
A Gene Co-Expression Network-Based Drug Repositioning Approach Identifies Candidates for Treatment of Hepatocellular Carcinoma

 , , , , and
, , , , and 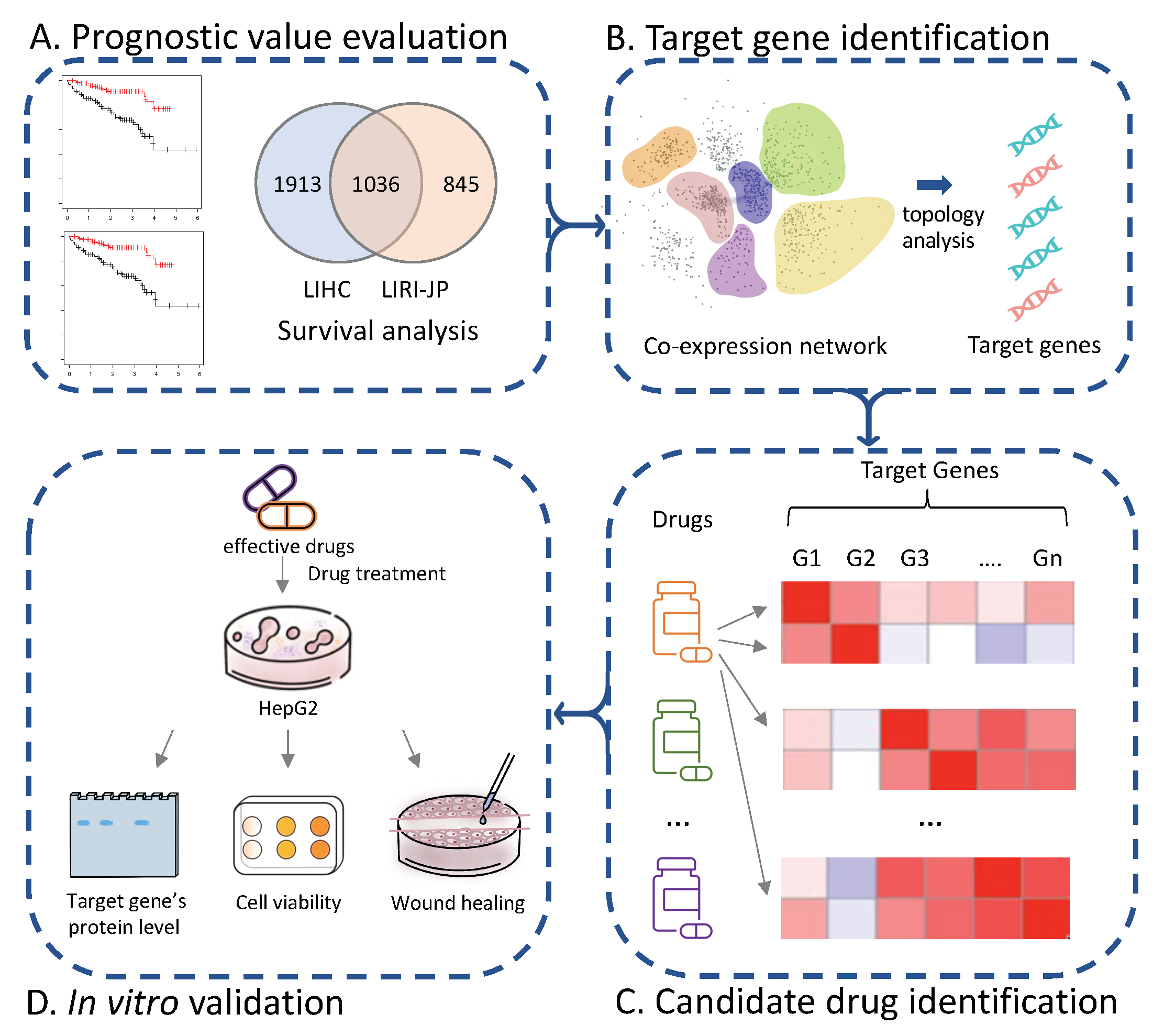{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data and Preprocessing
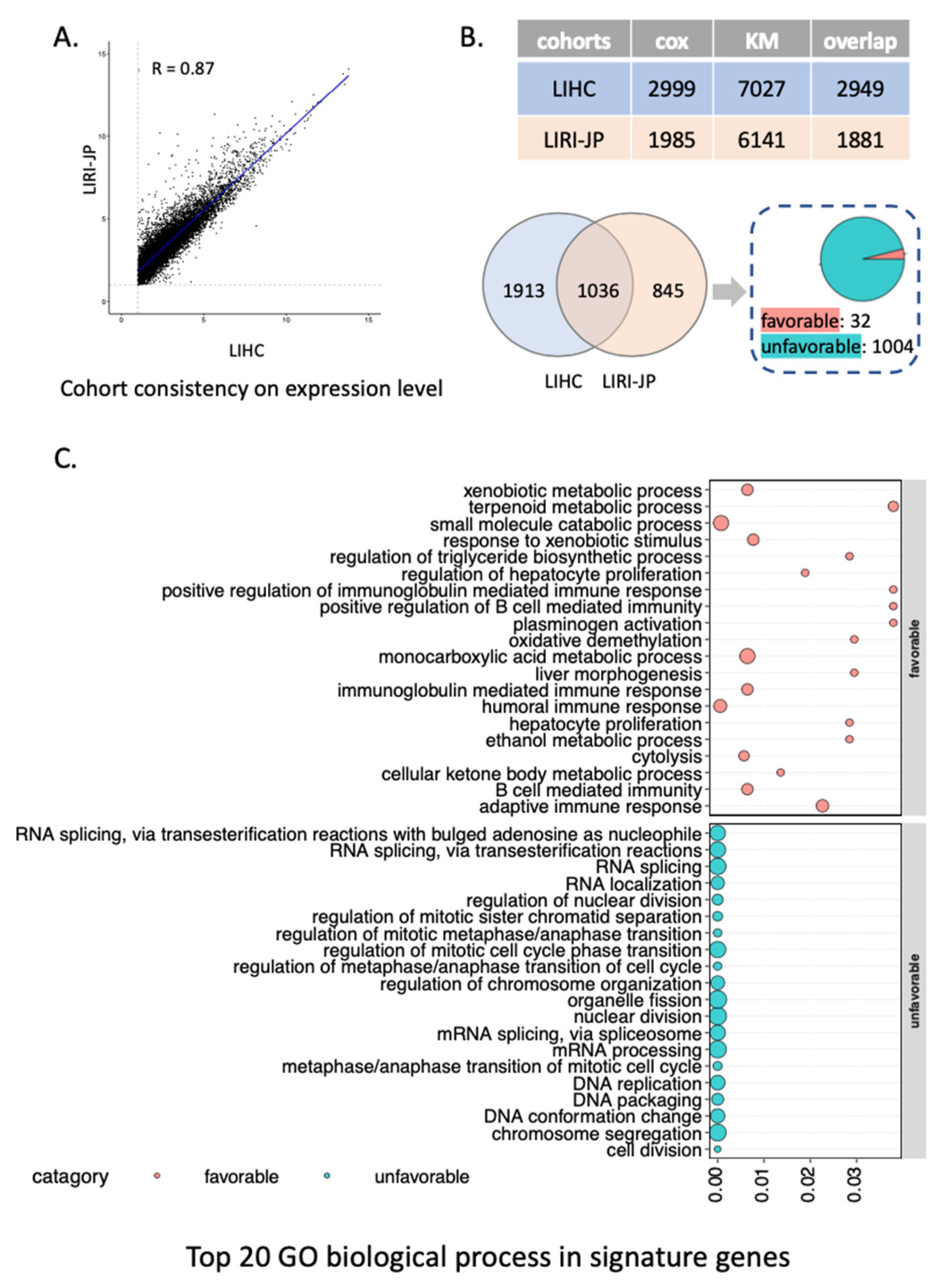
2.2. Survival Analysis
2.3. Functional Enrichment Analysis
2.4. Co-Expression Analysis and Module Identification
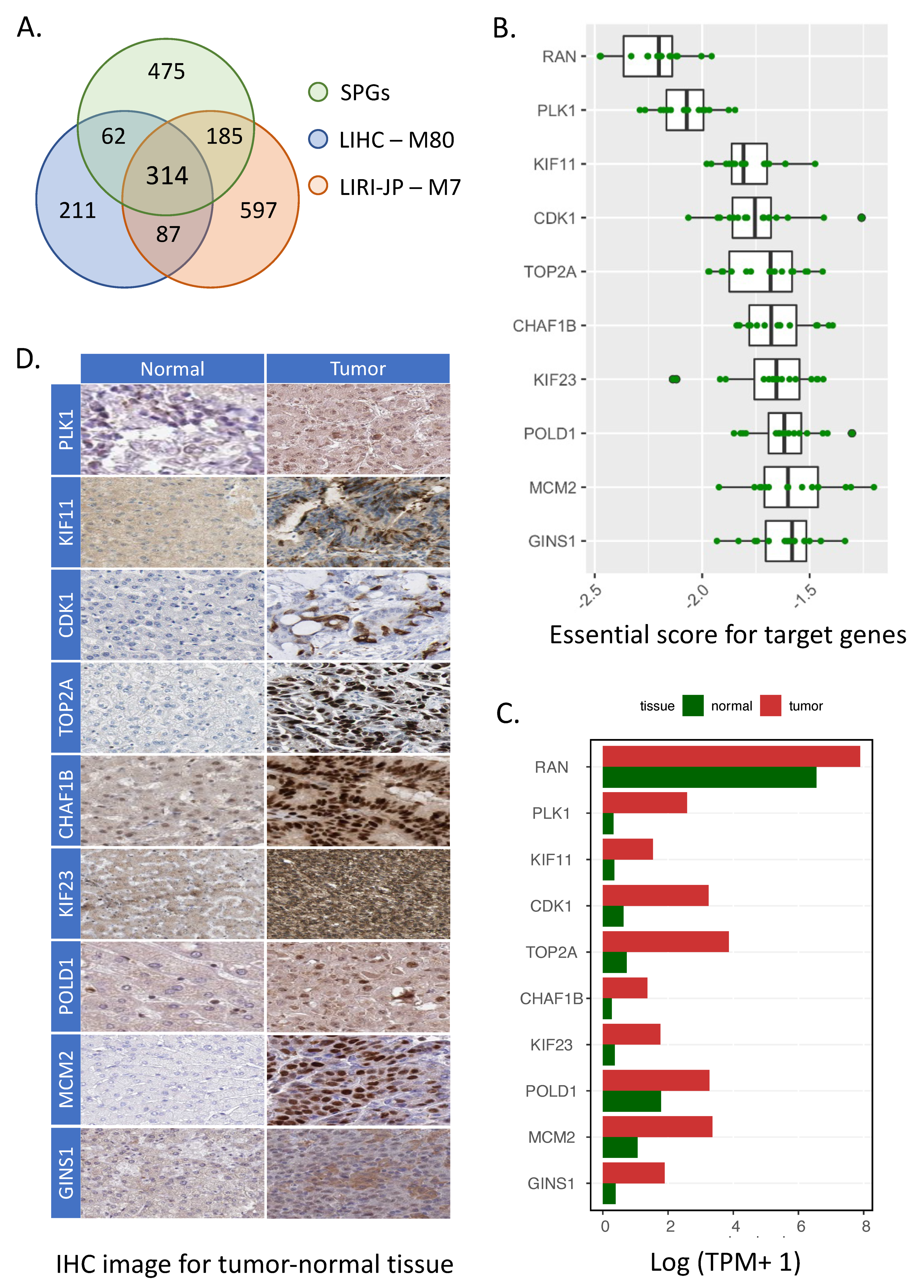
2.5. Identification of HCC Target Genes
2.6. Expression Differences of Target Genes
2.7. The scRNA-Seq Data Processing
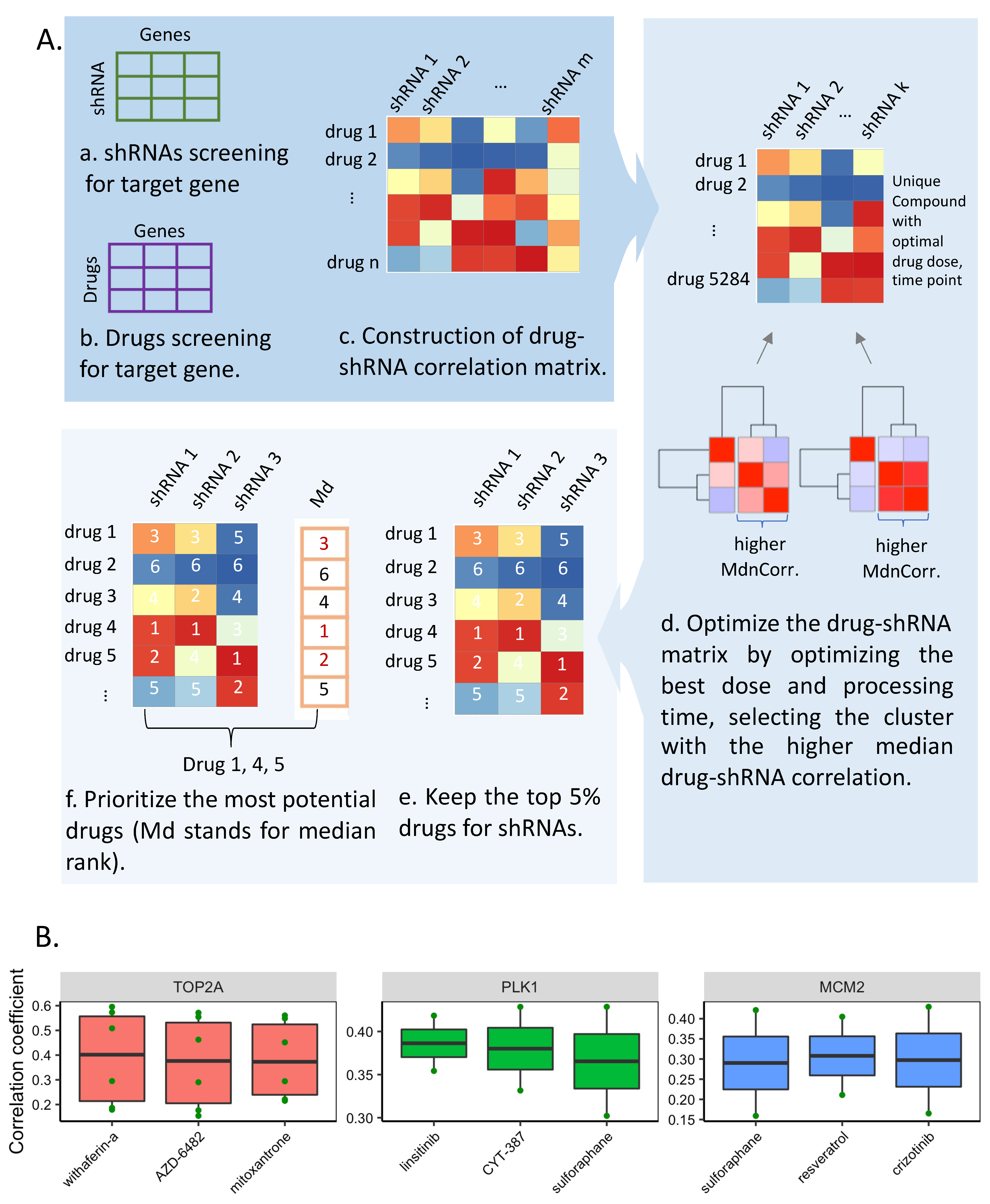
2.8. Drug Repositioning for HCC
2.9. In Vitro Validation
2.9.1. Cells Culture
2.9.2. Drug Treatment
2.9.3. Western Blots
2.9.4. Cell Viability Assay
2.9.5. Wound Healing Assay
2.10. Molecular Docking Analysis
3. Results
3.1. Survival Analysis Identifies Signature Prognostic Genes of HCC
3.2. Co-Expression Network Analysis Identifies Hub Modules of HCC
3.3. Identification of Target Genes in HCC
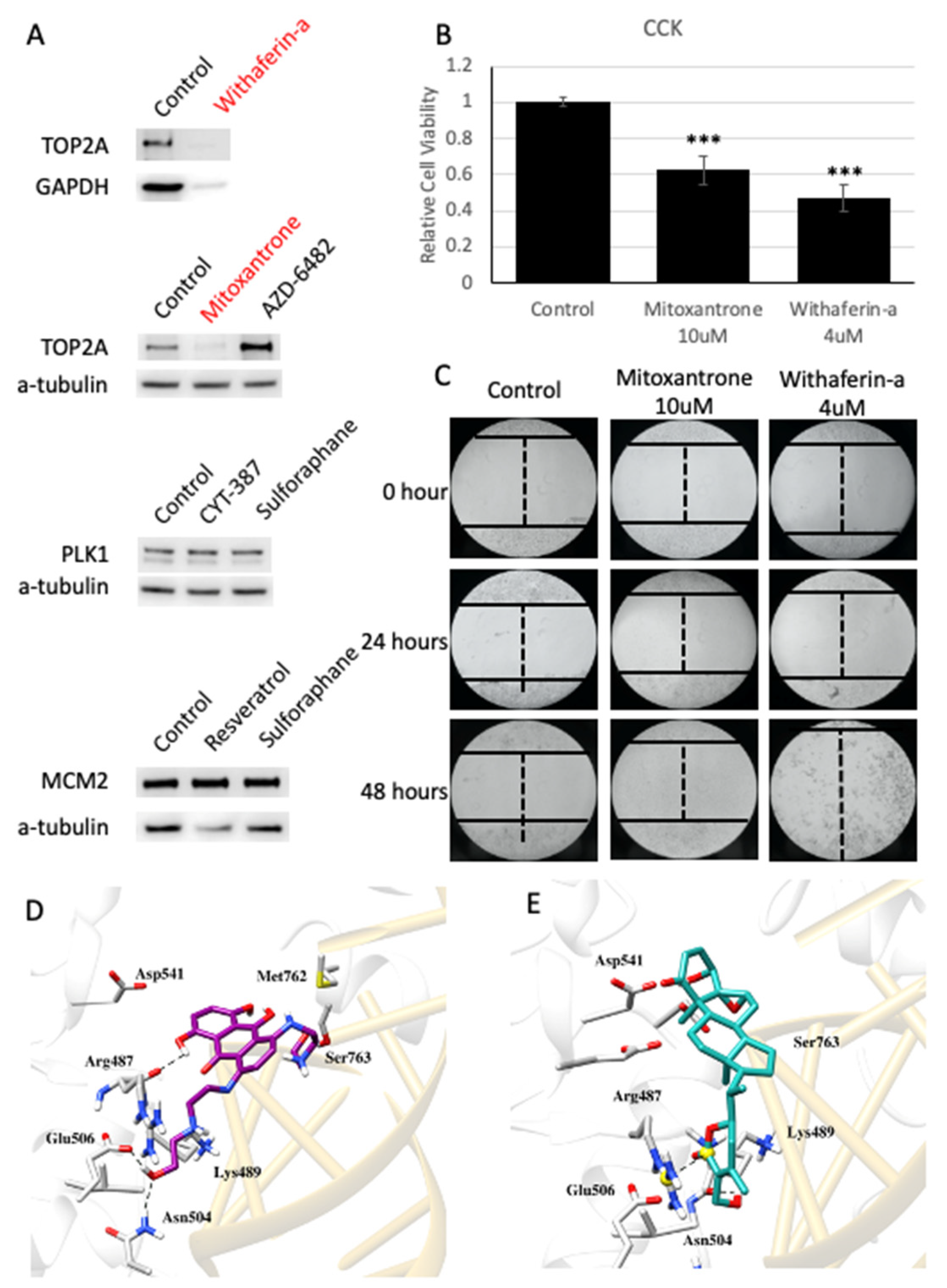
3.4. Drug Repositioning for HCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suresh, D.; Srinivas, A.N.; Kumar, D.P. Etiology of Hepatocellular Carcinoma: Special Focus on Fatty Liver Disease. Front. Oncol. 2020, 10, 2673. [Google Scholar] [CrossRef]
- Ryerson, A.B.; Eheman, C.R.; Altekruse, S.F.; Ward, J.W.; Jemal, A.; Sherman, R.L.; Henley, S.J.; Holtzman, D.; Lake, A.; Noone, A.-M.; et al. Annual Report to the Nation on the Status of Cancer, 1975-2012, featuring the increasing incidence of liver cancer. Cancer 2016, 122, 1312–1337. