
Unfolded Protein Response Is Activated by Aurora Kinase A in Esophageal Adenocarcinoma


Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. TCGA and GEO Datasets Analysis
2.3. RNA Sequencing and Gene Set Enrichment Analysis
2.4. qRT-PCR and Human Esophageal Tissue Samples
2.5. Cell Viability ATP-Glo and Clonogenic Cell Survival Assay
2.6. Western Blotting
2.7. Flow Cytometry Analysis of Cell Apoptosis/Death
2.8. Immunofluorescence
2.9. Animal Models
3. Results
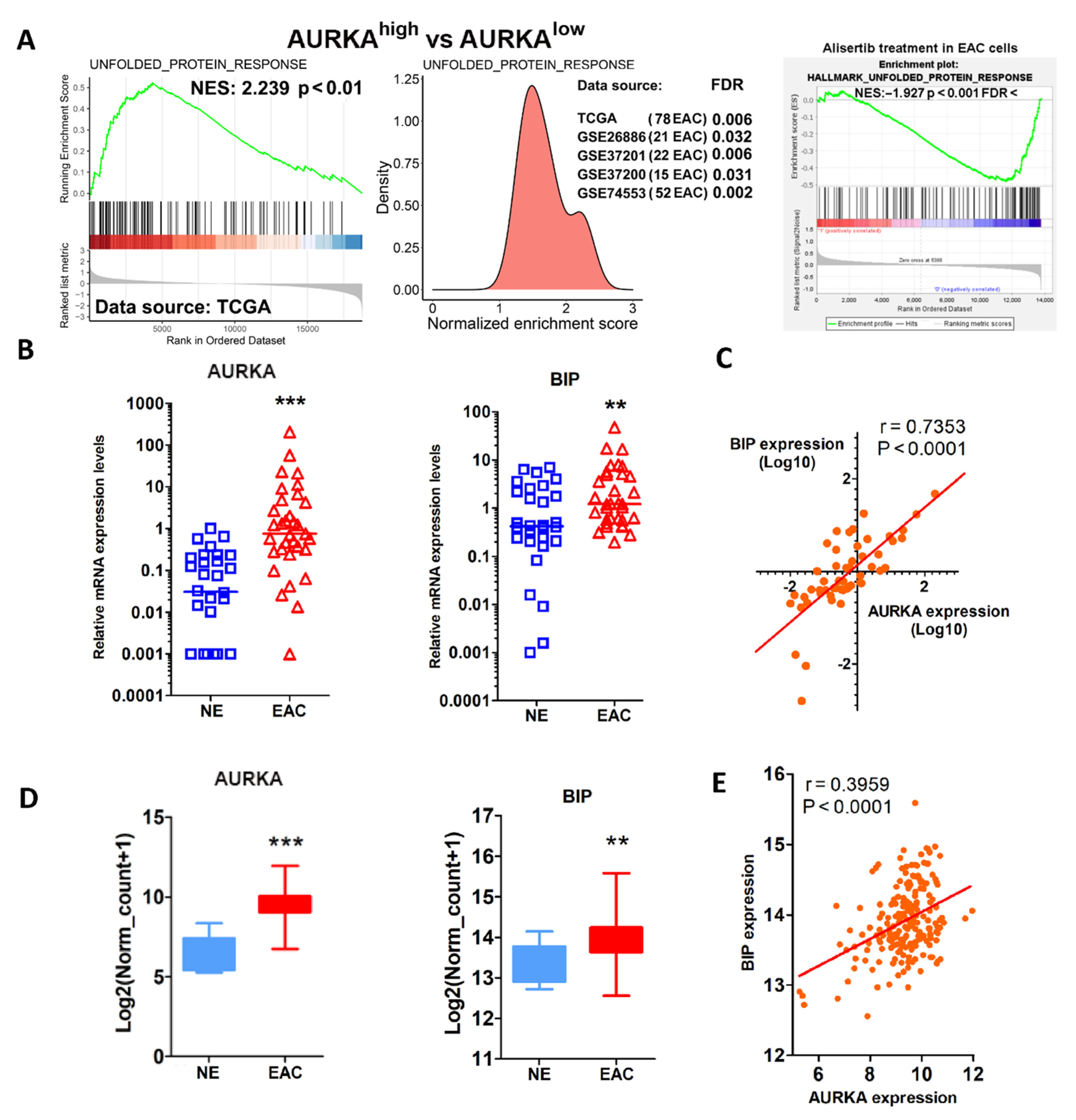
3.1. AURKA Overexpression Correlates with UPR Activation in EAC
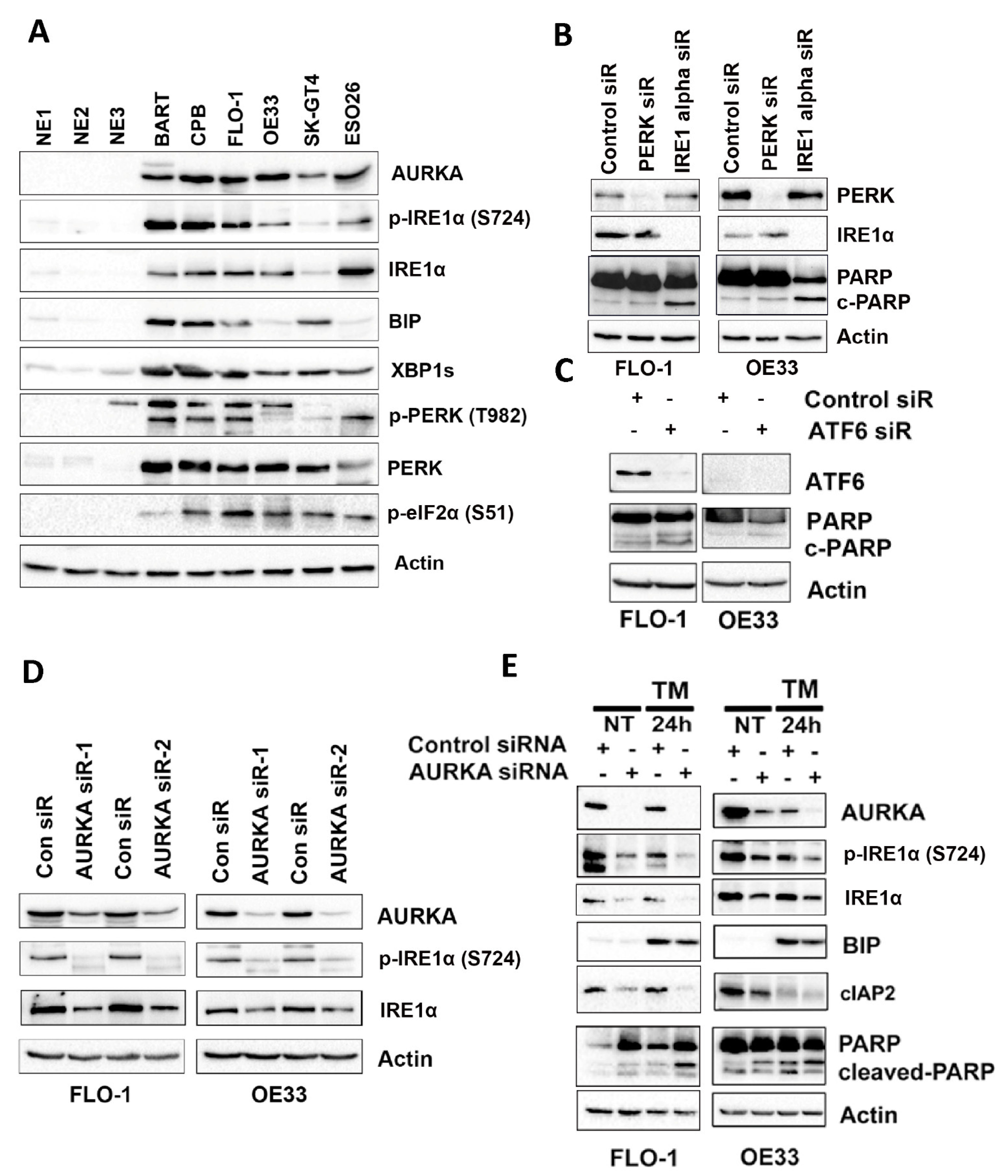
3.2. IRE1α Promotes EAC Cell Survival through AURKA
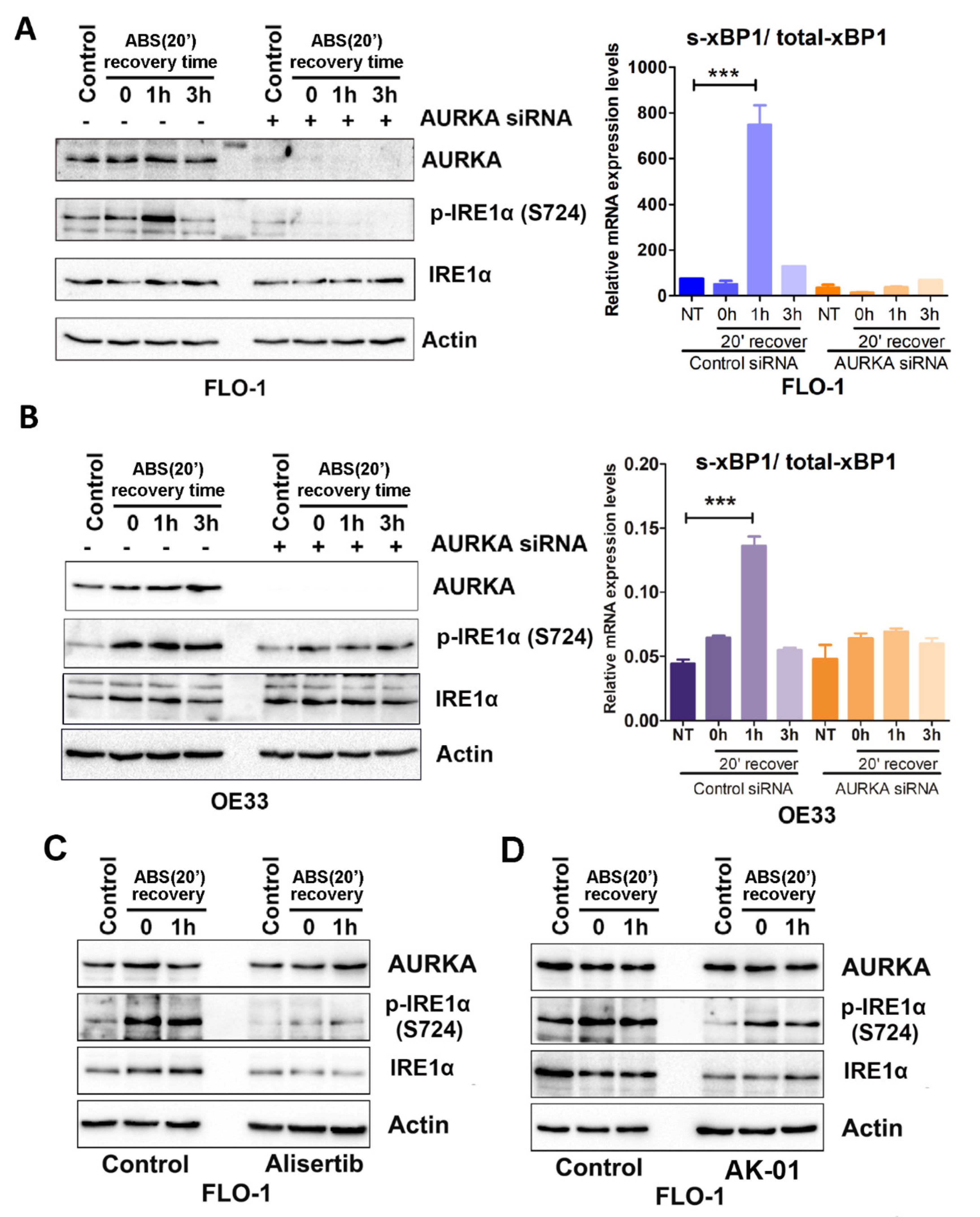
3.3. IRE1α Activation Is Dependent on AURKA in Response to Acidic Bile Salt Reflux Conditions in EAC Cells
3.4. AURKA Protects EAC Cells from ABS-Induced Apoptosis
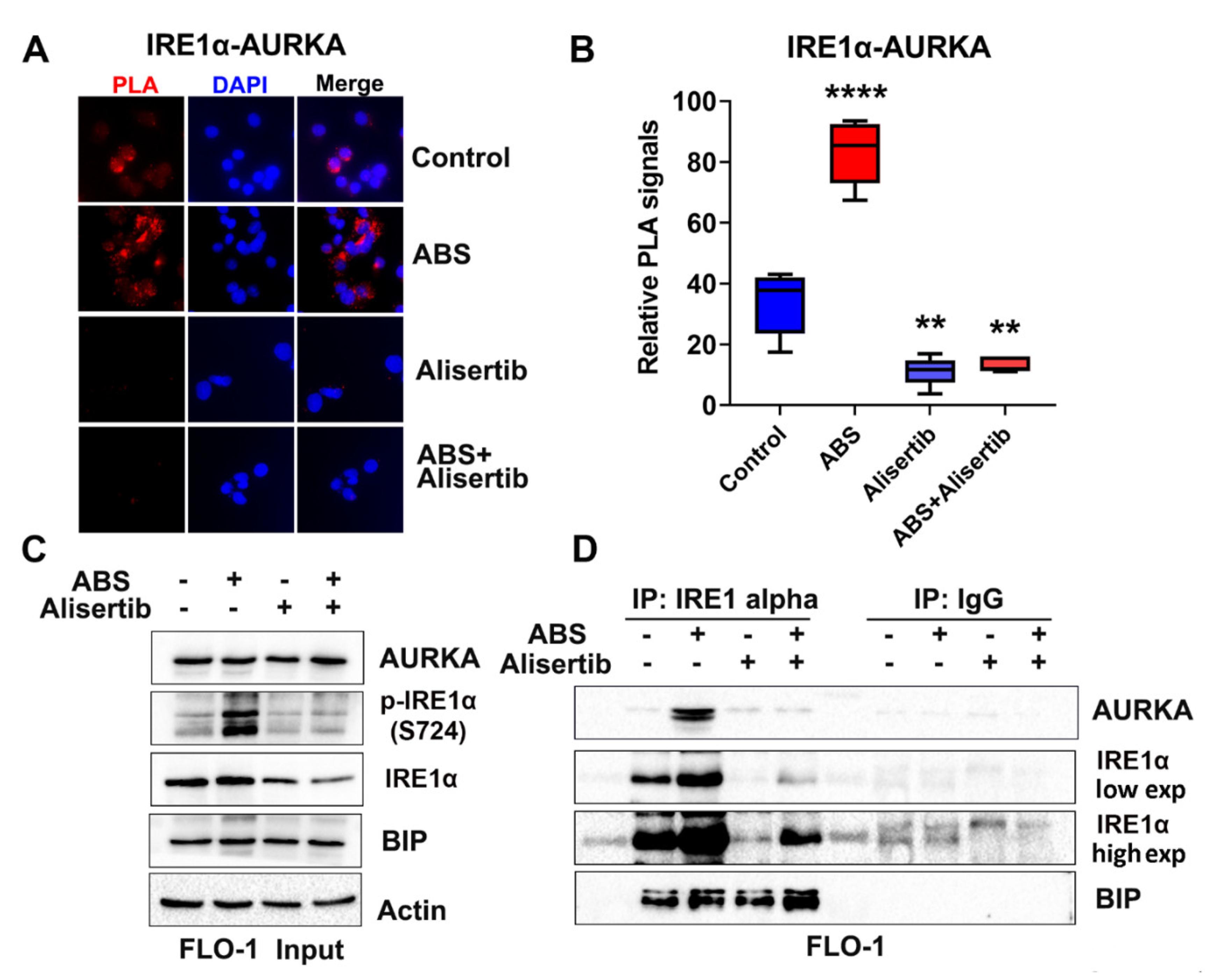
3.5. AURKA Binds to IRE1α in EAC Cells
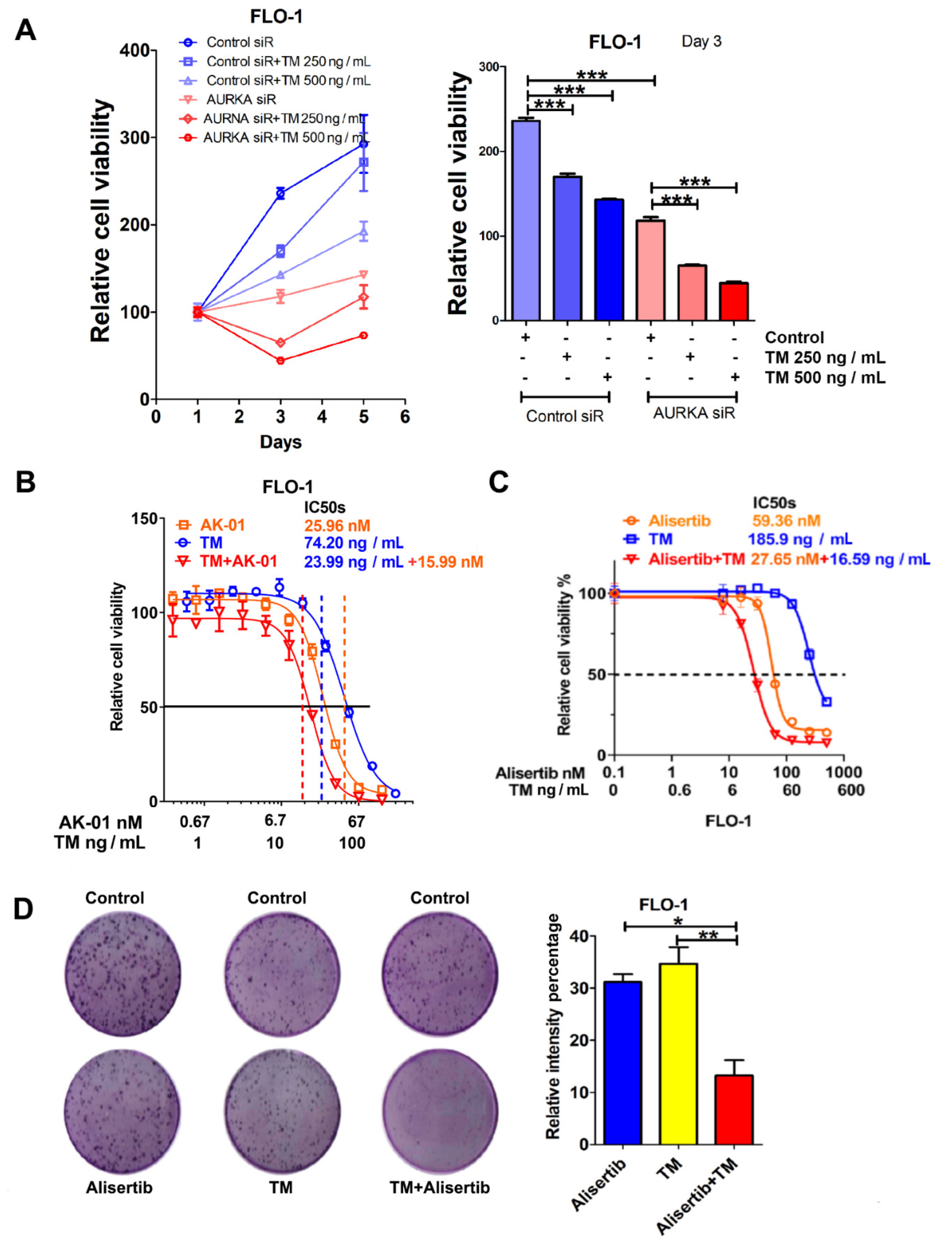
3.6. AURKA Promotes Survival under Tunicamycin-Induced ER Stress in EAC Cells
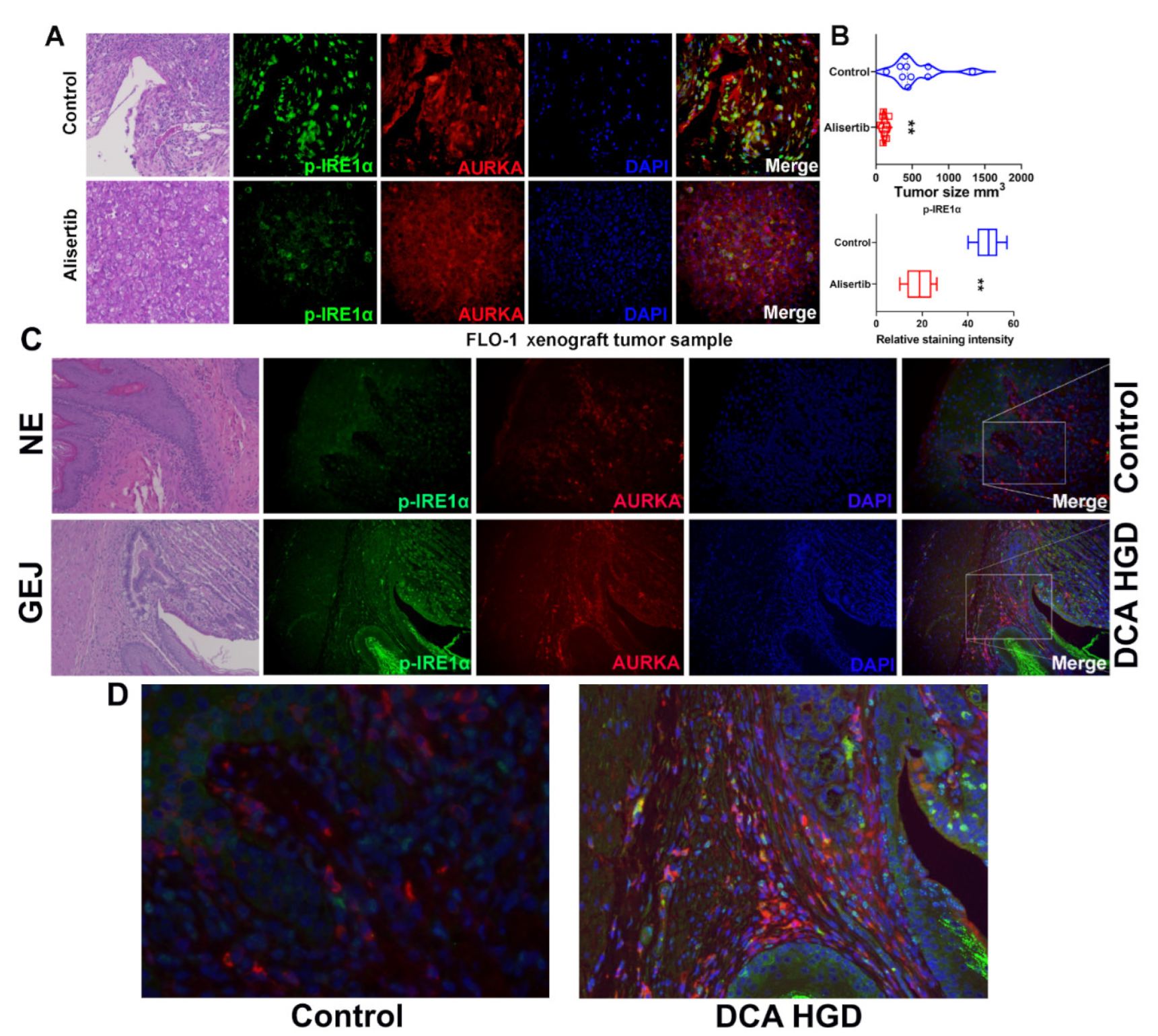
3.7. AURKA and ABS Promote IRE1α Phosphorylation In Vivo
4. Discussion
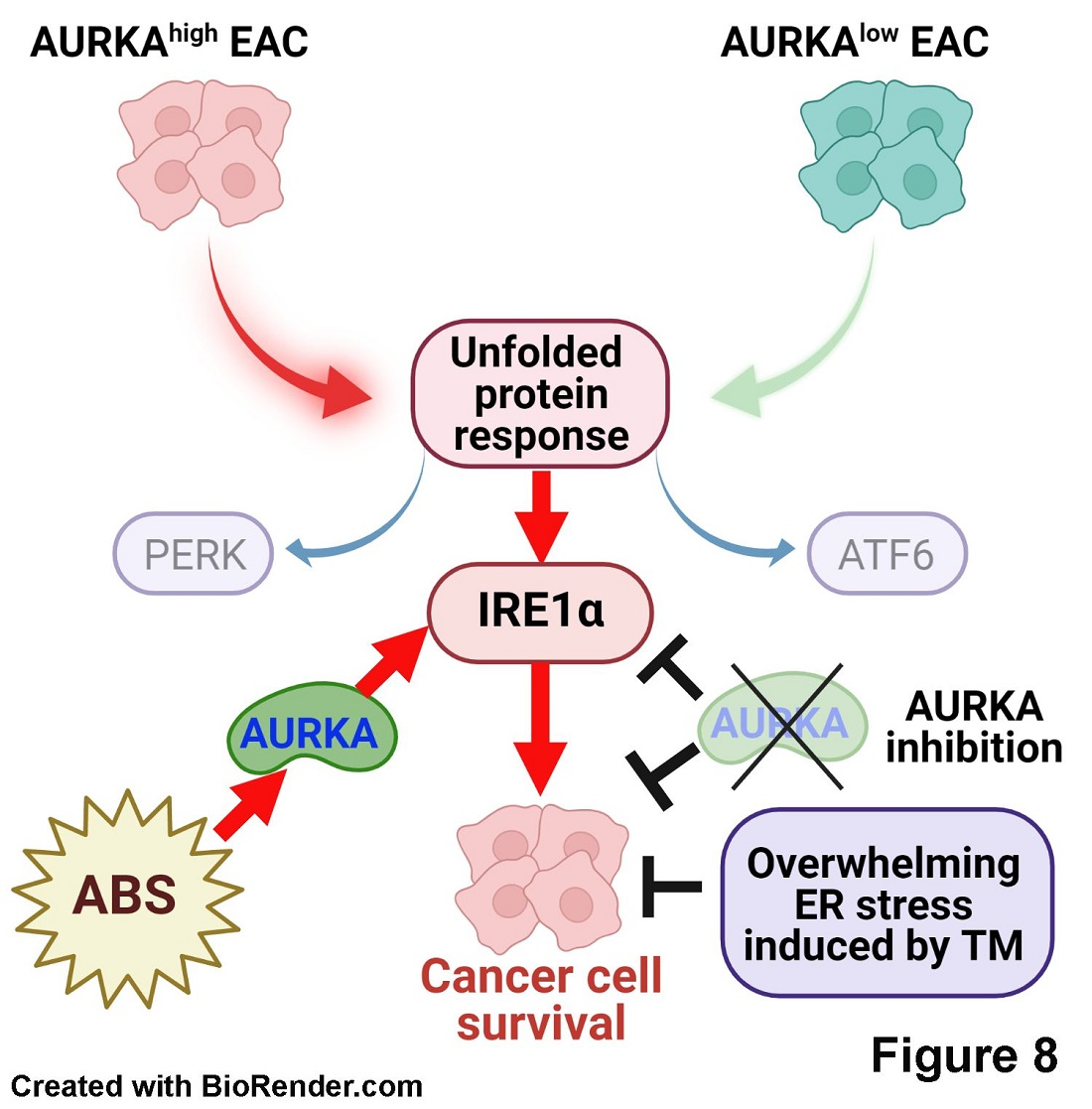
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, A.G.; Stevens, G.A.; Ezzati, M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R.; et al. