, R.E.; Grisham, R.N.; et al. Phase 2 study of LY3023414 in patients with advanced endometrial cancer harboring activating mutations in the PI3K pathway. Cancer 2020, 126, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Hung, C.Y.; Lusvarghi, S.; Huang, Y.H.; Tseng, P.J.; Hung, T.H.; Yu, J.S.; Ambudkar, S.V. Overexpression of ABCB1 and ABCG2 contributes to reduced efficacy of the PI3K/mTOR inhibitor samotolisib (LY3023414) in cancer cell lines. Biochem. Pharmacol. 2020, 180, 114137. [Google Scholar] [CrossRef] [PubMed]
- Kopa, P.; Macieja, A.; Gulbas, I.; Pastwa, E.; Poplawski, T. Inhibition of DNA-PK potentiates the synergistic effect of NK314 and etoposide combination on human glioblastoma cells. Mol. Biol. Rep. 2020, 47, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Ismail, I.H.; Mårtensson, S.; Moshinsky, D.; Rice, A.; Tang, C.; Howlett, A.; McMahon, G.; Hammarsten, O. SU11752 inhibits the DNA-dependent protein kinase and DNA double-strand break repair resulting in ionizing radiation sensitization. Oncogene 2004, 23, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, T.; Zhao, Z.; Qu, Y.; Zhang, M.; Wang, H.; Zhang, Z.; Zhou, W.; Fan, X.; Yu, C.; Zhan, Q.; et al. Targeting hyperactivated DNA-PKcs by KU0060648 inhibits glioma progression and enhances temozolomide therapy via suppression of AKT signaling. Oncotarget 2016, 7, 55555–55571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, L.; Sun, C.; Zhang, H.; Miao, G.; Di, C.X.; Zhou, X.; Zhou, R.; Wang, Z. DNA-PKcs deficiency inhibits glioblastoma cell-derived angiogenesis after ionizing radiation. J. Cell Physiol. 