
Molecular Landscape of Small Bowel Adenocarcinoma

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Molecular Characteristics
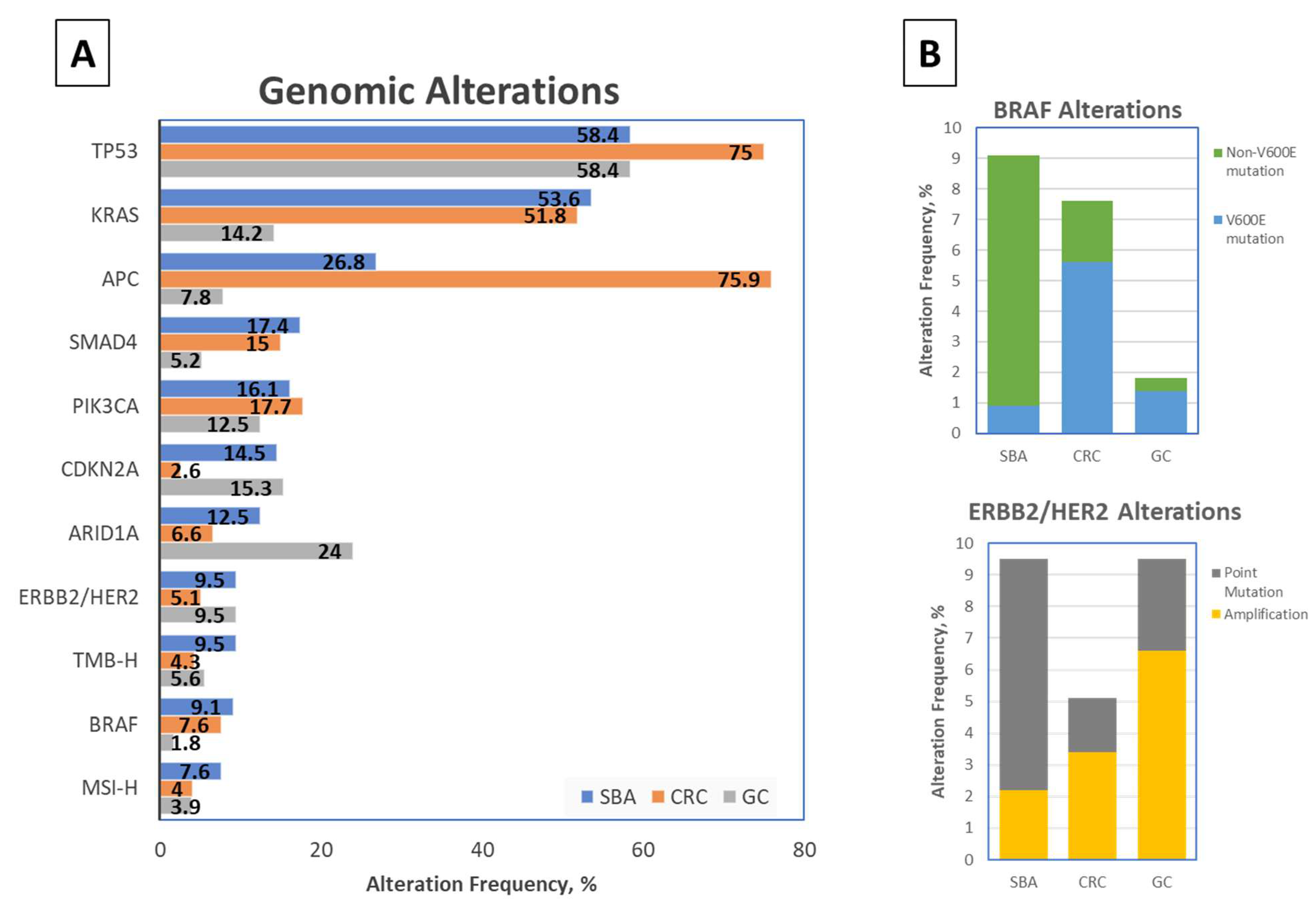
2.1. Comparison with Neighboring Intestinal Cancers
2.2. Small Bowel Subsite Comparison
2.3. Comparison across SBA Etiologies
3. Potentially Targetable Genomic Alterations
3.1. BRAF
3.2. ERBB2/HER2
3.3. Microsatellite Instability and Tumor Mutational Burden
3.4. EGFR
3.5. Other Targetable Genomic Alterations
3.6. Epigenetic Alterations and Non-Coding RNA
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Schrock, A.B.; Devoe, C.E.; McWilliams, R.; Sun, J.; Aparicio, T.; Stephens, P.J.; Ross, J.S.; Wilson, R.; Miller, V.A.; Ali, S.M.; et al. Genomic Profiling of Small-Bowel Adenocarcinoma. JAMA Oncol. 2017, 3, 1546–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, K.; Raghav, K.; Overman, M.J. Small Bowel Adenocarcinoma: Etiology, Presentation, and Molecular Alterations. J. Natl. Compr. Cancer Netw. 2019, 17, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Raghav, K.; Overman, M.J. Small bowel adenocarcinomas—Existing evidence and evolving paradigms. Nat. Rev. Clin. Oncol. 2013, 10, 534–544. [Google Scholar] [CrossRef]
- Neugut, A.I.; Jacobson, J.S.; Suh, S.; Mukherjee, R.; Arber, N. The epidemiology of cancer of the small bowel. Cancer Epidemiol. Biomark. Prev. 1998, 7, 243–251. [Google Scholar]
- Siegel, R.; Miller, K.D.; Jemal, A. Cancer statistics, 2021. CA Cancer J Clin. 2021, 70, 7–30. [Google Scholar] [CrossRef]
- DeSesso, J.; Jacobson, C. Anatomical and physiological parameters affecting gastrointestinal absorption in humans and rats. Food Chem. Toxicol. 2001, 39, 209–228. [Google Scholar] [CrossRef]
- Haselkorn, T.; Whittemore, A.S.; Lilienfeld, D.E. Incidence of Small Bowel Cancer in the United States and Worldwide: Geographic, Temporal, and Racial Differences. Cancer Causes Control 2005, 16, 781–787. [Google Scholar] [CrossRef]
- Overman, M.J.; Raghav, K.; Lieu, C.H.; Fournier, C.F. Small Bowel Cancer and Appendiceal Tumors. The MD Anderson Manual of Medical Oncology, 3rd ed.; Kantarjian, H.M., Wolff, R.A., Eds.; McGraw Hill: New York, NY, USA, 2016. [Google Scholar]