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Arnold, M.; Soerjomataram, I.; Ferlay, J.; Forman, D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015, 64, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Noble, F.; Mellows, T.; McCormick Matthews, L.H.; Bateman, A.C.; Harris, S.; Underwood, T.; Byrne, J.; Bailey, I.S.; Sharland, D.M.; Kelly, J.J.; et al. Tumour infiltrating lymphocytes correlate with improved survival in patients with oesophageal adenocarcinoma. Cancer Immunol. Immunother. 2016, 65, 651–662. [Google Scholar] [CrossRef]
- Leikin, J.B. Foreword: Gastroesophageal reflux disease (GERD). Dis. Mon. 2020, 66, 100858. [Google Scholar] [CrossRef]
- Veugelers, P.J.; Porter, G.A.; Guernsey, D.L.; Casson, A.G. Obesity and lifestyle risk factors for gastroesophageal reflux disease, Barrett esophagus and esophageal adenocarcinoma. Dis. Esophagus 2006, 19, 321–328. [Google Scholar] [CrossRef]
- Peng, D.; Lu, H.; Zhu, S.; Zhou, Z.; Hu, T.; Chen, Z.; Zaika, A.; El-Rifai, W. NRF2 antioxidant response protects against acidic bile salts-induced oxidative stress and DNA damage in esophageal cells. Cancer Lett. 2019, 458, 46–55. [Google Scholar] [CrossRef]
- Katsha, A.; Belkhiri, A.; Goff, L.; El-Rifai, W. Aurora kinase A in gastrointestinal cancers: Time to target. Mol. Cancer. 2015, 14, 106. [Google Scholar] [CrossRef] [Green Version]
- Kamran, M.; Long, Z.-J.; Xu, D.; Lv, S.-S.; Liu, B.; Wang, C.-L.; Xu, J.; Lam, E.; Liu, Q. Aurora kinase A regulates Survivin stability through targeting FBXL7 in gastric cancer drug resistance and prognosis. Oncogenesis. 2017, 6, e298. [Google Scholar] [CrossRef] [Green Version]
