
Tumor-Associated Macrophages in Gliomas—Basic Insights and Treatment Opportunities


Abstract
:Simple Summary
Abstract
1. Introduction
2. The Origin and Function of Macrophages and Microglia
3. Polarization of Microglia and Macrophages
4. Markers for TAM
5. Macrophages and Microglia in Glioma
5.1. The Function of TAMs in the TME
5.2. Spatial Distribution of Macrophages and Microglia in the TME
5.3. TAMs and the Interferon Response in Glioma
5.4. Prognostic Relevance of TAMs in Glioma
5.5. Experimental Models to Investigate the TME in Glioma
6. Therapeutic Options for Targeting TAMs
6.1. TAM Re-Education
6.1.1. Programmed Cell Death Protein 1 (PD-1)/PD-L1 Axis
6.1.2. Cytotoxic T-Lymphocyte-Associated Protein 4 (CTLA4)
6.1.3. CD47
6.1.4. CD73
6.1.5. STAT3
6.1.6. Macrophage Receptor with Collagenous Structure (MARCO)
6.1.7. CXCL16/CXCR6 Axis
6.1.8. WISP1
6.2. TAM Education
6.2.1. CD40/CD40L
6.2.2. Toll-like Receptor (TLR) Activation
6.3. TAM Depletion
6.3.1. P-Selectin
6.3.2. CCL2/CCR2
6.3.3. Colony Stimulating Factor 1 Receptor (CSF1R)
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Grech, N.; Dalli, T.; Mizzi, S.; Meilak, L.; Calleja, N.; Zrinzo, A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus 2020, 12, e8195. [Google Scholar] [CrossRef]
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Perus, L.J.M.; Walsh, L.A. Microenvironmental Heterogeneity in Brain Malignancies. Front. Immunol. 2019, 10, 2294. [Google Scholar] [CrossRef]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro-Oncology 2006, 8, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef]
- Urbantat, R.M.; Jelgersma, C.; Brandenburg, S.; Nieminen-Kelha, M.; Kremenetskaia, I.; Zollfrank, J.; Mueller, S.; Rubarth, K.; Koch, A.; Vajkoczy, P.; et al. Tumor-Associated Microglia/Macrophages as a Predictor for Survival in Glioblastoma and Temozolomide-Induced Changes in CXCR2 Signaling with New Resistance Overcoming Strategy by Combination Therapy. Int. J. Mol. Sci. 2021, 22, 11180. [Google Scholar] [CrossRef]
- Land, C.A.; Musich, P.R.; Haydar, D.; Krenciute, G.; Xie, Q. Chimeric antigen receptor T-cell therapy in glioblastoma: Charging the T cells to fight. J. Transl. Med. 2020, 18, 428. [Google Scholar] [CrossRef] [PubMed]
- Frederico, S.C.; Hancock, J.C.; Brettschneider, E.E.S.; Ratnam, N.M.; Gilbert, M.R.; Terabe, M. Making a Cold Tumor Hot: The Role of Vaccines in the Treatment of Glioblastoma. Front. Oncol. 2021, 11, 672508. [Google Scholar] [CrossRef] [PubMed]
- Ling, E.A.; Wong, W.C. The origin and nature of ramified and amoeboid microglia: A historical review and current concepts. Glia 1993, 7, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Alliot, F.; Godin, I.; Pessac, B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res. Dev. Brain Res. 1999, 117, 145–152. [Google Scholar] [CrossRef]
- Hoeffel, G.; Ginhoux, F. Ontogeny of Tissue-Resident Macrophages. Front. Immunol. 2015, 6, 486. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; de Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 2013, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, D.; Seo, N.; Hayashi, T.; Tahara, Y.; Fujii, K.; Tawara, I.; Miyahara, Y.; Okamori, K.; Yagita, H.; Imoto, S.; et al. Antigen delivery targeted to tumor-associated macrophages overcomes tumor immune resistance. J. Clin. Investig. 2019, 129, 1278–1294. [Google Scholar] [CrossRef]
- Tong, N.; He, Z.; Ma, Y.; Wang, Z.; Huang, Z.; Cao, H.; Xu, L.; Zou, Y.; Wang, W.; Yi, C.; et al. Tumor Associated Macrophages, as the Dominant Immune Cells, Are an Indispensable Target for Immunologically Cold Tumor-Glioma Therapy? Front. Cell Dev. Biol. 2021, 9, 706286. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Zou, X.B.; Chai, Y.F.; Yao, Y.M. Macrophage polarization in inflammatory diseases. Int. J. Biol. Sci. 2014, 10, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Xuan, W.; Lesniak, M.S.; James, C.D.; Heimberger, A.B.; Chen, P. Context-Dependent Glioblastoma-Macrophage/Microglia Symbiosis and Associated Mechanisms. Trends Immunol. 2021, 42, 280–292. [Google Scholar] [CrossRef]