- Aparicio, T.; Zaanan, A.; Svrcek, M.; Laurent-Puig, P.; Carrere, N.; Manfredi, S.; Locher, C.; Afchain, P. Small bowel adenocarcinoma: Epidemiology, risk factors, diagnosis and treatment. Dig. Liver Dis. 2014, 46, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Schottenfeld, D.; Beebe-Dimmer, J.L.; Vigneau, F.D. The Epidemiology and Pathogenesis of Neoplasia in the Small Intestine. Ann. Epidemiol. 2008, 19, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Sanders, L.M.; Henderson, C.E.; Hong, M.Y.; Barhoumi, R.; Burghardt, R.C.; Carroll, R.J.; Turner, N.D.; Chapkin, R.S.; Lupton, J.R. Pro-oxidant environment of the colon compared to the small intestine may contribute to greater cancer susceptibility. Cancer Lett. 2004, 208, 155–161. [Google Scholar] [CrossRef]
- Dennis, K.L.; Saadalla, A.; Blatner, N.R.; Wang, S.; Venkateswaran, V.; Gounari, F.; Cheroutre, H.; Weaver, C.T.; Roers, A.; Egilmez, N.K.; et al. T-cell Expression of IL10 Is Essential for Tumor Immune Surveillance in the Small Intestine. Cancer Immunol. Res. 2015, 3, 806–814. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.H.; Yu, M.C.; Mack, T.M. Smoking, alcohol use, dietary factors and risk of small intestinal adenocarcinoma. Int. J. Cancer 1997, 70, 512–517. [Google Scholar] [CrossRef]
- Kaerlev, L.; Teglbjaerg, P.S.; Sabroe, S.; Kolstad, H.A.; Ahrens, W.; Eriksson, M.; Guenel, P.; Hardell, L.; Launoy, G.; Merler, E.; et al. Is there an association between alcohol intake or smoking and small bowel adenocarcinoma? Results from a European multi-center case–control study. Cancer Causes Control 2000, 11, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.-H.; Linet, M.S.; McLaughlin, J.K.; Hsing, A.W.; Chien, H.T.C.; Blot, W.J. Risk factors for small intestine cancer. Cancer Causes Control 1993, 4, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Negri, E.; Bosetti, C.; La Vecchia, C.; Fioretti, F.; Conti, E.; Franceschi, S. Risk factors for adenocarcinoma of the small intestine. Int. J. Cancer 1999, 82, 171–174. [Google Scholar] [CrossRef]