- Katsha, A.; Wang, L.; Arras, J.; Omar, O.M.; Ecsedy, J.; Belkhiri, A.; El-Rifai, W. Activation of EIF4E by Aurora Kinase a Depicts a Novel Druggable Axis in Everolimus-Resistant Cancer Cells. Clin. Cancer Res. 2017, 23, 3756–3768. [Google Scholar] [CrossRef] [Green Version]
- Goff, L.W.; Azad, N.S.; Stein, S.; Whisenant, J.G.; Koyama, T.; Vaishampayan, U.; Hochster, H.; Connolly, R.; Weise, A.; Lorusso, P.M.; et al. Phase I study combining the aurora kinase a inhibitor alisertib with mFOLFOX in gastrointestinal cancer. Investig. New Drugs 2019, 37, 315–322. [Google Scholar] [CrossRef]
- Jiang, J.; Guo, Z.; Xu, J.; Sun, T.; Zheng, X. Identification of Aurora Kinase A as a Biomarker for Prognosis in Obesity Patients with Early Breast Cancer. OncoTargets Ther. 2020, 13, 4971–4985. [Google Scholar] [CrossRef]
- Wang-Bishop, L.; Chen, Z.; Gomaa, A.; Lockhart, A.C.; Salaria, S.; Wang, J.; Lewis, K.B.; Jeffrey Ecsedy, K.W.; Washington, K.; Beauchamp, R.D.; et al. Inhibition of AURKA Reduces Proliferation and Survival of Gastrointestinal Cancer Cells with Activated KRAS by Preventing Activation of RPS6KB1. Gastroenterology 2019, 156, 662–675.e667. [Google Scholar] [CrossRef]
- Wang, L.; Arras, J.; Katsha, A.; Hamdan, S.; Belkhiri, A.; Ecsedy, J.; El-Rifai, W. Cisplatin-resistant cancer cells are sensitive to Aurora kinase A inhibition by alisertib. Mol. Oncol. 2017, 11, 981–995. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [Green Version]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Maes, H.; van Vliet, A.R.; Agostinis, P. Targeting the hallmarks of cancer with therapy-induced endoplasmic reticulum (ER) stress. Mol. Cell Oncol. 