2015, 230, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Quiros, S.; Roos, W.P.; Kaina, B. Rad51 and BRCA2--New molecular targets for sensitizing glioma cells to alkylating anticancer drugs. PLoS ONE 2011, 6, e27183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poty, S.; Mandleywala, K.; O’Neill, E.; Knight, J.C.; Cornelissen, B.; Lewis, J.S. (89)Zr-PET imaging of DNA double-strand breaks for the early monitoring of response following α- and β-particle radioimmunotherapy in a mouse model of pancreatic ductal adenocarcinoma. Theranostics 2020, 10, 5802–5814. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.; Kersemans, V.; Allen, P.D.; Terry, S.Y.A.; Torres, J.B.; Mosley, M.; Smart, S.; Lee, B.Q.; Falzone, N.; Vallis, K.A.; et al. Imaging DNA Damage Repair In Vivo After (177)Lu-DOTATATE Therapy. J. Nucl. Med. 2020, 61, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, C.J.; Bajrami, I.; Lord, C.J. Synthetic Lethality and Cancer-Penetrance as the Major Barrier. Trends Cancer 2018, 4, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Galia, A.; Calogero, A.E.; Condorelli, R.; Fraggetta, F.; La Corte, A.; Ridolfo, F.; Bosco, P.; Castiglione, R.; Salemi, M. PARP-1 protein expression in glioblastoma multiforme. Eur. J. Histochem. 