- Cross, A.J.; Leitzmann, M.F.; Subar, A.F.; Thompson, F.E.; Hollenbeck, A.R.; Schatzkin, A. A Prospective Study of Meat and Fat Intake in Relation to Small Intestinal Cancer. Cancer Res. 2008, 68, 9274–9279. [Google Scholar] [CrossRef] [Green Version]
- Delaunoit, T.; Neczyporenko, F.; Limburg, P.J.; Erlichman, C. Pathogenesis and Risk Factors of Small Bowel Adenocarcinoma: A Colorectal Cancer Sibling? Am. J. Gastroenterol. 2005, 100, 703–710. [Google Scholar] [CrossRef]
- Rampertab, S.D.; Forde, K.A.; Green, P.H.R. Small bowel neoplasia in coeliac disease. Gut 2003, 52, 1211–1214. [Google Scholar] [CrossRef] [Green Version]
- Howdle, P.D.; Holmes, G.K.T. Small bowel malignancy in coeliac disease. Gut 2004, 53, 470. [Google Scholar]
- Caio, G.; Volta, U.; Ursini, F.; Manfredini, R.; de Giorgio, R. Small bowel adenocarcinoma as a complication of celiac disease: Clinical and diagnostic features. BMC Gastroenterol. 2019, 19, 45. [Google Scholar] [CrossRef] [Green Version]
- Green, P.H.; Fleischauer, A.T.; Bhagat, G.; Goyal, R.; Jabri, B.; Neugut, A.I. Risk of malignancy in patients with celiac disease. Am. J. Med. 2003, 115, 191–195. [Google Scholar] [CrossRef]
- Ciresi, D.L.; Scholten, D.J. The continuing clinical dilemma of primary tumors of the small intestine. Am. Surg. 1995, 61, 698–703. [Google Scholar] [PubMed]
- Talamonti, M.S.; Goetz, L.H.; Rao, S.; Joehl, R.J. Primary cancers of the small bowel: Analysis of prognostic factors and results of surgical management. Arch. Surg. 2003, 137, 564–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, M.J.; Hu, C.-Y.; Kopetz, S.; Abbruzzese, J.L.; Wolff, R.A.; Chang, G.J. A Population-Based Comparison of Adenocarcinoma of the Large and Small Intestine: Insights into a Rare Disease. Ann. Surg. Oncol. 2012, 19, 1439–1445. [Google Scholar] [CrossRef]
- Young, J.I.; Mongoue-Tchokote, S.; Wieghard, N.; Mori, M.; Vaccaro, G.M.; Sheppard, B.C.; Tsikitis, V.L. Treatment and survival of small bowel adenocarcinoma in the United States: A comparison with colon cancer. Dis. Colon Rectum 2016, 59, 306. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Small Bowel Adenocarcinoma (Version 2.2021). Available online: http://www.nccn.org/professionals/physician_gls/pdf/small_bowel.pdf (accessed on 19 December 2021).