2015, 2, e975089. [Google Scholar] [CrossRef] [Green Version]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Pellegrini, P.; Selvaraj, K.; Faustini, E.; Mofers, A.; Zhang, X.; Ternerot, J.; Schubert, A.; Linder, S.; D′arcy, P. Induction of ER Stress in Acute Lymphoblastic Leukemia Cells by the Deubiquitinase Inhibitor VLX1570. Int. J. Mol. Sci. 2020, 21, 4757. [Google Scholar] [CrossRef]
- Muñoz-Guardiola, P.; Casas, J.; Megías-Roda, E.; Solé, S.; Perez-Montoyo, H.; Yeste-Velasco, M.; Erazo, T.; Diéguez-Martínez, N.; Espinosa-Gil, S.; Muñoz-Pinedo, C.; et al. The anti-cancer drug ABTL0812 induces ER stress-mediated cytotoxic autophagy by increasing dihydroceramide levels in cancer cells. Autophagy 2020, 17, 1349–1366. [Google Scholar] [CrossRef]
- Muñoz-Guardiola, P.; Casas, J.; Megías-Roda, E.; Solé, S.; Perez-Montoyo, H.; Yeste-Velasco, M.; Erazo, T.; Diéguez-Martínez, N.; Espinosa-Gil, S.; Muñoz-Pinedo, C.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Z.; Soutto, M.; Wang, W.; Piazuelo, M.B.; Zhu, S.; Guo, Y.; Maturana, M.J.; Corvalan, A.; Chen, X.; et al. Integrated Analysis of Mouse and Human Gastric Neoplasms Identifies Conserved microRNA Networks in Gastric Carcinogenesis. Gastroenterology 2019, 156, 1127–1139.e1128. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Saad, R.; Jia, P.; Peng, D.; Zhu, S.; Washington, M.K.; Zhao, Z.; Xu, Z.; El-Rifai, W. Gastric adenocarcinoma has a unique microRNA signature not present in esophageal adenocarcinoma. Cancer 2013, 119, 1985–1993. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Belkhiri, A.; El-Rifai, W. DARPP-32 increases interactions between epidermal growth factor receptor and ERBB3 to promote tumor resistance to gefitinib. Gastroenterology 2011, 141, 1738–1748.e2. [Google Scholar] [CrossRef] [Green Version]
- Sehdev, V.; Peng, D.; Soutto, M.; Washington, M.K.; Revetta, F.; Ecsedy, J.; Zaika, A.; Rau, T.T.; Schneider-Stock, R.; Belkhiri, A.; et al. The aurora kinase A inhibitor MLN8237 enhances cisplatin-induced cell death in esophageal adenocarcinoma cells. Mol. Cancer Ther. 2012, 11, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef] [Green Version]
- Boukli, N.M.; Saiyed, Z.M.; Ricaurte, M.; Rodriguez, J.W.; Olivares, E.R.; Cubano, L.A.; Nair, M.P. Implications of ER stress, the unfolded protein response, and pro- and anti-apoptotic protein fingerprints in human monocyte-derived dendritic cells treated with alcohol. Alcohol Clin. Exp. Res. 2010, 34, 2081–2088. [Google Scholar] [CrossRef] [Green Version]
- Christis, C.; Fullaondo, A.; Schildknegt, D.; Mkrtchian, S.; Heck, A.J.; Braakman, I. Regulated increase in folding capacity prevents unfolded protein stress in the ER. J. Cell Sci. 2010, 123, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tan, Z.; Yang, Y. Negative feedback and modern anti-cancer strategies targeting the ER stress response. FEBS Lett. 2020, 594, 4247–4265. [Google Scholar] [CrossRef]