2012, 56, e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, A.; Carter, C.A.; Kelly, R.J.; Gutierrez, M.; Kummar, S.; Szabo, E.; Yancey, M.A.; Ji, J.; Mannargudi, B.; Woo, S.; et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin. Cancer Res. 2012, 18, 2344–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhawan, M.S.; Bartelink, I.H.; Aggarwal, R.R.; Leng, J.; Zhang, J.Z.; Pawlowska, N.; Terranova-Barberio, M.; Grabowsky, J.A.; Gewitz, A.; Chien, A.J.; et al. Differential Toxicity in Patients with and without DNA Repair Mutations: Phase I Study of Carboplatin and Talazoparib in Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 6400–6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Das, T.; Suman, S.K.; Kumar, C.; Sarma, H.D.; Dash, A. Targeted Tumor Therapy with Radiolabeled DNA Intercalator: A Possibility? Preclinical Investigations with (177)Lu-Acridine. Biomed. Res. Int. 2020, 2020, 9514357. [Google Scholar] [CrossRef] [PubMed]
- Kubalanza, K.; Konecny, G.E. Mechanisms of PARP inhibitor resistance in ovarian cancer. Curr. Opin. Obstet. Gynecol. 2020, 32, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Mayor-Ruiz, C.; Lafarga, V.; Murga, M.; Vega-Sendino, M.; Ortega, S.; Fernandez-Capetillo, O. A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 2016, 62, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Henssen, A.G.; Reed, C.; Jiang, E.; Garcia, H.D.; von Stebut, J.; MacArthur, I.C.; Hundsdoerfer, P.; Kim, J.H.; de Stanchina, E.; Kuwahara, Y.; et al. Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Sci. Transl. Med. 2017, 9, aam9078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinrichsen, I.; Ackermann, A.; Düding, T.; Graband, A.; Filmann, N.; Plotz, G.; Zeuzem, S.; Brieger, A. Loss of MLH1 sensitizes colon cancer cells to DNA-PKcs inhibitor KU60648. Mol. Carcinog. 