- Wu, S.Y.; Xing, F.; Sharma, S.; Wu, K.; Tyagi, A.; Liu, Y.; Zhao, D.; Deshpande, R.P.; Shiozawa, Y.; Ahmed, T.; et al. Nicotine promotes brain metastasis by polarizing microglia and suppressing innate immune function. J. Exp. Med. 2020, 217, e20191131. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walentynowicz, K.A.; Ochocka, N.; Pasierbinska, M.; Wojnicki, K.; Stepniak, K.; Mieczkowski, J.; Ciechomska, I.A.; Kaminska, B. In Search for Reliable Markers of Glioma-Induced Polarization of Microglia. Front. Immunol. 2018, 9, 1329. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Wisniewski, P.; Kijewska, M.; Gajdanowicz, P.; Pszczolkowska, D.; Przanowski, P.; Dabrowski, M.; Maleszewska, M.; Kaminska, B. Tumour-processed osteopontin and lactadherin drive the protumorigenic reprogramming of microglia and glioma progression. Oncogene 2016, 35, 6366–6377. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Dabrowski, M.; Lipko, M.; Sliwa, M.; Maleszewska, M.; Kaminska, B. Molecular definition of the pro-tumorigenic phenotype of glioma-activated microglia. Glia 2013, 61, 1178–1190. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Szulzewsky, F.; Arora, S.; de Witte, L.; Ulas, T.; Markovic, D.; Schultze, J.L.; Holland, E.C.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia 2016, 64, 1416–1436. [Google Scholar] [CrossRef] [PubMed]
- Roszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.G.; Lue, L.F. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimers Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Zeiner, P.S.; Preusse, C.; Golebiewska, A.; Zinke, J.; Iriondo, A.; Muller, A.; Kaoma, T.; Filipski, K.; Muller-Eschner, M.; Bernatz, S.; et al. Distribution and prognostic impact of microglia/macrophage subpopulations in gliomas. Brain Pathol. 2019, 29, 513–529. [Google Scholar] [CrossRef]
- Fu, B.; Tian, Z.; Wei, H. Subsets of human natural killer cells and their regulatory effects. Immunology 2014, 141, 483–489. [Google Scholar] [CrossRef]
- McFarland, H.I.; Nahill, S.R.; Maciaszek, J.W.; Welsh, R.M. CD11b (Mac-1): A marker for CD8+ cytotoxic T cell activation and memory in virus infection. J. Immunol. 1992, 149, 1326–1333. [Google Scholar]
- Christensen, J.E.; Andreasen, S.O.; Christensen, J.P.; Thomsen, A.R. CD11b expression as a marker to distinguish between recently activated effector CD8+ T cells and memory cells. Int. Immunol. 2001, 13, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Noy, R.; Swierczak, A.; Sugano, G.; Smith, H.; Wiechmann, L.; Pollard, J.W. Isolation of Mouse and Human Tumor-Associated Macrophages. Adv. Exp. Med. Biol. 2016, 899, 211–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.L.; Rios, E.; Duraes, C.; Ribeiro, R.; Machado, J.C.; Mantovani, A.; Barbosa, M.A.; Carneiro, F.; Oliveira, M.J. The Two Faces of Tumor-Associated Macrophages and Their Clinical Significance in Colorectal Cancer. Front. Immunol. 2019, 10, 1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [Green Version]
- Mildner, A.; Huang, H.; Radke, J.; Stenzel, W.; Priller, J. P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia 2017, 65, 375–387. [Google Scholar] [CrossRef]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Ohsawa, K.; Imai, Y.; Kanazawa, H.; Sasaki, Y.; Kohsaka, S. Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J. Cell Sci. 2000, 113, 3073–3084. [Google Scholar] [CrossRef]
- O’Malley, J.T.; Nadol, J.B., Jr.; McKenna, M.J. Anti CD163+, Iba1+, and CD68+ Cells in the Adult Human Inner Ear: Normal Distribution of an Unappreciated Class of Macrophages/Microglia and Implications for Inflammatory Otopathology in Humans. Otol. Neurotol. 2016, 37, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Rao, G.; Latha, K.; Ott, M.; Sabbagh, A.; Marisetty, A.; Ling, X.; Zamler, D.; Doucette, T.A.; Yang, Y.; Kong, L.Y.; et al. Anti-PD-1 Induces M1 Polarization in the Glioma Microenvironment and Exerts Therapeutic Efficacy in the Absence of CD8 Cytotoxic T Cells. Clin. Cancer Res. 2020, 26, 4699–4712. [Google Scholar] [CrossRef] [PubMed]
- Friebel, E.; Kapolou, K.; Unger, S.; Nunez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]
- Cai, X.; Yin, Y.; Li, N.; Zhu, D.; Zhang, J.; Zhang, C.Y.; Zen, K. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J. Mol. Cell Biol. 2012, 4, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Ciotti, G.M.; Braun, D.; Kalinin, S.; Curro, D.; Dello Russo, C.; Coli, A.; Mangiola, A.; Anile, C.; Feinstein, D.L.; et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 2017, 645, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, S.; Wang, Y.; Zhang, W.; Ma, K.; Hu, C.; Zhu, H.; Liang, S.; Liu, M.; Xu, N. IL-6 influences the polarization of macrophages and the formation and growth of colorectal tumor. Oncotarget 2018, 9, 17443–17454. [Google Scholar] [CrossRef] [Green Version]
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [Green Version]
