
TCR-Directed Therapy in the Treatment of Metastatic Uveal Melanoma



Abstract
:Simple Summary
Abstract
1. Introduction
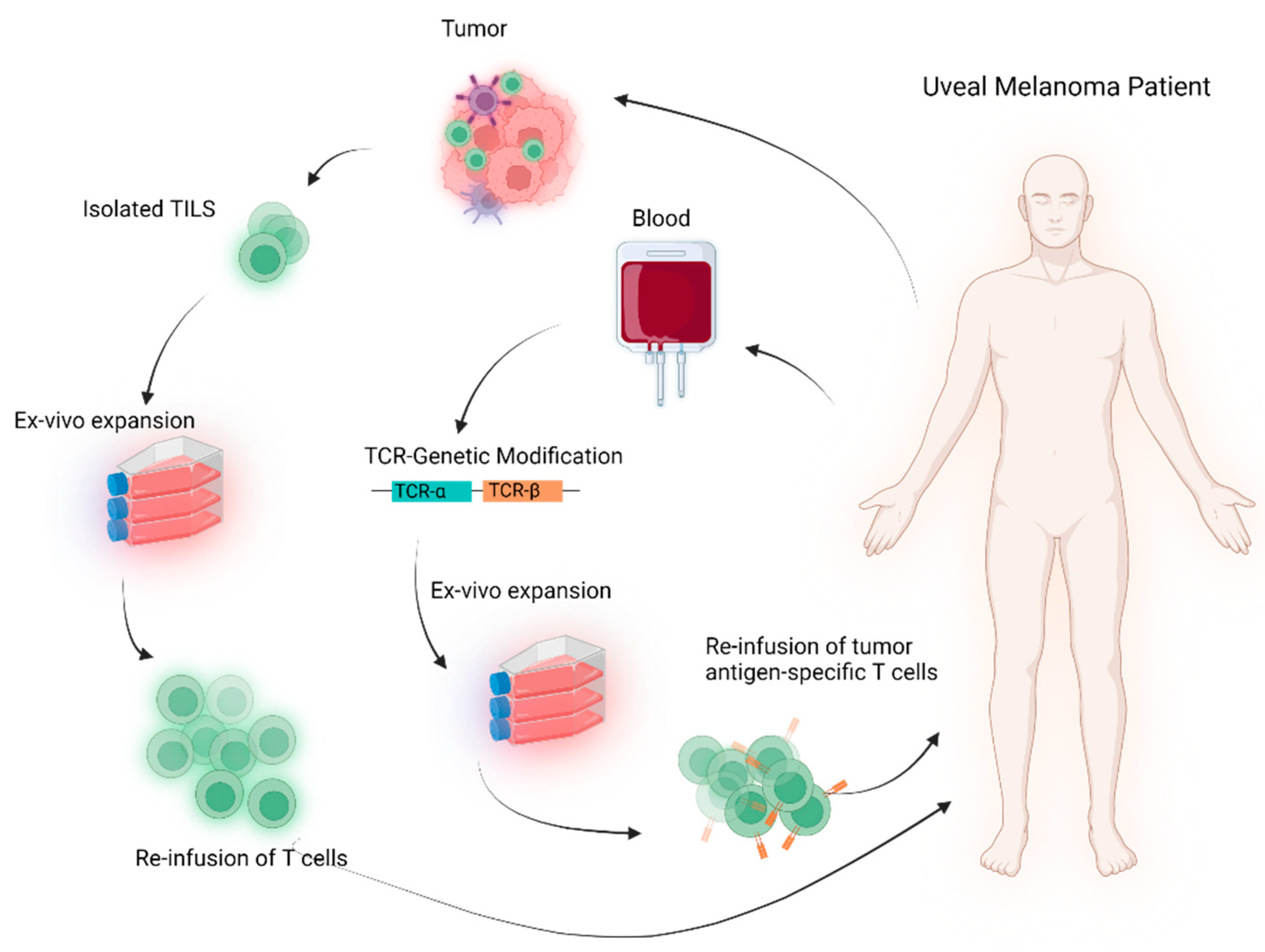
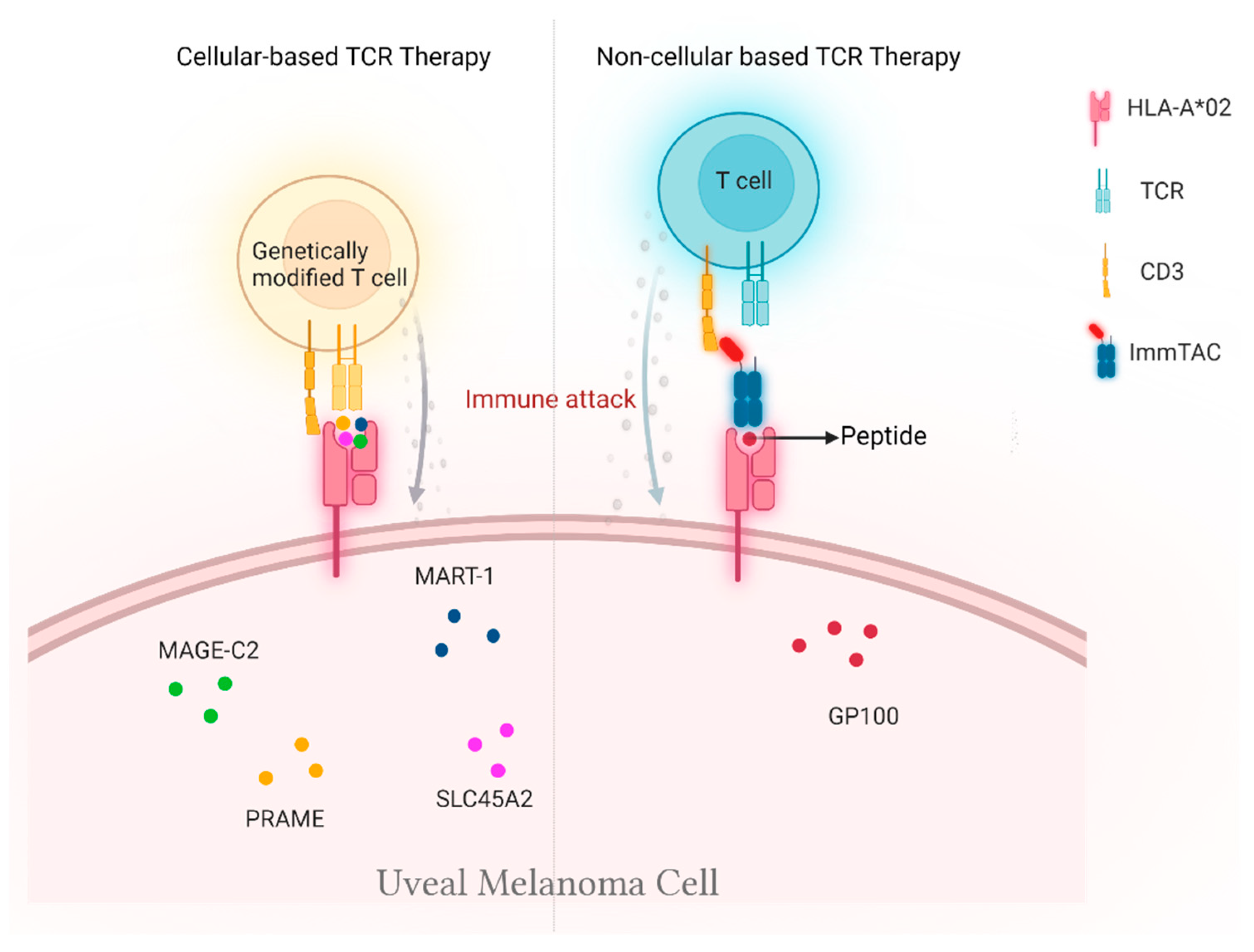
2. Cellular-Based TCR Therapy
2.1. Genetically Unmodified T Cells
2.2. Genetically Modified T Cells
3. Non-Cellular TCR-Based Therapy
4. Challenges and Opportunities
4.1. Immunosuppressive Microenvironment
4.2. HLA-Based Selection
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Hawkins, B.S.; Hayman, J.A.; Jaiyesimi, I.; Jampol, L.M.; et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch. Ophthalmol. 2005, 123, 1639–1643. [Google Scholar] [PubMed]
- Kujala, E.; Mäkitie, T.; Kivelä, T. Very long-term prognosis of patients with malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuk, D.; Shoushtari, A.N.; Barker, C.A.; Panageas, K.S.; Munhoz, R.R.; Momtaz, P.; Ariyan, C.E.; Brady, M.S.; Coit, D.G.; Bogatch, K.; et al. Prognosis of Mucosal, Uveal, Acral, Nonacral Cutaneous, and Unknown Primary Melanoma from the Time of First Metastasis. Oncologist 2016, 21, 848–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Kooij, M.K.; Speetjens, F.M.; van der Burg, S.H.; Kapiteijn, E. Uveal Versus Cutaneous Melanoma; Same Origin, Very Distinct Tumor Types. Cancers 2019, 11, 845. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, J.; Nilsson, L.M.; Mitra, S.; Alsén, S.; Shelke, G.V.; Sah, V.R.; Forsberg, E.M.V.; Stierner, U.; All-Eriksson, C.; Einarsdottir, B.; et al. Molecular profiling of driver events in metastatic uveal melanoma. Nat. Commun. 2020, 11, 1894. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [Green Version]
- Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [CrossRef] [Green Version]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Zimmer, L.; Vaubel, J.; Mohr, P.; Hauschild, A.; Utikal, J.; Simon, J.; Garbe, C.; Herbst, R.; Enk, A.; Kämpgen, E.; et al. Phase II DeCOG-Study of Ipilimumab in Pretreated and Treatment-Naïve Patients with Metastatic Uveal Melanoma. PLoS ONE 2015, 10, e0118564. [Google Scholar] [CrossRef] [PubMed]
- Algazi, A.P.; Tsai, K.K.; Shoushtari, A.N.; Munhoz, R.R.; Eroglu, Z.; Piulats, J.M.; Ott, P.A.; Johnson, D.B.; Hwang, J.; Daud, A.I.; et al. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer 2016, 122, 3344–3353. [Google Scholar] [CrossRef] [PubMed]