- Verfaillie, T.; Garg, A.D.; Agostinis, P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013, 332, 249–264. [Google Scholar] [CrossRef]
- Serrano-Negrón, J.; Zhang, Z.; Rivera-Ruiz, A.P.; Banerjee, A.; Romero-Nutz, E.C.; Sánchez-Torres, N.; Baksi, K.; Banerjee, D.K. Tunicamycin-induced ER stress in breast cancer cells neither expresses GRP78 on the surface nor secretes it into the media. Glycobiology 2019, 29, 599. [Google Scholar] [CrossRef]
- Taguchi, Y.; Horiuchi, Y.; Kano, F.; Murata, M. Novel prosurvival function of Yip1A in human cervical cancer cells: Constitutive activation of the IRE1 and PERK pathways of the unfolded protein response. Cell Death Dis. 2017, 8, e2718. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.Y.; Ou-Yang, F.; Hou, M.F.; Huang, H.W.; Wang, H.R.; Li, K.T.; Fayyazh, S.; Shui, C.-H.; Changdgj, H.-S. Oxidative stress-modulating drugs have preferential anti-cancer effects—Involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration. Semin. Cancer Biol. 2019, 58, 109–117. [Google Scholar] [CrossRef]
- Karakas, D.; Cevatemre, B.; Oral, A.Y.; Yilmaz, V.T.; Ulukaya, E. Unfolded Protein Response is Involved in Trans-Platinum (II) Complex-Induced Apoptosis in Prostate Cancer Cells via ROS Accumulation. Anti-cancer Agents Med. Chem. 2019, 19, 1184–1195. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.K.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1alpha. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Parasa, S.; Desai, M.; Vittal, A.; Chandrasekar, V.T.; Pervez, A.; Kennedy, K.F.; Gupta, N.; Shaheen, N.J.; Sharma, P. Estimating neoplasia detection rate (NDR) in patients with Barrett’s oesophagus based on index endoscopy: A systematic review and meta-analysis. Gut 2019, 68, 2122–2128. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Sehdev, V.; Katsha, A.; Arras, J.; Peng, D.; Soutto, M.; Ecsedy, J.; Zaika, A.; Belkhiri, A.; El-Rifai, W. HDM2 regulation by AURKA promotes cell survival in gastric cancer. Clin. Cancer Res. 2014, 20, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, A.; Bosch, L.J.W.; Kemp, S.M.-D.; Carvalho, B.; Sillars-Hardebol, A.H.; Dobson, R.J.; De Rinaldis, E.; Meijer, G.A.; Abeln, S.; Heringa, J.; et al. Aurora kinase A (AURKA) interaction with Wnt and Ras-MAPK signalling pathways in colorectal cancer. Sci. Rep. 2018, 8, 7522. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Mohamed, E.; Rodriguez, P.C. Unfolding anti-tumor immunity: ER stress responses sculpt tolerogenic myeloid cells in cancer. J. Immunother. Cancer 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Qumseya, B.J.; Bukannan, A.; Gendy, S.; Ahemd, Y.; Sultan, S.; Bain, P.; Gross, S.A.; Iyer, P.; Wani, S. Systematic review and meta-analysis of prevalence and risk factors for Barrett’s esophagus. Gastrointest. Endosc. 2019, 90, 707–717.e701. [Google Scholar] [CrossRef]