- Scholz, A.; Harter, P.N.; Cremer, S.; Yalcin, B.H.; Gurnik, S.; Yamaji, M.; Di Tacchio, M.; Sommer, K.; Baumgarten, P.; Bahr, O.; et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 2016, 8, 39–57. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Zhang, Y.; Wang, J.; Gu, J. Defects in Macrophage Reprogramming in Cancer Therapy: The Negative Impact of PD-L1/PD-1. Front. Immunol. 2021, 12, 690869. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieryng, A.; Pszczolkowska, D.; Bocian, K.; Dabrowski, M.; Rajan, W.D.; Kloss, M.; Mieczkowski, J.; Kaminska, B. Immune microenvironment of experimental rat C6 gliomas resembles human glioblastomas. Sci. Rep. 2017, 7, 17556. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.; Brandenburg, S.; Turkowski, K.; Muller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef]
- Brandenburg, S.; Blank, A.; Bungert, A.D.; Vajkoczy, P. Distinction of Microglia and Macrophages in Glioblastoma: Close Relatives, Different Tasks? Int. J. Mol. Sci. 2020, 22, 194. [Google Scholar] [CrossRef]
- Satoh, J.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016, 36, 39–49. [Google Scholar] [CrossRef]
- Chen, H.R.; Sun, Y.Y.; Chen, C.W.; Kuo, Y.M.; Kuan, I.S.; Tiger Li, Z.R.; Short-Miller, J.C.; Smucker, M.R.; Kuan, C.Y. Fate mapping via CCR2-CreER mice reveals monocyte-to-microglia transition in development and neonatal stroke. Sci. Adv. 2020, 6, eabb2119. [Google Scholar] [CrossRef]
- Konishi, H.; Kobayashi, M.; Kunisawa, T.; Imai, K.; Sayo, A.; Malissen, B.; Crocker, P.R.; Sato, K.; Kiyama, H. Siglec-H is a microglia-specific marker that discriminates microglia from CNS-associated macrophages and CNS-infiltrating monocytes. Glia 2017, 65, 1927–1943. [Google Scholar] [CrossRef]
- Buonfiglioli, A.; Hambardzumyan, D. Macrophages and microglia: The cerberus of glioblastoma. Acta Neuropathol. Commun. 2021, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Neidert, N.; von Ehr, A.; Zoller, T.; Spittau, B. Microglia-Specific Expression of Olfml3 Is Directly Regulated by Transforming Growth Factor beta1-Induced Smad2 Signaling. Front. Immunol. 2018, 9, 1728. [Google Scholar] [CrossRef] [Green Version]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjorgjevski, M.; Hannen, R.; Carl, B.; Li, Y.; Landmann, E.; Buchholz, M.; Bartsch, J.W.; Nimsky, C. Molecular profiling of the tumor microenvironment in glioblastoma patients: Correlation of microglia/macrophage polarization state with metalloprotease expression profiles and survival. Biosci. Rep. 2019, 39, BSR20182361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, C.C.; Gordon, P.M.K.; Liu, K.; Yang, R.; Sarkar, S.; Mirzaei, R.; Ahmad, S.T.; Hughes, M.L.; Yong, V.W.; Kelly, J.J.P. Differential microglia and macrophage profiles in human IDH-mutant and -wild type glioblastoma. Oncotarget 2019, 10, 3129–3143. [Google Scholar] [CrossRef] [Green Version]
- Gangoso, E.; Southgate, B.; Bradley, L.; Rus, S.; Galvez-Cancino, F.; McGivern, N.; Guc, E.; Kapourani, C.A.; Byron, A.; Ferguson, K.M.; et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 2021, 184, 2454–2470.e26. [Google Scholar] [CrossRef]
- Maire, C.L.; Mohme, M.; Bockmayr, M.; Fita, K.D.; Riecken, K.; Bornigen, D.; Alawi, M.; Failla, A.; Kolbe, K.; Zapf, S.; et al. Glioma escape signature and clonal development under immune pressure. J. Clin. Investig. 2020, 130, 5257–5271. [Google Scholar] [CrossRef]
- Dunn, G.P.; Fecci, P.E.; Curry, W.T. Cancer immunoediting in malignant glioma. Neurosurgery 2012, 71, 201–222; discussion 222–203. [Google Scholar] [CrossRef] [Green Version]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [Green Version]
- Amor, S.; McNamara, N.B.; Gerrits, E.; Marzin, M.C.; Kooistra, S.M.; Miron, V.E.; Nutma, E. White matter microglia heterogeneity in the CNS. Acta Neuropathol. 2021, 143, 125–141. [Google Scholar] [CrossRef]
- Sankowski, R.; Bottcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshie, O.; Matsushima, K. CCR4 and its ligands: From bench to bedside. Int. Immunol. 2015, 27, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Jin, J.; Huang, X.; Wu, F.; Huang, H.; Zhan, R. EMP3 mediates glioblastoma-associated macrophage infiltration to drive T cell exclusion. J. Exp. Clin. Cancer Res. 2021, 40, 160. [Google Scholar] [CrossRef]
- Petty, A.J.; Li, A.; Wang, X.; Dai, R.; Heyman, B.; Hsu, D.; Huang, X.; Yang, Y. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J. Clin. Investig. 2019, 129, 5151–5162. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11, 938. [Google Scholar] [CrossRef]