2017, 56, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Beebe, J.; Zhang, J.T. CC-115, a Dual Mammalian Target of Rapamycin/DNA-Dependent Protein Kinase Inhibitor in Clinical Trial, Is a Substrate of ATP-Binding Cassette G2, a Risk Factor for CC-115 Resistance. J. Pharmacol. Exp. Ther. 2019, 371, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Shirakami, Y.; Watabe, T.; Obata, H.; Kaneda, K.; Ooe, K.; Liu, Y.; Teramoto, T.; Toyoshima, A.; Shinohara, A.; Shimosegawa, E.; et al. Synthesis of [211At]4-astato-L-phenylalanine by dihydroxyboryl-astatine substitution reaction in aqueous solution. Sci. Rep. 2021, 11, 1–7. [Google Scholar] [CrossRef]
- Pozzi, O.R.; Zalutsky, M.R. Radiopharmaceutical chemistry of targeted radiotherapeutics, part 4: Strategies for (211)At labeling at high activities and radiation doses of (211)At α-particles. Nucl. Med. Biol. 2017, 46, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Filippone, N.R.; Reiner, T.; Roberts, S. Sensors and Inhibitors for the Detection of Ataxia Telangiectasia Mutated (ATM) Protein Kinase. Mol. Pharm. 2021, 18, 2470–2481. [Google Scholar] [CrossRef] [PubMed]
- Degorce, S.L.; Barlaam, B.; Cadogan, E.; Dishington, A.; Ducray, R.; Glossop, S.C.; Hassall, L.A.; Lach, F.; Lau, A.; McGuire, T.M.; et al. Discovery of Novel 3-Quinoline Carboxamides as Potent, Selective, and Orally Bioavailable Inhibitors of Ataxia Telangiectasia Mutated (ATM) Kinase. J. Med. Chem. 2016, 59, 6281–6292. [Google Scholar] [CrossRef] [PubMed]