- van der Kooij, M.K.; Joosse, A.; Speetjens, F.M.; Hospers, G.A.P.; Bisschop, C.; de Groot, J.W.B.; Koornstra, R.; Blank, C.U.; Kapiteijn, E. Anti-PD1 treatment in metastatic uveal melanoma in the Netherlands. Acta Oncol. 2017, 56, 101–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Qin, Y.; Bollin, K.; de Macedo, M.P.; Carapeto, F.; Kim, K.B.; Roszik, J.; Wani, K.M.; Reuben, A.; Reddy, S.T.; Williams, M.D.; et al. Immune profiling of uveal melanoma identifies a potential signature associated with response to immunotherapy. J. Immunother. Cancer 2020, 8, e000960. [Google Scholar] [CrossRef]
- Durante, M.A.; Rodriguez, D.A.; Kurtenbach, S.; Kuznetsov, J.N.; Sanchez, M.I.; Decatur, C.L.; Snyder, H.; Feun, L.G.; Livingstone, A.S.; Harbour, J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 2020, 11, 496. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Chandran, S.S.; Somerville, R.P.T.; Yang, J.C.; Sherry, R.M.; Klebanoff, C.A.; Goff, S.L.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.; Paria, B.C.; et al. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: A single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 792–802. [Google Scholar] [CrossRef]
- Rohaan, M.W. Results of a phase I trial with MART-1 T cell receptor modified T cells in patients with metastatic melanoma. Ann. Oncol. 2019, 30, v475–v532. [Google Scholar] [CrossRef]
- Wermke, M. Safety and anti-tumor activity of TCR-engineered autologous, PRAME-directed T cells across multiple advanced solid cancers at low doses–clinical update on the ACTengine® IMA203 trial. SITC 2021, 9. [Google Scholar] [CrossRef]
- Middleton, M.R.; McAlpine, C.; Woodcock, V.K.; Corrie, P.; Infante, J.R.; Steven, N.M.; Evans, T.R.J.; Anthoney, A.; Shoushtari, A.N.; Hamid, O.; et al. Tebentafusp, A TCR/Anti-CD3 Bispecific Fusion Protein Targeting gp100, Potently Activated Antitumor Immune Responses in Patients with Metastatic Melanoma. Clin. Cancer Res. 2020, 26, 5869–5878. [Google Scholar] [CrossRef]