- Tu, S.; Lin, X.; Qiu, J.; Zhou, J.; Wang, H.; Hu, S.; Yao, Y.; Wang, Y.; Deng, Y.; Zhou, Y.; et al. Crosstalk Between Tumor-Associated Microglia/Macrophages and CD8-Positive T Cells Plays a Key Role in Glioblastoma. Front. Immunol. 2021, 12, 650105. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutierrez-Vazquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Sun, L.; Mochizuki, A.Y.; Reynoso, J.G.; Orpilla, J.; Chow, F.; Kienzler, J.C.; Everson, R.G.; Nathanson, D.A.; Bensinger, S.J.; et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat. Commun. 2021, 12, 6938. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xiao, H.; Xu, M.; Zhou, C.; Yi, L.; Liang, H. Glioma-derived T cell immunoglobulin- and mucin domain-containing molecule-4 (TIM4) contributes to tumor tolerance. J. Biol. Chem. 2011, 286, 36694–36699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; DeKruyff, R.H. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef] [Green Version]
- Laoui, D.; Van Overmeire, E.; Di Conza, G.; Aldeni, C.; Keirsse, J.; Morias, Y.; Movahedi, K.; Houbracken, I.; Schouppe, E.; Elkrim, Y.; et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014, 74, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, J.; Zhang, Z.; Gao, Z.; Qi, Y.; Qiu, W.; Pan, Z.; Guo, Q.; Li, B.; Zhao, S.; et al. Hypoxic glioma-derived exosomes promote M2-like macrophage polarization by enhancing autophagy induction. Cell Death Dis. 2021, 12, 373. [Google Scholar] [CrossRef]
- Henze, A.T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016, 126, 3672–3679. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. IFNgamma: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- GuhaSarkar, D.; Su, Q.; Gao, G.; Sena-Esteves, M. Systemic AAV9-IFNbeta gene delivery treats highly invasive glioblastoma. Neuro-Oncology 2016, 18, 1508–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Happold, C.; Roth, P.; Silginer, M.; Florea, A.M.; Lamszus, K.; Frei, K.; Deenen, R.; Reifenberger, G.; Weller, M. Interferon-beta induces loss of spherogenicity and overcomes therapy resistance of glioblastoma stem cells. Mol. Cancer Ther. 2014, 13, 948–961. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Yang, R.; Mirzaei, R.; Rawji, K.; Poon, C.; Mishra, M.K.; Zemp, F.J.; Bose, P.; Kelly, J.; Dunn, J.F.; et al. Control of brain tumor growth by reactivating myeloid cells with niacin. Sci. Transl. Med. 2020, 12, eaay9924. [Google Scholar] [CrossRef]
- James, C.D.; He, J.; Carlbom, E.; Nordenskjold, M.; Cavenee, W.K.; Collins, V.P. Chromosome 9 deletion mapping reveals interferon alpha and interferon beta-1 gene deletions in human glial tumors. Cancer Res. 1991, 51, 1684–1688. [Google Scholar]
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017, 38, 542–557. [Google Scholar] [CrossRef]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro-Oncology 2013, 15, 1079–1087. [Google Scholar] [CrossRef]
- Prosniak, M.; Harshyne, L.A.; Andrews, D.W.; Kenyon, L.C.; Bedelbaeva, K.; Apanasovich, T.V.; Heber-Katz, E.; Curtis, M.T.; Cotzia, P.; Hooper, D.C. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin. Cancer Res. 2013, 19, 3776–3786. [Google Scholar] [CrossRef] [Green Version]
- Woolf, Z.; Swanson, M.E.V.; Smyth, L.C.; Mee, E.W.; Schweder, P.; Heppner, P.; Kim, B.J.H.; Turner, C.; Oldfield, R.L.; Curtis, M.A.; et al. Single-cell image analysis reveals a protective role for microglia in glioblastoma. Neurooncol. Adv. 2021, 3, vdab031. [Google Scholar] [CrossRef]
- Sorensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akter, F.; Simon, B.; de Boer, N.L.; Redjal, N.; Wakimoto, H.; Shah, K. Pre-clinical tumor models of primary brain tumors: Challenges and opportunities. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188458. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.; Fakurnejad, S.; Sayegh, E.T.; Clark, A.J.; Ivan, M.E.; Sun, M.Z.; Safaee, M.; Bloch, O.; James, C.D.; Parsa, A.T. Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl. Med. 2014, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Sin, W.C.; Aftab, Q.; Bechberger, J.F.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef]
- Khalsa, J.K.; Cheng, N.; Keegan, J.; Chaudry, A.; Driver, J.; Bi, W.L.; Lederer, J.; Shah, K. Immune phenotyping of diverse syngeneic murine brain tumors identifies immunologically distinct types. Nat. Commun. 2020, 11, 3912. [Google Scholar] [CrossRef] [PubMed]
- Genoud, V.; Marinari, E.; Nikolaev, S.I.; Castle, J.C.; Bukur, V.; Dietrich, P.Y.; Okada, H.; Walker, P.R. Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. Oncoimmunology 2018, 7, e1501137. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Q. Immune checkpoint inhibitors in GBM. J. Neurooncol. 2021, 155, 1–11. [Google Scholar] [CrossRef]
- Costa, B.; Fletcher, M.N.C.; Boskovic, P.; Ivanova, E.L.; Eisemann, T.; Lohr, S.; Bunse, L.; Lower, M.; Burchard, S.; Korshunov, A.; et al. A Set of Cell Lines Derived from a Genetic Murine Glioblastoma Model Recapitulates Molecular and Morphological Characteristics of Human Tumors. Cancers 2021, 13, 230. [Google Scholar] [CrossRef]