- Lee, H.J.; Lee, O.J.; Jang, K.T.; Bae, Y.K.; Chung, J.Y.; Eom, D.W.; Kim, J.M.; Yu, E.; Hong, S.M. Combined loss of E-cadherin and aberrant beta-catenin protein expression correlates with a poor prognosis for small intestinal adenocarcinoma. Am. J. Clin. Pathol. 2013, 139, 167–176. [Google Scholar] [CrossRef]
- Alvi, M.A.; McArt, D.; Kelly, P.; Fuchs, M.-A.; Alderdice, M.; McCabe, C.M.; Bingham, V.; Mcgready, C.; Tripathi, S.; Emmert-Streib, F.; et al. Comprehensive molecular pathology analysis of small bowel adenocarcinoma reveals novel targets with potential for clinical utility. Oncotarget 2015, 6, 20863–20874. [Google Scholar] [CrossRef] [Green Version]
- Laforest, A.; Aparicio, T.; Zaanan, A.; Silva, F.P.; Didelot, A.; Desbeaux, A.; Le Corre, D.; Benhaim, L.; Pallier, K.; Aust, D.; et al. ERBB2 gene as a potential therapeutic target in small bowel adenocarcinoma. Eur. J. Cancer 2014, 50, 1740–1746. [Google Scholar] [CrossRef]
- Hänninen, U.A.; Katainen, R.; Tanskanen, T.; Plaketti, R.-M.; Laine, R.; Hamberg, J.; Ristimäki, A.; Pukkala, E.; Taipale, M.; Mecklin, J.-P.; et al. Exome-wide somatic mutation characterization of small bowel adenocarcinoma. PLoS Genet. 2018, 14, e1007200. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, T.; Svrcek, M.; Zaanan, A.; Beohou, E.; Laforest, A.; Afchain, P.; Mitry, E.; Taieb, J.; Di Fiore, F.; Gornet, J.-M.; et al. Small bowel adenocarcinoma phenotyping, a clinicobiological prognostic study. Br. J. Cancer 2013, 109, 3057–3066. [Google Scholar] [CrossRef] [Green Version]
- Groves, C.J.; Saunders, B.P.; Spigelman, A.D.; Phillips, R.K.S. Duodenal cancer in patients with familial adenomatous polyposis (FAP): Results of a 10 year prospective study. Gut 2002, 50, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Jagelman, D.; Decosse, J.; Bussey, H. Upper gastrointestinal cancer in familial adenomatous polyposis. Lancet 1988, 331, 1149–1151. [Google Scholar] [CrossRef]
- Bülow, S.; Alm, T.; Fausa, O.; Hultcrantz, R.; Järvinen, H.; Vasen, H. DAF Project Group Duodenal adenomatosis in familial adenomatous polyposis. Int. J. Color. Dis. 1995, 10, 43–46. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Brensinger, J.D.; Tersmette, A.C.; Goodman, S.N.; Petersen, G.M.; Booker, S.V.; Cruz–Correa, M.; Offerhaus, J.A. Very high risk of cancer in familial Peutz–Jeghers syndrome. Gastroenterology 2000, 119, 1447–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haan, J.C.; Buffart, T.E.; Eijk, P.P.; van de Wiel, M.A.; van Wieringen, W.N.; Howdle, P.D.; Mulder, C.J.J.; van de Velde, C.J.; Quirke, P.; Nagtegaal, I.D.; et al. Small bowel adenocarcinoma copy number profiles are more closely related to colorectal than to gastric cancers. Ann. Oncol. 2012, 23, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Lowenfels, A. Why are small-bowel tumours so rare? Lancet 1973, 301, 24–26. [Google Scholar] [CrossRef]
- Calman, K.C. Why are small bowel tumours rare? An experimental model. Gut 1974, 15, 552–554. [Google Scholar] [CrossRef] [Green Version]
- Blaker, H.; Helmchen, B.; Bonisch, A.; Aulmann, S.; Penzel, R.; Otto, H.F.; Rieker, R.J. Mutational activation of the RAS-RAF-MAPK and the WNT pathway in small intestinal adenocarcinomas. Scand. J. Gastroenterol. 2004, 39, 748–753. [Google Scholar] [CrossRef]
- Miyaki, M.; Konishi, M.; Kikuchi-Yanoshita, R.; Enomoto, M.; Igari, T.; Tanaka, K.; Muraoka, M.; Takahashi, H.; Amada, Y.; Fukayama, M. Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors. Cancer Res. 1994, 54, 3011–3020. [Google Scholar]