- Sacco, J.J.; Carvajal, R.; Butler, M.O.; Shoushtari, A.; Hassel, J.C. A phase (ph) II, multi-center study of the safety and efficacy of tebentafusp (tebe) (IMCgp100) in patients (pts) with metastatic uveal melanoma (mUM). Ann. Oncol. 2020, 31, S1441–S1451. [Google Scholar] [CrossRef]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Forget, M.-A.; Chacon, J.; Bernatchez, C.; Haymaker, C.; Chen, J.Q.; Hwu, P.; Radvanyi, L.G. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: Current status and future outlook. Cancer J. 2012, 18, 160–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, T.J.; Trancikova, D.; Ruiter, D.J.; van Muijen, G.N. High expression of immunotherapy candidate proteins gp100, MART-1, tyrosinase and TRP-1 in uveal melanoma. Br. J. Cancer 1998, 78, 1156–1161. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.S.; Yu, Y.Y.; Dudley, M.E.; Zheng, Z.; Robbins, P.F.; Li, Y.; Wunderlich, J.; Hawley, R.G.; Moayeri, M.; Rosenberg, S.A.; et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum. Gene. Ther. 2005, 16, 457–472. [Google Scholar] [CrossRef]
- Field, M.G.; Decatur, C.L.; Kurtenbach, S.; Gezgin, G.; van der Velden, P.A.; Jager, M.J.; Kozak, K.N.; Harbour, J.W. PRAME as an Independent Biomarker for Metastasis in Uveal Melanoma. Clin. Cancer Res. 2016, 22, 1234–1242. [Google Scholar] [CrossRef] [Green Version]
- Gezgin, G.; Luk, S.J.; Cao, J.; Dogrusöz, M.; van der Steen, D.M.; Hagedoorn, R.S.; Krijgsman, D.; van der Velden, P.A.; Field, M.G.; Luyten, G.P.M.; et al. PRAME as a Potential Target for Immunotherapy in Metastatic Uveal Melanoma. JAMA Ophthalmol. 2017, 135, 541–549. [Google Scholar] [CrossRef]
- Ikeda, H.; Lethé, B.; Lehmann, F.; van Baren, N.; Baurain, J.F.; de Smet, C.; Chambost, H.; Vitale, M.; Moretta, A.; Boon, T.; et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity 1997, 6, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Bin, B.H.; Bhin, J.; Yang, S.H.; Shin, M.; Nam, Y.J.; Choi, D.H.; Shin, D.W.; Lee, A.Y.; Hwang, D.; Cho, E.G.; et al. Membrane-Associated Transporter Protein (MATP) Regulates Melanosomal pH and Influences Tyrosinase Activity. PLoS ONE 2015, 10, e0129273. [Google Scholar] [CrossRef] [PubMed]
- Guedj, M.; Bourillon, A.; Combadières, C.; Rodero, M.; Dieudé, P.; Descamps, V.; Dupin, N.; Wolkenstein, P.; Aegerter, P.; Lebbe, C.; et al. Variants of the MATP/SLC45A2 gene are protective for melanoma in the French population. Hum. Mutat. 2008, 29, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Li, Y.F.; El-Gamil, M.; Rosenberg, S.A.; Robbins, P.F. Use of an in Vitro Immunoselected Tumor Line to Identify Shared Melanoma Antigens Recognized by HLA-A*0201-restricted T Cells. Cancer Res. 2001, 61, 1089–1094. [Google Scholar] [PubMed]