- Cogels, M.M.; Rouas, R.; Ghanem, G.E.; Martinive, P.; Awada, A.; Van Gestel, D.; Krayem, M. Humanized Mice as a Valuable Pre-Clinical Model for Cancer Immunotherapy Research. Front. Oncol. 2021, 11, 784947. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPalpha antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications. Cancers 2014, 6, 376–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Ostling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepore, F.; D’Alessandro, G.; Antonangeli, F.; Santoro, A.; Esposito, V.; Limatola, C.; Trettel, F. CXCL16/CXCR6 Axis Drives Microglia/Macrophages Phenotype in Physiological Conditions and Plays a Crucial Role in Glioma. Front. Immunol. 2018, 9, 2750. [Google Scholar] [CrossRef] [Green Version]
- Tao, W.; Chu, C.; Zhou, W.; Huang, Z.; Zhai, K.; Fang, X.; Huang, Q.; Zhang, A.; Wang, X.; Yu, X.; et al. Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat. Commun. 2020, 11, 3015. [Google Scholar] [CrossRef]
- Walker, P.R.; Migliorini, D. The CD40/CD40L axis in glioma progression and therapy. Neuro-Oncology 2015, 17, 1428–1430. [Google Scholar] [CrossRef] [Green Version]
- Chonan, M.; Saito, R.; Shoji, T.; Shibahara, I.; Kanamori, M.; Sonoda, Y.; Watanabe, M.; Kikuchi, T.; Ishii, N.; Tominaga, T. CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro-Oncology 2015, 17, 1453–1462. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Zhu, S.; Qiao, Y.; Liu, Y.J.; Chen, W.; Zhao, G.; Chen, J. Recent advances in the role of toll-like receptors and TLR agonists in immunotherapy for human glioma. Protein Cell 2014, 5, 899–911. [Google Scholar] [CrossRef] [Green Version]
- Butowski, N.; Chang, S.M.; Junck, L.; DeAngelis, L.M.; Abrey, L.; Fink, K.; Cloughesy, T.; Lamborn, K.R.; Salazar, A.M.; Prados, M.D. A phase II clinical trial of poly-ICLC with radiation for adult patients with newly diagnosed supratentorial glioblastoma: A North American Brain Tumor Consortium (NABTC01-05). J. Neurooncol. 2009, 91, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Yeini, E.; Ofek, P.; Pozzi, S.; Albeck, N.; Ben-Shushan, D.; Tiram, G.; Golan, S.; Kleiner, R.; Sheinin, R.; Israeli Dangoor, S.; et al. P-selectin axis plays a key role in microglia immunophenotype and glioblastoma progression. Nat. Commun. 2021, 12, 1912. [Google Scholar] [CrossRef]
- Zhu, X.; Fujita, M.; Snyder, L.A.; Okada, H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J. Neurooncol. 2011, 104, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, G.; Barresi, V.; Indraccolo, S.; Simbolo, M.; Fassan, M.; Mandruzzato, S.; Simonelli, M.; Caccese, M.; Pizzi, M.; Fassina, A.; et al. Pembrolizumab Activity in Recurrent High-Grade Gliomas with Partial or Complete Loss of Mismatch Repair Protein Expression: A Monocentric, Observational and Prospective Pilot Study. Cancers 2020, 12, 2283. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Qi, Y.; Liu, B.; Sun, Q.; Xiong, X.; Chen, Q. Immune Checkpoint Targeted Therapy in Glioma: Status and Hopes. Front. Immunol. 2020, 11, 578877. [Google Scholar] [CrossRef]
- Andersen, B.M.; Faust Akl, C.; Wheeler, M.A.; Chiocca, E.A.; Reardon, D.A.; Quintana, F.J. Glial and myeloid heterogeneity in the brain tumour microenvironment. Nat. Rev. Cancer 2021, 21, 786–802. [Google Scholar] [CrossRef]
- Simonds, E.F.; Lu, E.D.; Badillo, O.; Karimi, S.; Liu, E.V.; Tamaki, W.; Rancan, C.; Downey, K.M.; Stultz, J.; Sinha, M.; et al. Deep immune profiling reveals targetable mechanisms of immune evasion in immune checkpoint inhibitor-refractory glioblastoma. J. Immunother. Cancer 2021, 9, e002181. [Google Scholar] [CrossRef]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, J.; Mills, L.; Malo, C.S.; Jin, F.; Kurokawa, C.; Geekiyanage, H.; Schroeder, M.; Sarkaria, J.; Johnson, A.J.; Galanis, E. Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro-Oncology 2017, 19, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.; Ferreira, T.; Bretscher, C.; Grewenig, A.; El-Andaloussi, N.; Bonifati, S.; Marttila, T.; Palissot, V.; Hossain, J.A.; Azuaje, F.; et al. Oncolytic H-1 parvovirus binds to sialic acid on laminins for cell attachment and entry. Nat. Commun. 2021, 12, 3834. [Google Scholar] [CrossRef] [PubMed]
- Hossain, J.A.; Marchini, A.; Fehse, B.; Bjerkvig, R.; Miletic, H. Suicide gene therapy for the treatment of high-grade glioma: Past lessons, present trends, and future prospects. Neurooncol. Adv. 2020, 2, vdaa013. [Google Scholar] [CrossRef] [PubMed]
- Hossain, J.A.; Latif, M.A.; Ystaas, L.A.R.; Ninzima, S.; Riecken, K.; Muller, A.; Azuaje, F.; Joseph, J.V.; Talasila, K.M.; Ghimire, J.; et al. Long-term treatment with valganciclovir improves lentiviral suicide gene therapy of glioblastoma. Neuro-Oncology 2019, 21, 890–900. [Google Scholar] [CrossRef]