- Pike, K.G.; Barlaam, B.; Cadogan, E.; Campbell, A.; Chen, Y.; Colclough, N.; Davies, N.L.; de-Almeida, C.; Degorce, S.L.; Didelot, M.; et al. The Identification of Potent, Selective, and Orally Available Inhibitors of Ataxia Telangiectasia Mutated (ATM) Kinase: The Discovery of AZD0156 (8-{6-[3-(Dimethylamino)propoxy]pyridin-3-yl}-3-methyl-1-(tetrahydro-2H-pyran-4-yl)-1,3-dihydro-2H-imidazo[4,5-c]quinolin-2-one). J. Med. Chem. 2018, 61, 3823–3841. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Thomas, S.E.; Chaplin, A.K.; Hardwick, S.W.; Chirgadze, D.Y.; Blundell, T.L. Structural insights into inhibitor regulation of the DNA repair protein DNA-PKcs. Nature 2022, 601, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Walton, M.I.; Eve, P.D.; Hayes, A.; Valenti, M.; De Haven Brandon, A.; Box, G.; Boxall, K.J.; Aherne, G.W.; Eccles, S.A.; Raynaud, F.I.; et al. The preclinical pharmacology and therapeutic activity of the novel CHK1 inhibitor SAR-020106. Mol. Cancer Ther. 2010, 9, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Yamamori, T.; Bo, T.; Sakai, Y.; Inanami, O. MK-8776, a novel Chk1 inhibitor, exhibits an improved radiosensitizing effect compared to UCN-01 by exacerbating radiation-induced aberrant mitosis. Transl. Oncol. 2017, 10, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Guzi, T.J.; Paruch, K.; Dwyer, M.P.; Labroli, M.; Shanahan, F.; Davis, N.; Taricani, L.; Wiswell, D.; Seghezzi, W.; Penaflor, E.; et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol. Cancer Ther. 2011, 10, 