- Park, J.; Talukder, A.H.; Lim, S.A.; Kim, K.; Pan, K.; Melendez, B.; Bradley, S.D.; Jackson, K.R.; Khalili, J.S.; Wang, J.; et al. SLC45A2: A Melanoma Antigen with High Tumor Selectivity and Reduced Potential for Autoimmune Toxicity. Cancer Immunol. Res. 2017, 5, 618–629. [Google Scholar] [CrossRef] [Green Version]
- Oates, J.; Jakobsen, B.K. ImmTACs: Novel bi-specific agents for targeted cancer therapy. Oncoimmunology 2013, 2, e22891. [Google Scholar] [CrossRef] [Green Version]
- Strobel, S.B.; Machiraju, D.; Hülsmeyer, I.; Becker, J.C.; Paschen, A.; Jäger, D.; Wels, W.S.; Bachmann, M.; Hassel, J.C. Expression of Potential Targets for Cell-Based Therapies on Melanoma Cells. Life 2021, 11, 269. [Google Scholar] [CrossRef] [PubMed]
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 2012, 18, 980–987. [Google Scholar] [CrossRef]
- Harper, J.; Adams, K.J.; Bossi, G.; Wright, D.E.; Stacey, A.R.; Bedke, N.; Martinez-Hague, R.; Blat, D.; Humbert, L.; Buchanan, H.; et al. An approved in vitro approach to preclinical safety and efficacy evaluation of engineered T cell receptor anti-CD3 bispecific (ImmTAC) molecules. PLoS ONE 2018, 13, e0205491. [Google Scholar] [CrossRef]
- Boudousquie, C.; Bossi, G.; Hurst, J.M.; Rygiel, K.A.; Jakobsen, B.K.; Hassan, N.J. Polyfunctional response by ImmTAC (IMCgp100) redirected CD8(+) and CD4(+) T cells. Immunology 2017, 152, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouw, N.; Treffers-Westerlaken, E.; Kraan, J.; Wittink, F.; ten Hagen, T.; Verweij, J.; Debets, R. Combination of IL-21 and IL-15 enhances tumour-specific cytotoxicity and cytokine production of TCR-transduced primary T cells. Cancer Immunol. Immunother. 2010, 59, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Souri, Z.; Wierenga, A.P.A.; Kroes, W.G.M.; van der Velden, P.A.; Verdijk, R.M.; Eikmans, M.; Luyten, G.P.M.; Jager, M.J. LAG3 and Its Ligands Show Increased Expression in High-Risk Uveal Melanoma. Cancers 2021, 13, 4445. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Singh, M.K.; Kumar, N.; Jha, J.; Lomi, N.; Sen, S.; Kashyap, S. 189P Prognostic significance of lymphocyte activation gene-3 (LAG3 gene) in uveal melanoma patients. Ann. Oncol. 2021, 32, S1463–S1464. [Google Scholar] [CrossRef]