- Adam, L.; Lucas, F.A.S.; Fowler, R.; Yu, Y.; Wu, W.; Liu, Y.; Wang, H.; Menter, D.G.; Tetzlaff, M.T.; Ensor, J.E.; et al. DNA Sequencing of Small Bowel Adenocarcinomas Identifies Targetable Recurrent Mutations in the ERBB2 Signaling Pathway. Clin. Cancer Res. 2019, 25, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Kelsey, C.R.; Nelson, J.W.; Willett, C.G.; Chino, J.P.; Clough, R.W.; Bendell, J.C.; Tyler, D.S.; Hurwitz, H.I.; Morse, M.A.; Clary, B.M.; et al. Duodenal Adenocarcinoma: Patterns of Failure After Resection and the Role of Chemoradiotherapy. Int. J. Radiat. Oncol. 2007, 69, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Aarnio, M.; Mecklin, J.-P.; Aaltonen, L.A.; Nyström-Lahti, M.; Järvinen, H.J. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (hnpcc) syndrome. Int. J. Cancer 1995, 64, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Vasen, J.F.; Wijnen, J.T.; Menko, F.H.; Kleibeuker, J.H.; Taal, B.G.; Griffioen, G.; Nagengast, F.M.; Meijers-Heijboer, E.H.; Bertario, L.; Varesco, L.; et al. Cancer risk in families with hereditary nonpolyposis colorectal cancer diagnosed by mutation analysis. Gastroenterology 1996, 110, 1020. [Google Scholar] [CrossRef] [PubMed]
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.-J.; Caron, O.; et al. Cancer Risks Associated with Germline Mutations in MLH1, MSH2, and MSH6 Genes in Lynch Syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef]
- Rodriguez-Bigas, M.A.; Vasen, H.F.; Lynch, H.T.; Watson, P.; Myrhøj, T.; Järvinen, H.J.; Mecklin, J.P.; Macrae, F.; St John, D.J.; Bertario, L.; et al. Characteristics of small bowel carcinoma in hereditary nonpolyposis colorectal carcinoma. International Collaborative Group on HNPCC. Cancer 1998, 83, 240–244. [Google Scholar]
- Palascak-Juif, V.; Bouvier, A.M.; Cosnes, J.; Flourié, B.; Bouché, O.; Cadiot, G.; Lémann, M.; Bonaz, B.; Denet, C.; Marteau, P.; et al. Small bowel adenocarcinoma in patients with Crohn’s disease compared with small bowel adenocarcinoma de novo. Inflamm Bowel Dis. 2005, 11, 828. [Google Scholar] [CrossRef]
- Von Roon, A.C.; Reese, G.; Teare, J.; Constantinides, V.; Darzi, A.W.; Tekkis, P.P. The risk of cancer in patients with Crohn’s disease. Dis. Colon Rectum 2007, 50, 839–855. [Google Scholar] [CrossRef]
- Cahill, C.; Gordon, P.H.; Petrucci, A.; Boutros, M. Small bowel adenocarcinoma and Crohn’s disease: Any further ahead than 50 years ago? World J. Gastroenterol. 2014, 20, 11486–11495. [Google Scholar] [CrossRef]
- Yaeger, R.; Shah, M.A.; Miller, V.A.; Kelsen, J.R.; Wang, K.; Heins, Z.J.; Ross, J.S.; He, Y.; Sanford, E.; Yantiss, R.K.; et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct from Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology 2016, 151, 278–287.e6. [Google Scholar] [CrossRef] [Green Version]
- Diosdado, B.; Buffart, T.E.; Watkins, R.; Carvalho, B.; Ylstra, B.; Tijssen, M.; Bolijn, A.S.; Lewis, F.; Maude, K.; Verbeke, C.; et al. High-Resolution Array Comparative Genomic Hybridization in Sporadic and Celiac Disease–Related Small Bowel Adenocarcinomas. Clin. Cancer Res. 2010, 16, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Potter, D.D.; Murray, J.A.; Donohue, J.H.; Burgart, L.J.; Nagorney, D.M.; Van Heerden, J.A.; Plevak, M.F.; Zinsmeister, A.R.; Thibodeau, S.N. The Role of Defective Mismatch Repair in Small Bowel Adenocarcinoma in Celiac Disease. Cancer Res. 2004, 64, 7073–7077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Society of Clinical Oncology. Programmed death-1 blockade in mismatch repair deficient cancer independent of tumor histology. J. Clin. Oncol. 2016, 34, 3003. Available online: http://meetinglibrary.asco.org/content/170754-176 (accessed on 9 December 2021). [CrossRef]
- García-Foncillas, J.; Sunakawa, Y.; Aderka, D.; Wainberg, Z.; Ronga, P.; Witzler, P.; Stintzing, S. Distinguishing Features of Cetuximab and Panitumumab in Colorectal Cancer and Other Solid Tumors. Front. Oncol. 2019, 9, 849. [Google Scholar] [CrossRef]
- Overman, M.J.; Wolff, R.A.; Wang, H. Reply: Cetuximab in small bowel adenocarcinoma: A new friend? Br. J. Cancer 2010, 103, 1306. [Google Scholar] [CrossRef] [Green Version]