- Pilanc, P.; Wojnicki, K.; Roura, A.J.; Cyranowski, S.; Ellert-Miklaszewska, A.; Ochocka, N.; Gielniewski, B.; Grzybowski, M.M.; Blaszczyk, R.; Stanczak, P.S.; et al. A Novel Oral Arginase 1/2 Inhibitor Enhances the Antitumor Effect of PD-1 Inhibition in Murine Experimental Gliomas by Altering the Immunosuppressive Environment. Front. Oncol. 2021, 11, 703465. [Google Scholar] [CrossRef]
- Lukas, R.V.; Rodon, J.; Becker, K.; Wong, E.T.; Shih, K.; Touat, M.; Fasso, M.; Osborne, S.; Molinero, L.; O’Hear, C.; et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J. Neurooncol. 2018, 140, 317–328. [Google Scholar] [CrossRef]
- Litak, J.; Mazurek, M.; Grochowski, C.; Kamieniak, P.; Rolinski, J. PD-L1/PD-1 Axis in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5347. [Google Scholar] [CrossRef] [Green Version]
- Jacques, F.H.; Nicholas, G.; Lorimer, I.A.J.; Sikati Foko, V.; Prevost, J.; Dumais, N.; Milne, K.; Nelson, B.H.; Woulfe, J.; Jansen, G.; et al. Avelumab in newly diagnosed glioblastoma. Neurooncol. Adv. 2021, 3, vdab118. [Google Scholar] [CrossRef]
- Wang, X.B.; Fan, Z.Z.; Anton, D.; Vollenhoven, A.V.; Ni, Z.H.; Chen, X.F.; Lefvert, A.K. CTLA4 is expressed on mature dendritic cells derived from human monocytes and influences their maturation and antigen presentation. BMC Immunol. 2011, 12, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barclay, A.N.; Van den Berg, T.K. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: Structure, function, and therapeutic target. Annu. Rev. Immunol. 2014, 32, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Sun, H.; Yu, J.; Tian, W.; Song, Y. Targeting CD47 for cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P.; et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef]
- Russ, A.; Hua, A.B.; Montfort, W.R.; Rahman, B.; Riaz, I.B.; Khalid, M.U.; Carew, J.S.; Nawrocki, S.T.; Persky, D.; Anwer, F. Blocking “don’t eat me” signal of CD47-SIRPalpha in hematological malignancies, an in-depth review. Blood Rev. 2018, 32, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Horrigan, S.K.; Reproducibility Project: Cancer, B. Replication Study: The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. eLife 2017, 6, e18173. [Google Scholar] [CrossRef]
- Kamber, R.A.; Nishiga, Y.; Morton, B.; Banuelos, A.M.; Barkal, A.A.; Vences-Catalan, F.; Gu, M.; Fernandez, D.; Seoane, J.A.; Yao, D.; et al. Inter-cellular CRISPR screens reveal regulators of cancer cell phagocytosis. Nature 2021, 597, 549–554. [Google Scholar] [CrossRef]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- von Roemeling, C.A.; Wang, Y.; Qie, Y.; Yuan, H.; Zhao, H.; Liu, X.; Yang, Z.; Yang, M.; Deng, W.; Bruno, K.A.; et al. Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nat. Commun. 2020, 11, 1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Lv, B.; Liu, Y.; Hua, T.; Han, J.; Sun, C.; Xu, L.; Zhang, Z.; Feng, Z.; Cai, Y.; et al. Blocking the CD47-SIRPalpha axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology 2018, 7, e1391973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Leiss, L.; Yang, N.; Rygh, C.B.; Mitra, S.S.; Cheshier, S.H.; Weissman, I.L.; Huang, B.; Miletic, H.; Bjerkvig, R.; et al. Surgical debulking promotes recruitment of macrophages and triggers glioblastoma phagocytosis in combination with CD47 blocking immunotherapy. Oncotarget 2017, 8, 12145–12157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azambuja, J.H.; Gelsleichter, N.E.; Beckenkamp, L.R.; Iser, I.C.; Fernandes, M.C.; Figueiro, F.; Battastini, A.M.O.; Scholl, J.N.; de Oliveira, F.H.; Spanevello, R.M.; et al. CD73 Downregulation Decreases In Vitro and In Vivo Glioblastoma Growth. Mol. Neurobiol. 2019, 56, 3260–3279. [Google Scholar] [CrossRef] [PubMed]
- de Leve, S.; Wirsdorfer, F.; Jendrossek, V. Targeting the Immunomodulatory CD73/Adenosine System to Improve the Therapeutic Gain of Radiotherapy. Front. Immunol. 2019, 10, 698. [Google Scholar] [CrossRef] [Green Version]
- Eichin, D.; Laurila, J.P.; Jalkanen, S.; Salmi, M. CD73 Activity is Dispensable for the Polarization of M2 Macrophages. PLoS ONE 2015, 10, e0134721. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Novitskiy, S.V.; Sachsenmeier, K.F.; Fornai, M.; Blandizzi, C.; Hasko, G. Switching off CD73: A way to boost the activity of conventional and targeted antineoplastic therapies. Drug Discov. Today 2017, 22, 1686–1696. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Komohara, Y.; Kudo, R.; Tsurushima, K.; Ohnishi, K.; Ikeda, T.; Takeya, M. Oleanolic acid inhibits macrophage differentiation into the M2 phenotype and glioblastoma cell proliferation by suppressing the activation of STAT3. Oncol. Rep. 2011, 26, 1533–1537. [Google Scholar] [CrossRef]