- Lipson, E.J.; Tawbi, H.A.-H.; Schadendorf, D.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.; Lao, C.D.; Menezes, J.J.; et al. Relatlimab (RELA) plus nivolumab (NIVO) versus NIVO in first-line advanced melanoma: Primary phase III results from RELATIVITY-047 (CA224-047). J. Clin. Oncol. 2021, 39, 9503. [Google Scholar] [CrossRef]
- Lepore, M.; Mori, L.; De Libero, G. The Conventional Nature of Non-MHC-Restricted T Cells. Front. Immunol. 2018, 9, 1365. [Google Scholar] [CrossRef] [Green Version]
- Capsomidis, A.; Benthall, G.; Van Acker, H.H.; Fisher, J.; Kramer, A.M.; Abeln, Z.; Majani, Y.; Gileadi, T.; Wallace, R.; Gustafsson, K.; et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. 2018, 26, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Nussbaumer, O.; Koslowski, M. The emerging role of γδ T cells in cancer immunotherapy. Immuno-Oncol. Technol. 2019, 1, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.; Anderson, J. Engineering Approaches in Human Gamma Delta T Cells for Cancer Immunotherapy. Front. Immunol. 2018, 9, 1409. [Google Scholar] [CrossRef]
- Silva-Santos, B.; Serre, K.; Norell, H. γδ T cells in cancer. Nat. Rev. Immunol 2015, 15, 683–691. [Google Scholar] [CrossRef]
- Xiao, L.; Chen, C.; Li, Z.; Zhu, S.; Tay, J.C.; Zhang, X.; Zha, S.; Zeng, J.; Tan, W.K.; Liu, X.; et al. Large-scale expansion of Vγ9Vδ2 T cells with engineered K562 feeder cells in G-Rex vessels and their use as chimeric antigen receptor-modified effector cells. Cytotherapy 2018, 20, 420–435. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| NCT No. | Phase | Trial | Target | Sponsor/Collaborators | Status | Study Years | |
|---|---|---|---|---|---|---|---|
| HLA-Independent | Cellular-Based TCR Therapy | ||||||
| Genetically Unmodified T cells | |||||||
| NCT01814046 | II | Non-myeloablative lymohocyte depletion followed by transfer of autologous TILs with or without high-dose aldesleukin | Non-specific | National Institutes of Health Clinical Center (CC) | Results published [19] | March 2013–July 2017 | |
| NCT03467516 | II | Non-myeloablative lymohocyte depletion followed by transfer of autologous TILs with high-dose aldesleukin | Non-Specific | Udai Kammula; University of Pittsburgh | Recruiting (Results pending) | May 2018–December 2023 | |