591–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.R.; Yang, Z.Z.; Wang, S.J.; Zhang, L.; Luo, J.R.; Feng, Y.; Yu, X.L.; Chen, X.X.; Guo, X.M. The Chk1 inhibitor MK-8776 increases the radiosensitivity of human triple-negative breast cancer by inhibiting autophagy. Acta Pharmacol. Sin. 2017, 38, 513–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labroli, M.A.; Dwyer, M.P.; Poker, C.; Keertikar, K.M.; Rossman, R.; Guzi, T.J. A convergent preparation of the CHK1 inhibitor MK-8776 (SCH 900776). Tetrahedron. Lett. 2016, 57, 2601–2603. [Google Scholar] [CrossRef]







{kind=link}
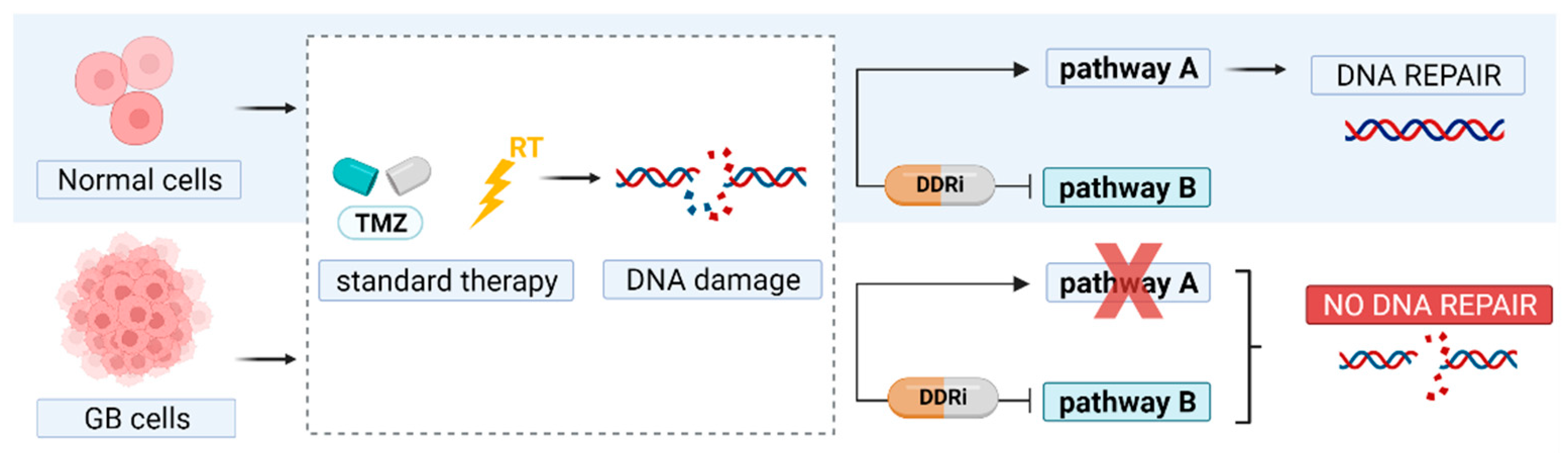{kind=link}
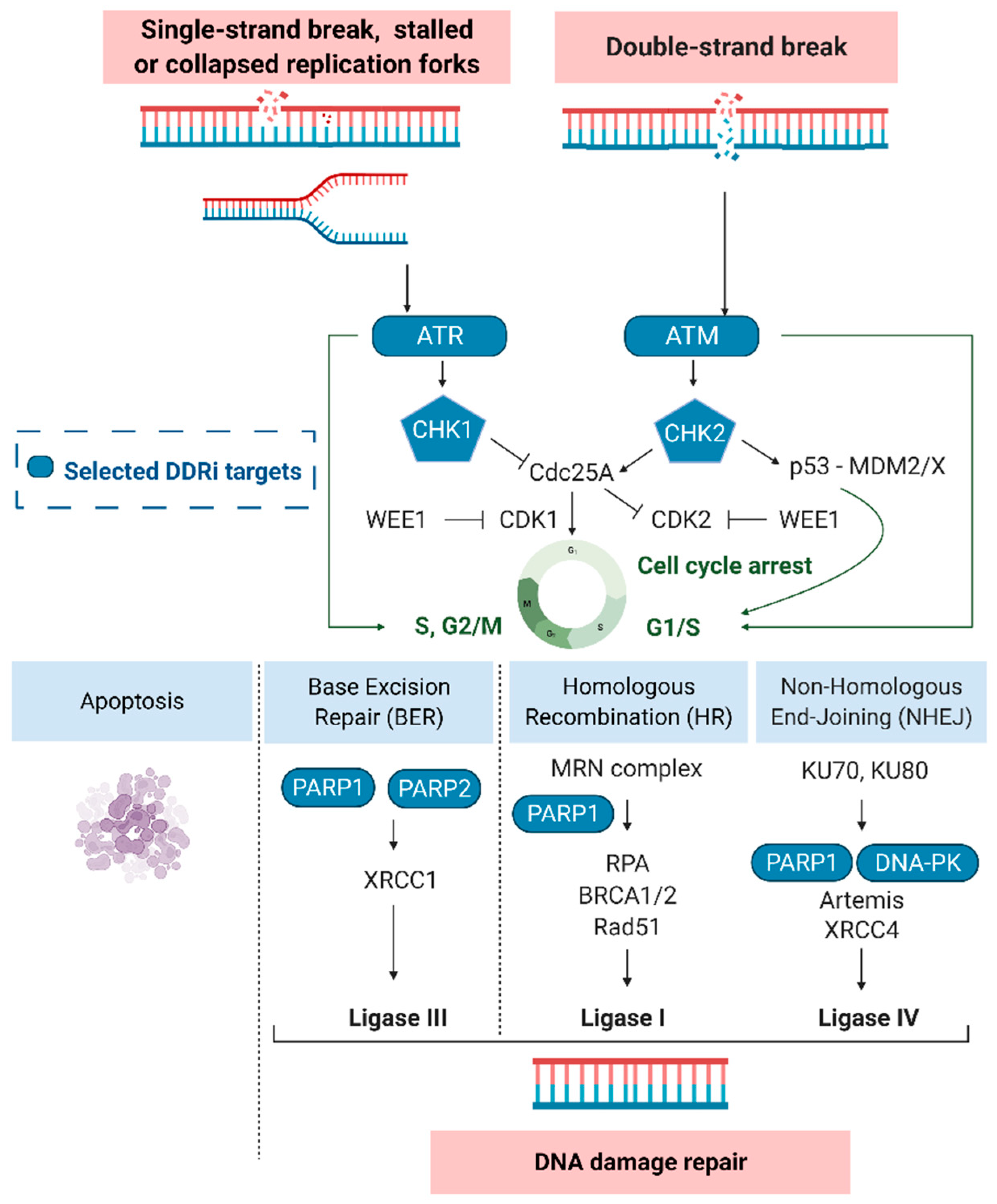{kind=link}
{kind=link}
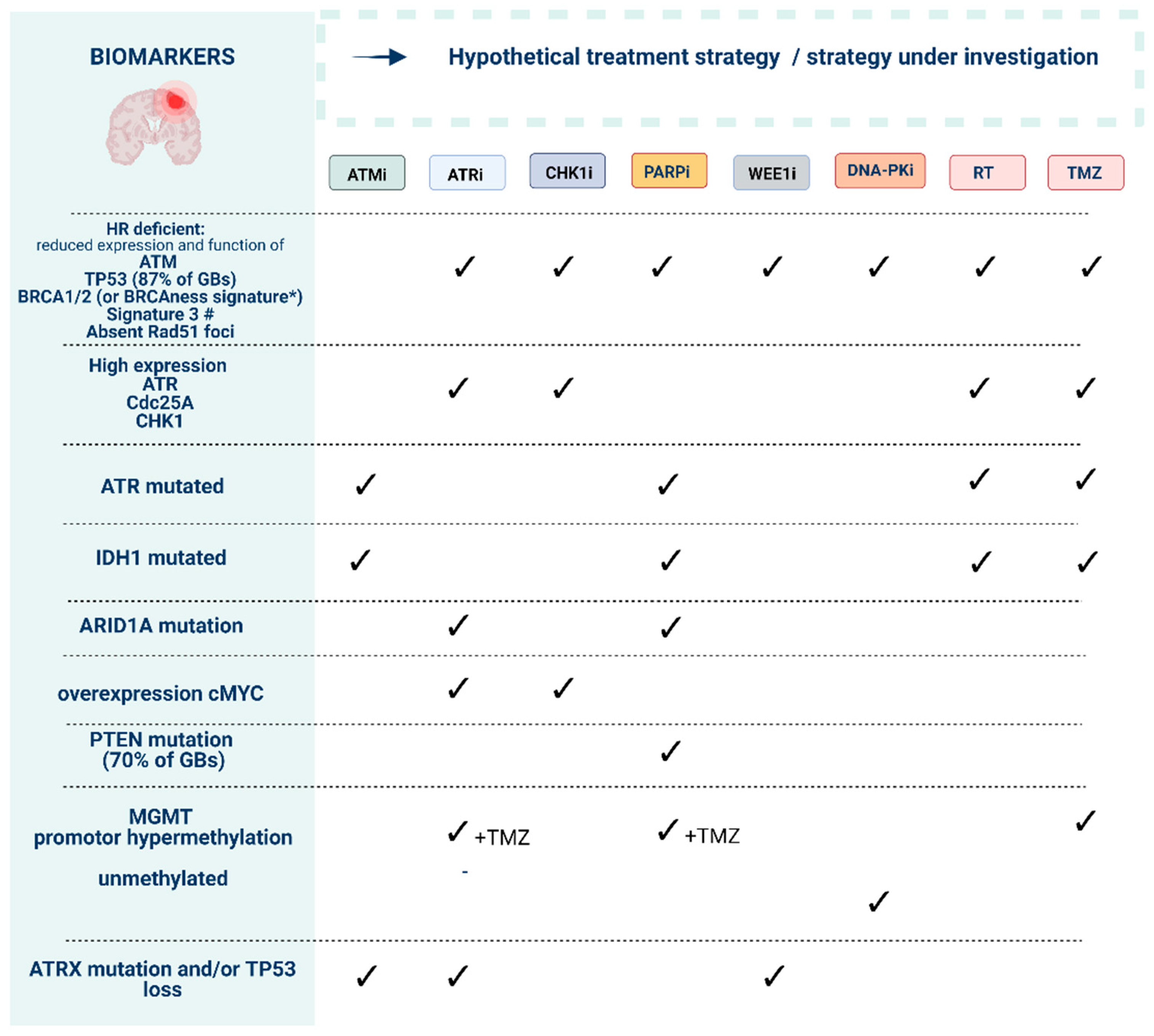{kind=link}
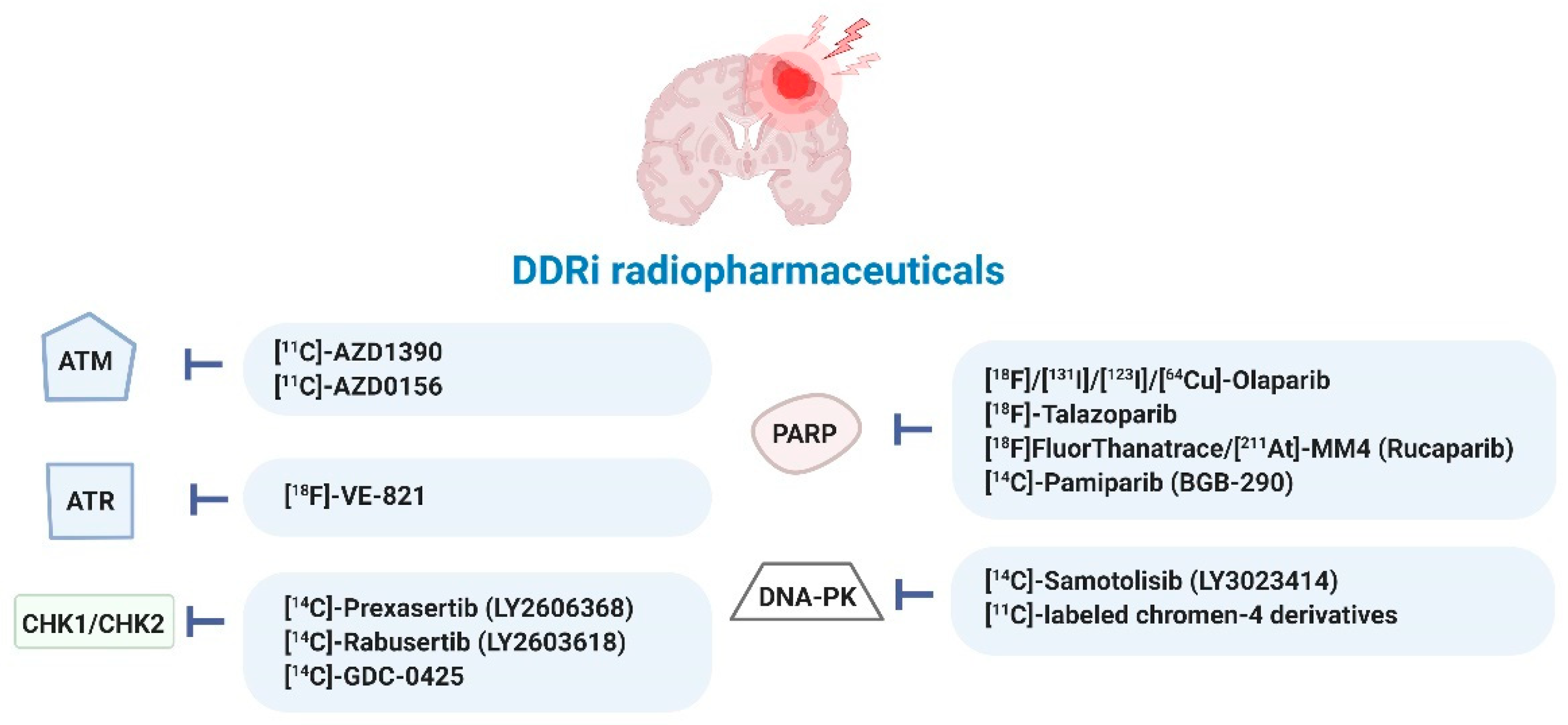{kind=link}
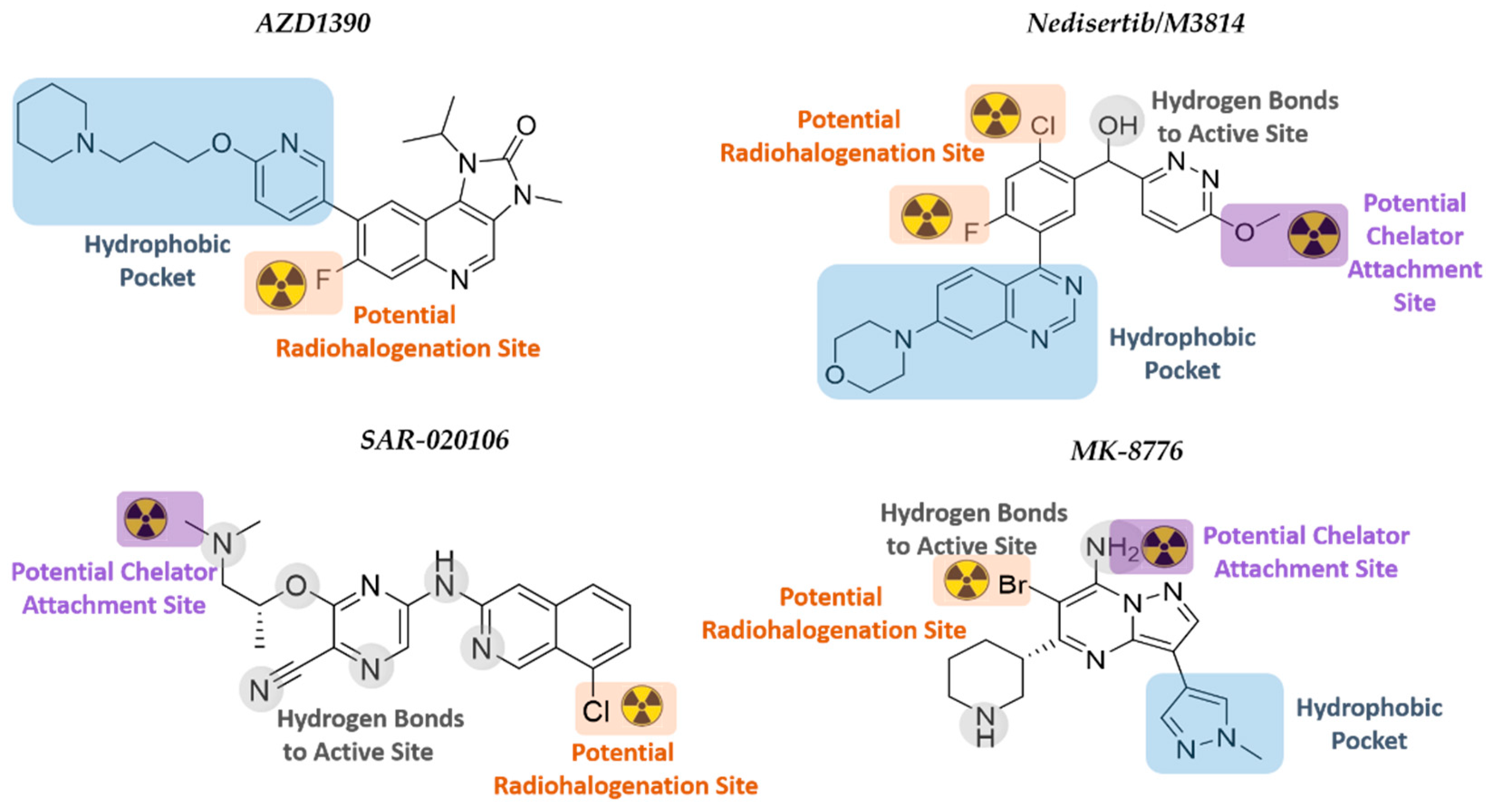{kind=link}
| Target | Drug | Combined Therapy * | Clinical Phase | Glioma Type | Biomarker Selection/Evaluation | Trial Number (Status) Reference |
|---|---|---|---|---|---|---|
| ATM | AZD1390 | RT | I | nd/rec GB, malignant brain neoplasms | - | NCT03423628 (r) |
| ATM | AZD0156 | Olaparib/Irinotecan/Fluorouracil/Folonic acid | I | AST (incl glioma) | - | NCT02588105 (anr) |
| PARP1/2/3 | Olaparib | TMZ/RT | II | advanced | IDH1/2 mutations | NCT03212274 (r) |
| TMZ/RT | II | rec HGG | IDH mutant | NCT03561870 (unknown) | ||
| Cediranib/vs Bevacizumab | II | rec GB | Angiogenesis-DNA repair | NCT02974621 (anr) | ||
| TMZ | I | rec GB | - | NCT01390571 (c) [129] | ||
| Pamiparib/TMZ/RT | 0/I | nd/rec GB | - | NCT04614909 (r) | ||