- Jensen, K.V.; Cseh, O.; Aman, A.; Weiss, S.; Luchman, H.A. The JAK2/STAT3 inhibitor pacritinib effectively inhibits patient-derived GBM brain tumor initiating cells in vitro and when used in combination with temozolomide increases survival in an orthotopic xenograft model. PLoS ONE 2017, 12, e0189670. [Google Scholar] [CrossRef] [Green Version]
- Sa, J.K.; Chang, N.; Lee, H.W.; Cho, H.J.; Ceccarelli, M.; Cerulo, L.; Yin, J.; Kim, S.S.; Caruso, F.P.; Lee, M.; et al. Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol. 2020, 21, 216. [Google Scholar] [CrossRef] [PubMed]
- Rosito, M.; Deflorio, C.; Limatola, C.; Trettel, F. CXCL16 orchestrates adenosine A3 receptor and MCP-1/CCL2 activity to protect neurons from excitotoxic cell death in the CNS. J. Neurosci. 2012, 32, 3154–3163. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Schulte, A.; Schnack, C.; Hundhausen, C.; Reiss, K.; Brodway, N.; Held-Feindt, J.; Mentlein, R. Enhanced expression and shedding of the transmembrane chemokine CXCL16 by reactive astrocytes and glioma cells. J. Neurochem. 2005, 93, 1293–1303. [Google Scholar] [CrossRef]
- Gurbuz, I.; Chiquet-Ehrismann, R. CCN4/WISP1 (WNT1 inducible signaling pathway protein 1): A focus on its role in cancer. Int. J. Biochem. Cell Biol. 2015, 62, 142–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, D.; Zhang, Q.; Yu, H.; Zhao, Y.; Shen, L. Identification of WISP1 as a novel oncogene in glioblastoma. Int. J. Oncol. 2017, 51, 1261–1270. [Google Scholar] [CrossRef]
- Shao, N.; Mao, J.; Xue, L.; Wang, R.; Zhi, F.; Lan, Q. Carnosic acid potentiates the anticancer effect of temozolomide by inducing apoptosis and autophagy in glioma. J. Neurooncol. 2019, 141, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Ponomarev, E.D.; Shriver, L.P.; Dittel, B.N. CD40 expression by microglial cells is required for their completion of a two-step activation process during central nervous system autoimmune inflammation. J. Immunol. 2006, 176, 1402–1410. [Google Scholar] [CrossRef] [Green Version]
- Korniluk, A.; Kemona, H.; Dymicka-Piekarska, V. Multifunctional CD40L: Pro- and anti-neoplastic activity. Tumour Biol. 2014, 35, 9447–9457. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Dutcher, J.P.; Anderson, J.E.; Eckhardt, S.G.; Stephans, K.F.; Razvillas, B.; Garl, S.; Butine, M.D.; Perry, V.P.; Armitage, R.J.; et al. Phase I study of recombinant human CD40 ligand in cancer patients. J. Clin. Oncol. 2001, 19, 3280–3287. [Google Scholar] [CrossRef]
- Medina-Echeverz, J.; Hinterberger, M.; Testori, M.; Geiger, M.; Giessel, R.; Bathke, B.; Kassub, R.; Grabnitz, F.; Fiore, G.; Wennier, S.T.; et al. Synergistic cancer immunotherapy combines MVA-CD40L induced innate and adaptive immunity with tumor targeting antibodies. Nat. Commun. 2019, 10, 5041. [Google Scholar] [CrossRef]
- Saellstrom, S.; Sadeghi, A.; Eriksson, E.; Segall, T.; Dimopoulou, M.; Korsgren, O.; Loskog, A.S.; Totterman, T.H.; Hemminki, A.; Ronnberg, H. Adenoviral CD40 Ligand Immunotherapy in 32 Canine Malignant Melanomas-Long-Term Follow Up. Front. Vet. Sci. 2021, 8, 695222. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, A.; Ohkuri, T.; Okada, H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol. Immunother. 2014, 63, 847–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Xun, Y.; Yang, H.; Kaminska, B.; You, H. Toll-like receptors and toll-like receptor-targeted immunotherapy against glioma. J. Hematol. Oncol. 2021, 14, 176. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, Q.; Lubas, M.; Yuan, Y.; Yalcin, F.; Efe, I.E.; Xia, P.; Motta, E.; Buonfiglioli, A.; Lehnardt, S.; et al. Synergistic Toll-like Receptor 3/9 Signaling Affects Properties and Impairs Glioma-Promoting Activity of Microglia. J. Neurosci. 2020, 40, 6428–6443. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Chamberlain, M.C.; Grossman, S.A.; Peereboom, D.M.; Lesser, G.J.; Batchelor, T.T.; Desideri, S.; Salazar, A.M.; Ye, X. A multi-institution phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro-Oncology 2010, 12, 1071–1077. [Google Scholar] [CrossRef]
- Garzon-Muvdi, T.; Theodros, D.; Luksik, A.S.; Maxwell, R.; Kim, E.; Jackson, C.M.; Belcaid, Z.; Ganguly, S.; Tyler, B.; Brem, H.; et al. Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget 2018, 9, 20681–20697. [Google Scholar] [CrossRef] [Green Version]
- Ferber, S.; Tiram, G.; Sousa-Herves, A.; Eldar-Boock, A.; Krivitsky, A.; Scomparin, A.; Yeini, E.; Ofek, P.; Ben-Shushan, D.; Vossen, L.I.; et al. Co-targeting the tumor endothelium and P-selectin-expressing glioblastoma cells leads to a remarkable therapeutic outcome. eLife 2017, 6, e25281. [Google Scholar] [CrossRef]