| NCT04812470 | I | Preconditioning chemotherapy with melphlan followed by the transfer of autologous TILs administered via hepatic arterial infusion in addition with IL2 | Non-Specific | Sahlgrenska University Hospital; Vastra Gotaland Region; Miltenyi Biomedicine GmbH | Not yet recruiting | November 2021–March 2028 | |
| HLA-Dependent | Genetically Modified T cells | ||||||
| NCT02654821 | I/II | Non-myeloablative lymohocyte depletion followed by transfer of autologous TILs | MART-1 | The Netherlands Cancer Institute | Results presented [20] | March 2012–January 2020 | |
| NCT02743611 | I/II | Transfer of autologous TCR-engineered T cells in addition to rimiducid | PRAME | Bellicum Pharmaceuticals | Results presented [21] | April 2017–July 2020 | |
| NCT03068624 | I | Transfer of autologous TCR-engineered T cells in addition to cyclophosphamide, aldesleukin, ipilimumab | SLC45A2 | MD Anderson Cancer Center; NCI | Recruiting (Results pending) | September 2017–September 2021 | |
| NCT04729543 | I/II | Pre-treatment with valproic acid and 5′ azacytidine followed by transfer of autologous TCR-engineered T cells | MAGE-C2 | Erasmus Medical Center | Recruiting (Results pending) | October 2020-October 2027 | |
| Non-Cellular TCR-Based Therapy | |||||||
| NCT01211262 | I | IMCgp100, a monoclonal T cell receptor anti-CD3 scFv fusion protein | Gp100 | Immunocore Ltd. | Completed [22] | September 2010-July 2020 | |
| NCT02570308 | I/II | IMCgp100 using the intra-patient escalation dosing regimen | Gp100 | Immunocore Ltd. | Results presented [23] | February 2016–January 2021 | |
| NCT03070392 | III | Tebentafusp (IMCgp100) versus investigator choice (dacarbazine, ipilimumab, or pembrolizumab) in mUM | Gp100 | Immunocore Ltd. | Results published [24] | October 2017–March 2023 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strobel, S.B.; Machiraju, D.; Hassel, J.C. TCR-Directed Therapy in the Treatment of Metastatic Uveal Melanoma. Cancers 2022, 14, 1215. https://doi.org/10.3390/cancers14051215
Strobel SB, Machiraju D, Hassel JC. TCR-Directed Therapy in the Treatment of Metastatic Uveal Melanoma. Cancers. 2022; 14(5):1215. https://doi.org/10.3390/cancers14051215
Chicago/Turabian StyleStrobel, Sophia B., Devayani Machiraju, and Jessica C. Hassel. 2022. "TCR-Directed Therapy in the Treatment of Metastatic Uveal Melanoma" Cancers 14, no. 5: 1215. https://doi.org/10.3390/cancers14051215