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; De Baere, T.; Kulik, L.; Haber, P.K.; Greten, T.F.; Meyer, T.; Lencioni, R. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 293–313. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. New Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [Green Version]
- Cainap, C.; Qin, S.; Huang, W.-T.; Chung, I.J.; Pan, H.; Cheng, Y.; Kudo, M.; Kang, Y.-K.; Chen, P.-J.; Toh, H.-C.; et al. Linifanib Versus Sorafenib in Patients With Advanced Hepatocellular Carcinoma: Results of a Randomized Phase III Trial. J. Clin. Oncol. 2015, 33, 172–179. [Google Scholar] [CrossRef]
- Jindal, A.; Thadi, A.; Shailubhai, K. Hepatocellular Carcinoma: Etiology and Current and Future Drugs. J. Clin. Exp. Hepatol. 2019, 9, 221–232. [Google Scholar] [CrossRef]
- Kim, I.-W.; Jang, H.; Kim, J.H.; Kim, M.G.; Kim, S.; Oh, J.M. Computational Drug Repositioning for Gastric Cancer using Reversal Gene Expression Profiles. Sci Rep 2019, 9, 2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraj, A.B.; Wang, Q.Q.; Joseph, P.; Zheng, C.; Chen, Y.; Kovalenko, O.; Singh, S.; Armstrong, A.; Resnick, K.; Zanotti, K.; et al. Using a novel computational drug-repositioning approach (DrugPredict) to rapidly identify potent drug candidates for cancer treatment. Oncogene 2018, 37, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Turanli, B.; Altay, O.; Borén, J.; Turkez, H.; Nielsen, J.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Systems biology based drug repositioning for development of cancer therapy. Semin. Cancer Biol. 2021, 68, 47–58. [Google Scholar] [CrossRef]