- Tinoco, R.; Carrette, F.; Barraza, M.L.; Otero, D.C.; Magana, J.; Bosenberg, M.W.; Swain, S.L.; Bradley, L.M. PSGL-1 Is an Immune Checkpoint Regulator that Promotes T Cell Exhaustion. Immunity 2016, 44, 1190–1203. [Google Scholar] [CrossRef] [Green Version]
- Karki, N.R.; Kutlar, A. P-Selectin Blockade in the Treatment of Painful Vaso-Occlusive Crises in Sickle Cell Disease: A Spotlight on Crizanlizumab. J. Pain Res. 2021, 14, 849–856. [Google Scholar] [CrossRef]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Kretz, A.; Naumann, U.; Aulwurm, S.; Egashira, K.; Isenmann, S.; Weller, M. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann. Neurol. 2003, 54, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ao, J.Y.; Zhu, X.D.; Chai, Z.T.; Cai, H.; Zhang, Y.Y.; Zhang, K.Z.; Kong, L.Q.; Zhang, N.; Ye, B.G.; Ma, D.N.; et al. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 1544–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strachan, D.C.; Ruffell, B.; Oei, Y.; Bissell, M.J.; Coussens, L.M.; Pryer, N.; Daniel, D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology 2013, 2, e26968. [Google Scholar] [CrossRef] [Green Version]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Ruttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, D.; Chow, A.; Greter, M.; Saenger, Y.; Kwan, W.H.; Leboeuf, M.; Ginhoux, F.; Ochando, J.C.; Kunisaki, Y.; van Rooijen, N.; et al. Pretransplant CSF-1 therapy expands recipient macrophages and ameliorates GVHD after allogeneic hematopoietic cell transplantation. J. Exp. Med. 2011, 208, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef] [PubMed]


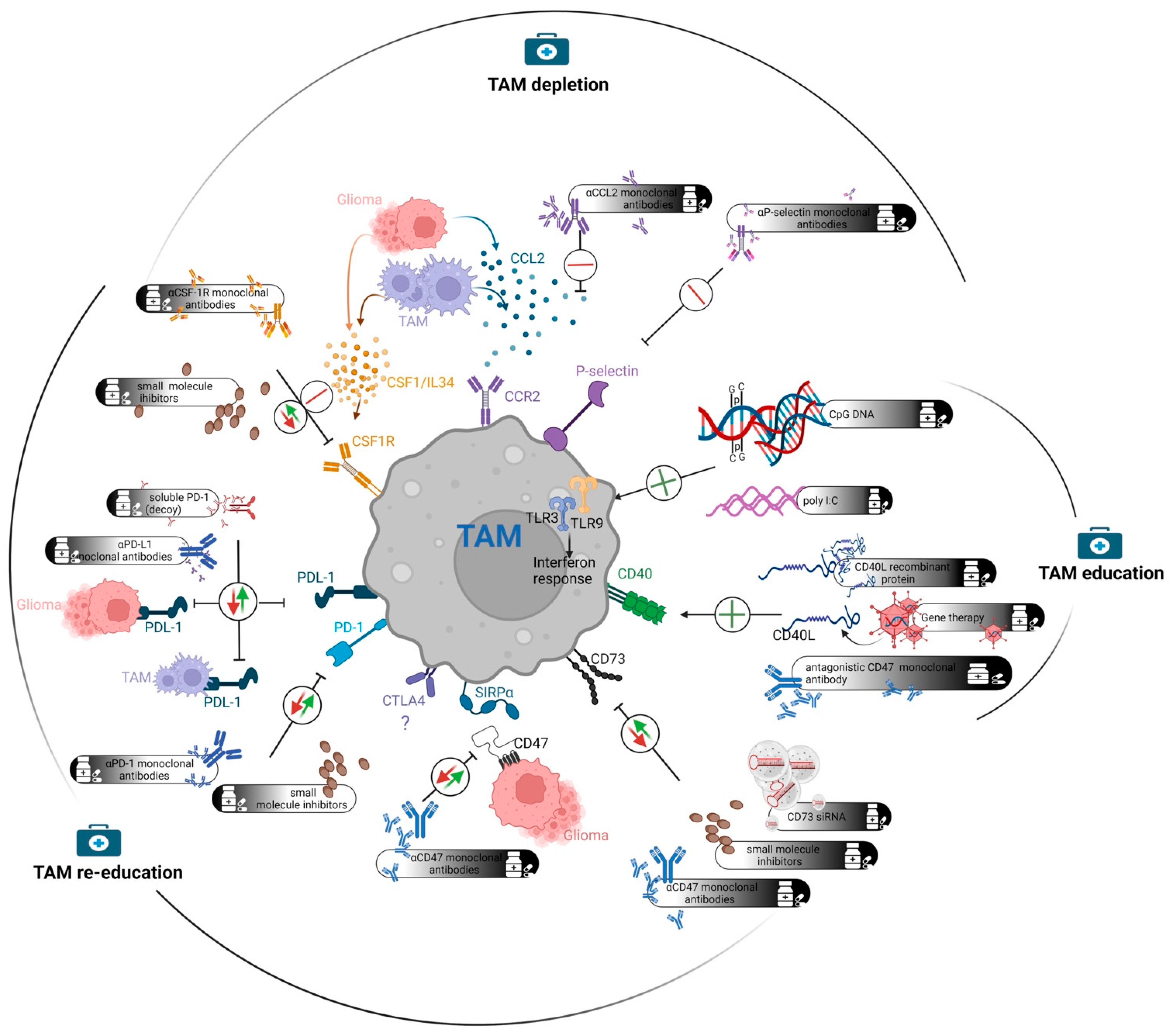{kind=link}
| Target | Mechanism | Effect of Treatment |
|---|---|---|
| TAM Re-education | ||
| PD-1/PD-L1 | PD-L1 binding to PD-1 receptors serves as inhibitory signaling for T cell activation and TAMs [120]. | Inhibition: Increased T-cell proliferation, activity and killing by both T cells and TAMs. |
| CTLA4 | Negative regulator of T-cell activation [56]. | Inhibition: Increased T-cell activation. Uncertain effects on TAMs. |
| CD47 | CD47 binding to SIRPα on TAMs serves as a “don’t eat me” signal [121]. | Inhibition: Increased phagocytic activity. Reduced M2 markers. |
| CD73 | Converts ATP to adenosine which stimulates immunosuppression [46]. | Inhibition: Reduced tumor growth and increased M1 polarization. |
| STAT3 | STAT3 activation stimulates tumor growth and reduces TAM activation [122]. | Inhibition: Decreased tumor progression and increased immune response. |
| MARCO | MARCO stimulation drives GBM towards mesenchymal shift. Induce immunosuppressive TAMs [123]. | Inhibition: Increased pro-inflammatory TAMs. Increased effect of CTLA2 in melanoma and breast cancer. |
| CXCL16 | CXCL16 binding to its receptor CXCR6 promotes tumor progression and M2 polarization in microglia [124]. | Inhibition: Increased M1 markers. Reduced tumor growth. |
| WISP1 | WISP1 binding to α6β1-receptor induces immunosuppression and tumor progression [125]. | Inhibition: Reduced immunosuppression. Reduced tumor growth. |
| TAM Education | ||
| CD40/CD40L | Increased antigen-presenting function and immune response [126,127]. | Activation: M1-like polarization and increased survival. |
| Toll-like receptor | Activates innate immune response [128,129]. | Activation: Limited success in the treatment of glioma. |
| TAM Depletion | ||
| SELP | SELP-PSGL-1 axis increases M2 shift [130]. | Inhibition: Reduced immunosuppression. Reduced tumor growth. |
| CCL2/CCR2 | Monocyte chemoattractant and promotes efflux of monocytes [131]. | Inhibition: Reduced influx of infiltrating TAMs. Increased survival in animal models. |
| CSF1R * | CSF1R stimulation induces differentiation and survival of TAMs [132]. | Inhibition: Reduced immunosuppression. Reduced tumor growth. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersen, J.K.; Miletic, H.; Hossain, J.A. Tumor-Associated Macrophages in Gliomas—Basic Insights and Treatment Opportunities. Cancers 2022, 14, 1319. https://doi.org/10.3390/cancers14051319
Andersen JK, Miletic H, Hossain JA. Tumor-Associated Macrophages in Gliomas—Basic Insights and Treatment Opportunities. Cancers. 2022; 14(5):1319. https://doi.org/10.3390/cancers14051319
Chicago/Turabian StyleAndersen, Johannes K., Hrvoje Miletic, and Jubayer A. Hossain. 2022. "Tumor-Associated Macrophages in Gliomas—Basic Insights and Treatment Opportunities" Cancers 14, no. 5: 1319. https://doi.org/10.3390/cancers14051319