- Poddar, N.; Raza, S.; Sharma, B.; Liu, M.; Gohari, A.; Kalavar, M. Small Bowel Adenocarcinoma Presenting with Refractory Iron Deficiency Anemia—Case Report and Review of Literature. Case Rep. Oncol. 2011, 4, 458–463. [Google Scholar] [CrossRef]
- Santini, D.; Fratto, M.E.; Spoto, C.; Russo, A.D.; Galluzzo, S.; Zoccoli, A.; Vincenzi, B.; Tonini, G. Cetuximab in small bowel adenocarcinoma: A new friend? Br. J. Cancer 2010, 103, 1305. [Google Scholar] [CrossRef]
- Gulhati, P.; Raghav, K.; Shroff, R.; Varadhachary, G.; Javle, M.; Qiao, W.; Wang, H.; Morris, J.; Wolff, R.; Overman, M.J. Phase II study of panitumumab in RAS wild-type metastatic adenocarcinoma of small bowel or ampulla of vater. Oncologist 2018, 23, 277-e26. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Li, H.; Aerbajinai, W.; Botos, I.; Rodgers, G.P. OLFM4-RET fusion is an oncogenic driver in small intestine adenocarcinoma. Oncogene 2021, 41, 72–82. [Google Scholar] [CrossRef]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Loregger, A.; Grandl, M.; Mejías-Luque, R.; Allgäuer, M.; Degenhart, K.; Haselmann, V.; Oikonomou, C.; Hatzis, P.; Janssen, K.P.; Nitsche, U.; et al. The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated b-catenin by sequestering TCF4 to the nuclear membrane. Sci. Signal. 2015, 8, ra90. [Google Scholar] [CrossRef]
- Lannagan, T.R.M.; Lee, Y.K.; Wang, T.; Roper, J.; Bettington, M.L.; Fennell, L.; Vrbanac, L.; Jonavicius, L.; Somashekar, R.; Gieniec, K.; et al. Genetic editing of colonic organoids provides a molecularly distinct and orthotopic preclinical model of serrated carcinogenesis. Gut 2019, 68, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Spit, M.; Fenderico, N.; Jordens, I.; Radaszkiewicz, T.; Lindeboom, R.G.; Bugter, J.M.; Cristobal, A.; Ootes, L.; van Osch, M.; Janssen, E.; et al. RNF43 truncations trap CK1 to drive niche-independent self-renewal in cancer. EMBO J. 2020, 39, e103932. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Dodge, M.E.; Tang, W.; Lu, J.; Ma, Z.; Fan, C.W.; Wei, S.; Hao, W.; Kilgore, J.; Williams, N.S.; et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, B.-K.; van Es, J.H.; Born, M.V.D.; Clevers, H. Porcupine inhibitor suppresses paracrine Wnt-driven growth of Rnf43;Znrf3-mutant neoplasia. Proc. Natl. Acad. Sci. USA 2015, 112, 7548–7550. [Google Scholar] [CrossRef] [Green Version]
- Jass, J.R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007, 50, 113–130. [Google Scholar] [CrossRef]
- Bariol, C.; Suter, C.; Cheong, K.; Ku, S.L.; Meagher, A.; Hawkins, N.; Ward, R. The relationship between hypomethylation and CpG island methylation in colorectal neoplasia. Am. J. Pathol. 2003, 162, 1361–1371. [Google Scholar] [CrossRef] [Green Version]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.-P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.G. Hypermethylation of tumor suppressor genes in cancer. Semin. Cancer Biol. 1999, 9, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P. Colon cancer: It’s CIN or CIMP. Clin. Cancer Res. 2008, 14, 5939–5940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warth, A.; Kloor, M.; Schirmacher, P.; Bläker, H. Genetics and epigenetics of small bowel adenocarcinoma: The interactions of CIN, MSI, and CIMP. Mod. Pathol. 2011, 24, 564–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Hirata, Y.; Suzuki, N.; Ihara, S.; Sakitani, K.; Kobayashi, Y.; Kinoshita, H.; Hayakawa, Y.; Yamada, A.; Watabe, H.; et al. Characterization of a New Small Bowel Adenocarcinoma Cell Line and Screening of Anti-Cancer Drug against Small Bowel Adenocarcinoma. Am. J. Pathol. 2015, 185, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, Y.H.; Kim, S.E.; Kim, N.G.; Noh, S.H.; Kim, H. Concerted promoter hypermethylation of hMLH1, p16INK4A, and E-cadherin in gastric carcinomas with microsatellite instability. J. Pathol. 2003, 200, 23–31. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Esteller, M.; Wang, S.; Tamura, G.; Suzuki, H.; Yin, J.; Zou, T.T.; Abraham, J.M.; Kong, D.; Smolinski, K.N.; et al. Hypermethylation of the hMLH1 gene promoter in human gastric cancers with microsatellite instability. Cancer Res. 1999, 59, 1090–1095. [Google Scholar]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef]