- Luo, H.; Li, M.; Yang, M.; Wu, F.-X.; Li, Y.; Wang, J. Biomedical data and computational models for drug repositioning: A comprehensive review. Brief. Bioinform. 2020, 22, 1604–1619. [Google Scholar] [CrossRef]
- Keenan, A.B.; Jenkins, S.L.; Jagodnik, K.M.; Koplev, S.; He, E.; Torre, D.; Wang, Z.; Dohlman, A.B.; Silverstein, M.C.; Lachmann, A.; et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018, 6, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Eskandari, E.; Mahjoubi, F.; Motalebzadeh, J. An integrated study on TFs and miRNAs in colorectal cancer metastasis and evaluation of three co-regulated candidate genes as prognostic markers. Gene 2018, 679, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Alshabi, A.M.; Vastrad, B.; Shaikh, I.A.; Vastrad, C. Identification of important invasion and proliferation related genes in adrenocortical carcinoma. Med. Oncol. 2019, 36, 73. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Zhang, C.; Liu, Z.; Klevstig, M.; Mukhopadhyay, B.; Bergentall, M.; Cinar, R.; Ståhlman, M.; Sikanic, N.; Park, J.K.; et al. Network analyses identify liver-specific targets for treating liver diseases. Mol. Syst. Biol. 2017, 13, 938. [Google Scholar] [CrossRef]
- Bidkhori, G.; Benfeitas, R.; Elmas, E.; Kararoudi, M.N.; Arif, M.; Uhlen, M.; Nielsen, J.; Mardinoglu, A. Metabolic Network-Based Identification and Prioritization of Anticancer Targets Based on Expression Data in Hepatocellular Carcinoma. Front. Physiol. 2018, 9, 916. [Google Scholar] [CrossRef]