- Shin, G.C.; Moon, S.U.; Kang, H.S.; Choi, H.S.; Han, H.D.; Kim, K.H. PRKCSH contributes to tumorigenesis by selective boosting of IRE1 signaling pathway. Nat. Commun. 2019, 10, 3185. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Meng, Y.; Sheng, X.; Guan, Y.; Zhang, F.; Han, Z.; Kang, Y.; Tai, G.; Zhou, Y.; Cheng, H. Tunicamycin enhances human colon cancer cells to TRAIL-induced apoptosis by JNK-CHOP-mediated DR5 upregulation and the inhibition of the EGFR pathway. Anti-Cancer Drugs 2017, 28, 66–74. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; He, Z.; Deng, J.; Zhang, Z.; Liu, L.; Ye, W.; Liu, S. Tunicamycin induces ER stress and inhibits tumorigenesis of head and neck cancer cells by inhibiting N-glycosylation. Am. J. Transl. Res. 2020, 12, 541–550. [Google Scholar]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; Gomaa, A.; Wang-Bishop, L.; Ballout, F.; Hu, T.; McDonald, O.; Washington, M.K.; Livingstone, A.S.; Wang, T.C.; Peng, D.; et al. Unfolded Protein Response Is Activated by Aurora Kinase A in Esophageal Adenocarcinoma. Cancers 2022, 14, 1401. https://doi.org/10.3390/cancers14061401
Lu H, Gomaa A, Wang-Bishop L, Ballout F, Hu T, McDonald O, Washington MK, Livingstone AS, Wang TC, Peng D, et al. Unfolded Protein Response Is Activated by Aurora Kinase A in Esophageal Adenocarcinoma. Cancers. 2022; 14(6):1401. https://doi.org/10.3390/cancers14061401
Chicago/Turabian StyleLu, Heng, Ahmed Gomaa, Lihong Wang-Bishop, Farah Ballout, Tianling Hu, Oliver McDonald, Mary Kay Washington, Alan S. Livingstone, Timothy C. Wang, Dunfa Peng, and et al. 2022. "Unfolded Protein Response Is Activated by Aurora Kinase A in Esophageal Adenocarcinoma" Cancers 14, no. 6: 1401. https://doi.org/10.3390/cancers14061401