- Tang, R.; Changchien, C.R.; Wu, M.-C.; Fan, C.-W.; Liu, K.-W.; Chen, J.-S.; Chien, H.-T.; Hsieh, L.-L. Colorectal cancer without high microsatellite instability and chromosomal instability-an alternative genetic pathway to human colorectal cancer. Carcinogenesis 2004, 25, 841–846. [Google Scholar] [CrossRef]
- Hawkins, N.J.; Tomlinson, I.; Meagher, A.; Ward, R.L. Microsatellite-stable diploid carcinoma: A biologically distinct and aggressive subset of sporadic colorectal cancer. Br. J. Cancer 2001, 84, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Eu, K.W.; Seow-Choen, F.; Vijayan, V.; Cheah, P.Y. Microsatellite instability and aneuploidy rate in young colorectal-cancer patients do not differ significantly from those in older patients. Int. J. Cancer 1999, 80, 667–670. [Google Scholar] [CrossRef]
- Chan, T.L.; Curtis, L.C.; Leung, S.Y.; Farrington, S.M.; Ho, J.W.; Chan, A.S.; Lam, P.W.; Tse, C.W.; Dunlop, M.G.; Wyllie, A.H.; et al. Early-onset colorectal cancer with stable microsatellite DNA and near-diploid chromosomes. Oncogene 2001, 20, 4871–4876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.W.; Pincas, H.; Bacolod, M.D.; Schemmann, G.; Giardina, S.F.; Huang, J.; Barral, S.; Idrees, K.; Khan, S.A.; Zeng, Z.; et al. CpG island methylator phenotype associates with low-degree chromosomal abnormalities in colorectal cancer. Clin. Cancer Res. 2008, 14, 6005–6013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, A.; Nagasaka, T.; Arnold, C.N.; Inoue, T.; Hamilton, C.; Niedzwiecki, D.; Compton, C.; Mayer, R.J.; Goldberg, R.; Bertagnolli, M.M.; et al. The CpG Island Methylator Phenotype and Chromosomal Instability Are Inversely Correlated in Sporadic Colorectal Cancer. Gastroenterology 2007, 132, 127–138. [Google Scholar] [CrossRef] [PubMed]
- El Bairi, K.; Tariq, K.; Himri, I.; Jaafari, A.; Smaili, W.; Kandhro, A.H.; Gouri, A.; Ghazi, B. Decoding colorectal cancer epigenomics. Cancer Genet. 2018, 220, 49–76. [Google Scholar] [CrossRef]
- Porcellini, E.; Laprovitera, N.; Riefolo, M.; Ravaioli, M.; Garajova, I.; Ferracin, M. Epigenetic and epitranscriptomic changes in colorectal cancer: Diagnostic, prognostic, and treatment implications. Cancer Lett. 2018, 419, 84–95. [Google Scholar] [CrossRef]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef]
- Migliore, L.; Migheli, F.; Spisni, R.; Coppedè, F. Genetics, Cytogenetics, and Epigenetics of Colorectal Cancer. J. Biomed. Biotechnol. 2011, 2011, 792362. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Xu, Z.; Liu, D. Small non-coding RNA and colorectal cancer. J. Cell. Mol. Med. 2019, 23, 3050–3057. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016, 17, 1206. [Google Scholar] [CrossRef]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.C.; Dou, Y. Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Kay Li, Q.; et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-B.; Chen, Z.-L.; Li, J.-G.; Hu, X.-D.; Shi, X.-J.; Sun, Z.-M.; Zhang, F.; Zhao, Z.-R.; Li, Z.-T.; Liu, Z.-Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Akhtar-Zaidi, B.; Cowper-Sallari, R.; Corradin, O.; Saiakhova, A.; Bartels, C.F.; Balasubramanian, D.; Myeroff, L.; Lutterbaugh, J.; Jarrar, A.; Kalady, M.F.; et al. Epigenomic Enhancer Profiling Defines a Signature of Colon Cancer. Science 2012, 336, 736–739. [Google Scholar] [CrossRef] [Green Version]