- Pornputtapong, N.; Nookaew, I.; Nielsen, J. Human metabolic atlas: An online resource for human metabolism. Database 2015, 2015, bav068. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, B.; Ding, J.; Qi, J.; Gu, Z. Characteristics and prognostic value of potential dependency genes in clear cell renal cell carcinoma based on a large-scale CRISPR-Cas9 and RNAi screening database DepMap. Int. J. Med. Sci. 2021, 18, 2063–2075. [Google Scholar] [CrossRef] [PubMed]
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A.; et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Zhao, Q.; An, L.; Jiao, S.; Li, R.; Sang, Y.; Liao, J.; Nie, P.; Wen, F.; Ju, J.; et al. A TNFR2-hnRNPK Axis Promotes Primary Liver Cancer Development via Activation of YAP Signaling in Hepatic Progenitor Cells. Cancer Res. 2021, 81, 3036–3050. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single–cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kim, W.; Juszczak, K.; Arif, M.; Sato, Y.; Kume, H.; Ogawa, S.; Türkez, H.; Boren, J.; Nielsen, J.; et al. Drug Repositioning for Clear Cell Renal Cell Carcinoma Based on Stratification of Patients. SSRN Electron. J. 2021, 24. [Google Scholar] [CrossRef]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The Structure of DNA-Bound Human Topoisomerase II Alpha: Conformational Mechanisms for Coordinating Inter-Subunit Interactions with DNA Cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ccgi, M. Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group Inc.: Montreal, QC, USA, 2016. [Google Scholar]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, M.; Mwenifumbo, J.C.; Cheung, P.Y.; Paul, J.E.; Pugh, T.J.; Tang, M.J.; Chittaranjan, S.; Morin, R.D.; Asano, J.K.; Ally, A.A.; et al. Novel mRNA isoforms and mutations of uridine monophosphate synthetase and 5-fluorouracil resistance in colorectal cancer. Pharm. J. 2013, 13, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Arif, M.; Meng, Y.; Li, X.; Shong, K.E.; Turkez, H.; Nielsen, J.; Uhlén, M.; Borén, J.; Zhang, C. A network-based approach reveals the dysregulated transcriptional regulation in non-alcohol fatty liver disease. Iscience 2021, 24, 103222. [Google Scholar] [CrossRef] [PubMed]
- Tenen, D.G.; Chai, L.; Tan, J.L. Metabolic alterations and vulnerabilities in hepatocellular carcinoma. Gastroenterol. Rep. 2020, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.W.; Alaei-Mahabadi, B.; Samuelsson, T.; Lindh, M.; Larsson, E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 2013, 4, 2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Geel, R.M.J.M.; Hendrikx, J.J.M.A.; Vahl, J.E.; van Leerdam, M.E.; van den Broek, D.; Huitema, A.D.R.; Beijnen, J.H.; Schellens, J.H.M.; Burgers, S.A. Crizotinib-induced fatal fulminant liver failure. Lung Cancer 2016, 93, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.N.; Kim, J.O.; Lee, S.M.; Park, H.; Lee, J.H.; Rim, K.S.; Hwang, S.G.; Kim, N.K. Variation in the Dicer and RAN Genes Are Associated with Survival in Patients with Hepatocellular Carcinoma. PLoS ONE 2016, 11, e0162279. [Google Scholar] [CrossRef]