| PARP1/2 | Veliparib (ABT-888) | TMZ | II/III | nd GB | methylated MGMT-DDR genes-MGMT-PARP1 | NCT02152982 (anr) |
| TMZ/RT | II | nd GB | unmethylated MGMT | [130] | ||
| TMZ/RT | II | nd grade III-IV | no H3 K27M/BRAFV600 mutations | NCT03581292 (anr) | ||
| TMZ | I/II | rec GB | - | [131] | ||
| TMZ/RT | I | nd GB | plasma proteomic evaluation | NCT00770471 (c) | ||
| PARP1/2 | Talazoparib | Carboplatin | II | rec GB | IDH/PTEN mutation “BRCAness” signature * | NCT04740190 (r) |
| PARP1/2 | Niraparib (MK-4827) | RT | II | rec GB | - | NCT04715620 (r) |
| Tumor-Treating Fields | II | rec GB | MGMT | NCT04221503 (r) | ||
| TMZ | I | advanced cancer, incl GB | - | NCT01294735 (c) [132] | ||
| PARP1/2 | Pamiparib (BGB-290) | TMZ/RT | I/II | nd/rec GB | MGMT | NCT03150862 (c) |
| TMZ | I/II | rec grade II-IV | IDH1/2-mutant | NCT03914742 (r) | ||
| TMZ | I | nd/rec grade I-IV | IDH1/2-mutant | NCT03749187 (r) | ||
| TMZ/RT | 0/I | nd/rec GB | - | NCT04614909 (r) | ||
| DNA-PK | Nedisertib (M3814) | TMZ/RT | I | nd GB | MGMT unmethylated | NCT04555577 (r) |
| PI3K/mTOR/DNA-PK | Samotolisib (LY3023414) | - | II | paediatric CNS tumors | PI3K/mTOR mutations | NCT03213678 (r) |
| Inclusion Criteria |
| 1. The DDRi was studied preclinically or in clinical trials in GB. 2. The DDRi is a small molecule that: A. contains a halogen which indicates a position that can potentially be radio-iodinated or -astatinated; and/or B. has a potential site for attachment of a chelator. 3. The DDRi has already been radiolabeled with a diagnostic isotope and was studied in GB. |