- Kantidakis, T.; Saponaro, M.; Mitter, R.; Horswell, S.; Kranz, A.; Boeing, S.; Aygün, O.; Kelly, G.P.; Matthews, N.; Stewart, A.; et al. Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev. 2016, 30, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Romano, G.; Veneziano, D.; Acunzo, M.; Croce, C.M. Small non-coding RNA and cancer. Carcinogenesis 2017, 38, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Bao, Y.; Yang, W. Regulatory miRNAs in Colorectal Carcinogenesis and Metastasis. Int. J. Mol. Sci. 2017, 18, 890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Zhu, H.; Lv, L.; Zhou, Y.; Huo, J. MiRNA s in oesophageal squamous cancer. Neth. J. Med. 2013, 71, 69–75. [Google Scholar] [PubMed]
- Catela Ivkovic, T.; Voss, G.; Cornella, H.; Ceder, Y. microRNAs as cancer therapeutics: A step closer to clinical application. Cancer Lett. 2017, 407, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Orellana, E.; Kasinski, A.L. MicroRNAs in Cancer: A Historical Perspective on the Path from Discovery to Therapy. Cancers 2015, 7, 1388–1405. [Google Scholar] [CrossRef] [PubMed]
- Pichler, M.; Calin, A.G. MicroRNAs in cancer: From developmental genes in worms to their clinical application in patients. Br. J. Cancer 2015, 113, 569–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfossi, S.; Fu, X.; Nagvekar, R.; Calin, G.A. MicroRNAs, Regulatory Messengers Inside and Outside Cancer Cells. Adv. Exp. Med. Biol. 2018, 1056, 87–108. [Google Scholar] [CrossRef]
- Oom, A.L.; Humphries, B.A.; Yang, C. MicroRNAs: Novel Players in Cancer Diagnosis and Therapies. BioMed Res. Int. 2014, 2014, 959461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandino, G.; Fazi, F.; Donzelli, S.; Kedmi, M.; Sas-Chen, A.; Muti, P.; Strano, S.; Yarden, Y. Tumor suppressor microRNAs: A novel non-coding alliance against cancer. FEBS Lett. 2014, 588, 2639–2652. [Google Scholar] [CrossRef] [Green Version]
- Orang, A.V.; Barzegari, A. MicroRNAs in Colorectal Cancer: From Diagnosis to Targeted Therapy. Asian Pac. J. Cancer Prev. 2014, 15, 6989–6999. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, S.; Kaur, K.; Huang, R.; Zhang, Q.; Kaur, P.; Yazdani, H.O.; Bilal, M.U.; Zheng, J.; Zheng, L.; Wang, X.-S. MicroRNAs in colorectal cancer: Role in metastasis and clinical perspectives. World J. Gastroenterol. 2014, 20, 17011–17019. [Google Scholar] [CrossRef]
- Mohammadi, A.; Mansoori, B.; Baradaran, B. The role of microRNAs in colorectal cancer. Biomed. Pharmacother. 2016, 84, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Schetter, A.J.; Okayama, H.; Harris, C.C. The Role of MicroRNAs in Colorectal Cancer. Cancer J. 2012, 18, 244–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, H.; Frampton, A.E.; Malczewska, A.; Ottaviani, S.; Stronach, E.A.; Flora, R.; Kaemmerer, D.; Schwach, G.; Pfragner, R.; Faiz, O.; et al. MicroRNAs associated with small bowel neuroendocrine tumours and their metastases. Endocr.-Relat. Cancer 2016, 23, 711–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malczewska, A.; Frampton, A.E.; Mato Prado, M.; Ameri, S.; Dabrowska, A.F.; Zagorac, S.; Clift, A.K.; Kos-Kudła, B.; Faiz, O.; Stebbing, J.; et al. Circulating MicroRNAs in Small-bowel Neuroendocrine Tumors: A Potential Tool for Diagnosis and Assessment of Effectiveness of Surgical Resection. Ann. Surg. 2021, 274, e1–e9. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandya, K.; Overman, M.J.; Gulhati, P. Molecular Landscape of Small Bowel Adenocarcinoma. Cancers 2022, 14, 1287. https://doi.org/10.3390/cancers14051287
Pandya K, Overman MJ, Gulhati P. Molecular Landscape of Small Bowel Adenocarcinoma. Cancers. 2022; 14(5):1287. https://doi.org/10.3390/cancers14051287
Chicago/Turabian StylePandya, Karan, Michael J. Overman, and Pat Gulhati. 2022. "Molecular Landscape of Small Bowel Adenocarcinoma" Cancers 14, no. 5: 1287. https://doi.org/10.3390/cancers14051287