- He, Z.; Deng, W.; Jiang, B.; Liu, S.; Tang, M.; Liu, Y.; Zhang, J. Hsa-let-7b inhibits cell proliferation by targeting PLK1 in HCC. Gene 2018, 673, 46–55. [Google Scholar] [CrossRef]
- Dai, Z.; Wang, K.; Gao, Y. The critical role of B4GALT4 in promoting microtubule spindle assembly in HCC through the regulation of PLK1 and RHAMM expression. J. Cell. Physiol. 2021, 237, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Xu, H.; Li, W.; Xu, X.; Liu, H.; Wei, F. LINC00346 Acts as a Competing Endogenous RNA Regulating Development of Hepatocellular Carcinoma via Modulating CDK1/CCNB1 Axis. Front. Bioeng. Biotechnol. 2020, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Wang, B.; Wang, X.; Hou, D.; Su, H.; Huang, H. Nine hub genes related to the prognosis of HBV-positive hepatocellular carcinoma identified by protein interaction analysis. Ann. Transl. Med. 2020, 8, 478. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yuan, J.H.; Huang, J.F.; Yang, F.; Wang, T.T.; Ma, J.Z.; Zhang, L.; Zhou, C.C.; Wang, F.; Yu, J.; et al. Long noncoding RNA FTX inhibits hepatocellular carcinoma proliferation and metastasis by binding MCM2 and miR-374a. Oncogene 2016, 35, 5422–5434. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, L.; Zhang, H.; Liu, X.; Wang, Z.; Dong, B.; Cao, G. GINS1 Induced Sorafenib Resistance by Promoting Cancer Stem Properties in Human Hepatocellular Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.-C.; Zhang, L.; Liao, W.-J.; Ao, L.; Lin, Z.-M.; Kang, W.; Chen, W.-N.; Lin, X. Screening and Identification of Potential Biomarkers in Hepatitis B Virus-Related Hepatocellular Carcinoma by Bioinformatics Analysis. Front. Genet. 