| Exclusion Criteria |
| 1. Clinical trials results indicate candidate exclusion by way of: A. findings in GB patients revealed unwanted safety/tolerability issues (single agent), serious adverse events that were irreversible or responsible for treatment discontinuation, and/or B. occurrence of unfavorable pharmacokinetic properties. 2. The DDRi does not contain a halogen or any possible site for chelator attachment. 3. The DDRi has already been radiolabeled (diagnostic and/or therapeutic radionuclide) but was not studied in GB. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Everix, L.; Nair, S.; Driver, C.H.S.; Goethals, I.; Sathekge, M.M.; Ebenhan, T.; Vandevoorde, C.; Bolcaen, J. Perspective on the Use of DNA Repair Inhibitors as a Tool for Imaging and Radionuclide Therapy of Glioblastoma. Cancers 2022, 14, 1821. https://doi.org/10.3390/cancers14071821
Everix L, Nair S, Driver CHS, Goethals I, Sathekge MM, Ebenhan T, Vandevoorde C, Bolcaen J. Perspective on the Use of DNA Repair Inhibitors as a Tool for Imaging and Radionuclide Therapy of Glioblastoma. Cancers. 2022; 14(7):1821. https://doi.org/10.3390/cancers14071821
Chicago/Turabian StyleEverix, Liesbeth, Shankari Nair, Cathryn H. S. Driver, Ingeborg Goethals, Mike M. Sathekge, Thomas Ebenhan, Charlot Vandevoorde, and Julie Bolcaen. 2022. "Perspective on the Use of DNA Repair Inhibitors as a Tool for Imaging and Radionuclide Therapy of Glioblastoma" Cancers 14, no. 7: 1821. https://doi.org/10.3390/cancers14071821