2020, 11, 555537. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Fu, H.; Yin, J.; Zhao, Q. CHAF1B knockdown blocks migration in a hepatocellular carcinoma model. Oncol. Rep. 2018, 40, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jin, Z.; Song, X.; Wang, J.; Li, Y.; Qian, X.; zhang, Y.; Yin, Y. Evaluation of KIF23 variant 1 expression and relevance as a novel prognostic factor in patients with hepatocellular carcinoma. BMC Cancer 2015, 15, 961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Wu, H.; Sun, F.; Qi, J.; Zhu, Q. A novel four-gene of iron metabolism-related and methylated for prognosis prediction of hepatocellular carcinoma. Bioengineered 2021, 12, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; He, X.; Guo, G.; Zhang, X.; Li, J.; Liu, J.; Lin, Y. High-throughput screening identified mitoxantrone to induce death of hepatocellular carcinoma cells with autophagy involvement. Biochem. Biophys. Res. Commun. 2020, 521, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-R.; He, Q. Study on liver targeting and hepatocytes permeable valaciclovir polybutylcyanoacrylate nanoparticles. World J. Gastroenterol. 1999, 5, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Shiragannavar, V.D.; Gowda, N.G.S.; Kumar, D.P.; Mirshahi, F.; Santhekadur, P.K. Withaferin A Acts as a Novel Regulator of Liver X Receptor-α in HCC. Front. Oncol. 2021, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.-B.; Hui, B.; Shi, Y.-H.; Zhou, J.; Peng, Y.-F.; Gu, C.-Y.; Yang, H.; Shi, G.-M.; Ke, A.-W.; Wang, X.-Y. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin. Cancer Res. 2011, 17, 6229–6238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.-H.; Ding, Z.-B.; Zhou, J.; Hui, B.; Shi, G.-M.; Ke, A.-W.; Wang, X.-Y.; Dai, Z.; Peng, Y.-F.; Gu, C.-Y. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011, 7, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sun, X.; Zeng, L.; Liu, J.; Zhang, Z. A randomized multicenter phase II clinical trial of mitoxantrone-loaded nanoparticles in the treatment of 108 patients with unresected hepatocellular carcinoma. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 419–423. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, M.; Shong, K.; Li, X.; Ashraf, S.; Shi, M.; Kim, W.; Nielsen, J.; Turkez, H.; Shoaie, S.; Uhlen, M.; et al. A Gene Co-Expression Network-Based Drug Repositioning Approach Identifies Candidates for Treatment of Hepatocellular Carcinoma. Cancers 2022, 14, 1573. https://doi.org/10.3390/cancers14061573
Yuan M, Shong K, Li X, Ashraf S, Shi M, Kim W, Nielsen J, Turkez H, Shoaie S, Uhlen M, et al. A Gene Co-Expression Network-Based Drug Repositioning Approach Identifies Candidates for Treatment of Hepatocellular Carcinoma. Cancers. 2022; 14(6):1573. https://doi.org/10.3390/cancers14061573
Chicago/Turabian StyleYuan, Meng, Koeun Shong, Xiangyu Li, Sajda Ashraf, Mengnan Shi, Woonghee Kim, Jens Nielsen, Hasan Turkez, Saeed Shoaie, Mathias Uhlen, and et al. 2022. "A Gene Co-Expression Network-Based Drug Repositioning Approach Identifies Candidates for Treatment of Hepatocellular Carcinoma" Cancers 14, no. 6: 1573. https://doi.org/10.3390/cancers14061573