
Impact of Exogenous Treatment with Histidine on Hepatocellular Carcinoma Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
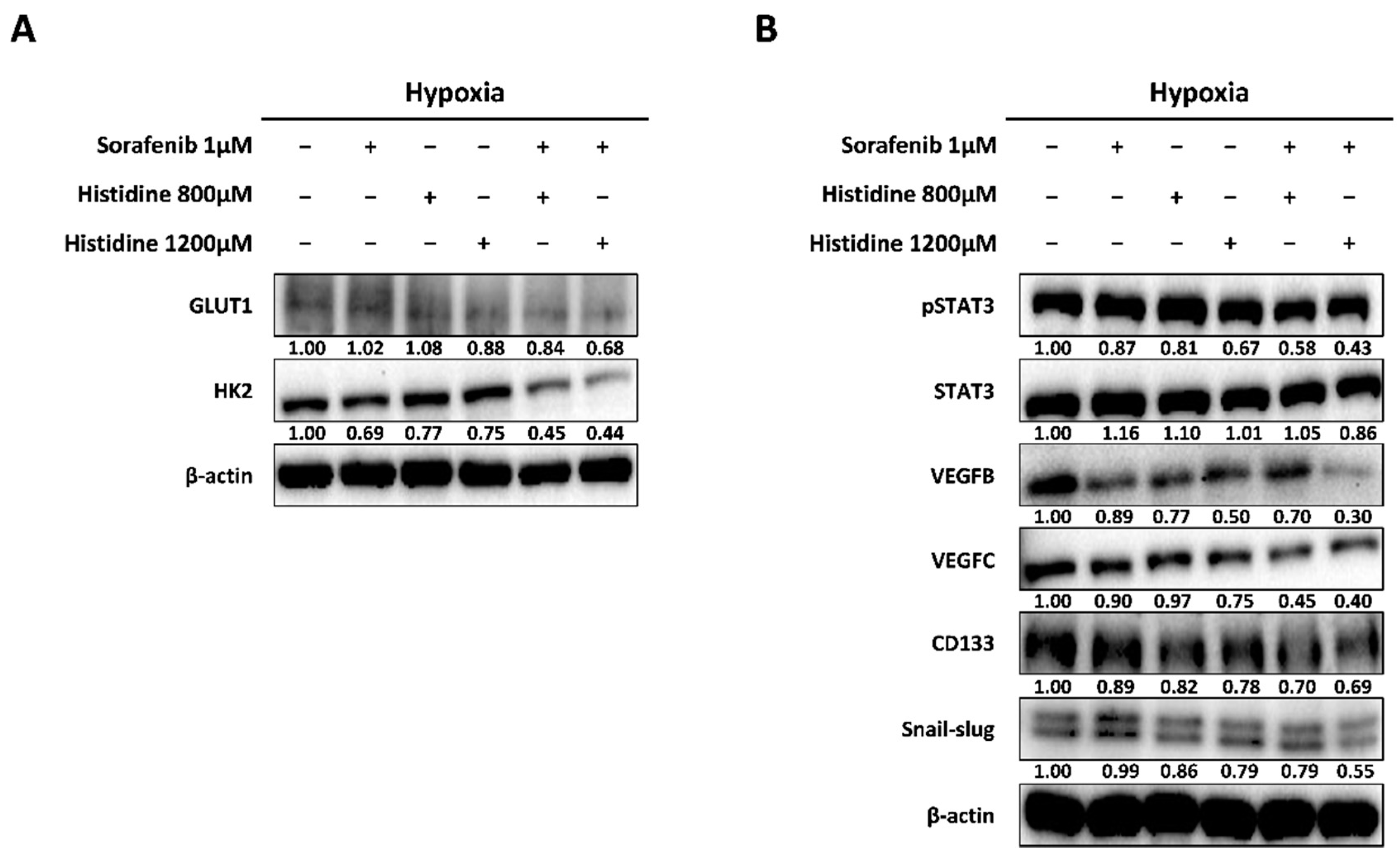{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture and Treatment
2.3. Western Blotting
2.4. RNA Sequencing and Real-Time PCR (RT-PCR)
2.5. Migration and Cell Viability Assay
2.6. Immunostaining
2.7. Statistical Analysis
3. Results
3.1. Effect of Exogenous Hist Idine Treatment on HepG2 Cells
3.2. Expression of SLC7A5/LAT1 in HCC Cell Lines with Differences in Glycolysis
3.3. Expression of SLC7A5/LAT1 in Patients with HCC
3.4. Increased Anti-Cancer Effect after Combined Treatment with Sorafenib and Histidine
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Balogh, J.; Victor, D., 3rd; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P. Hepatocellular Carcinoma: A Review. J. Hepato cell Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruix, J.; Sherman, M. Practice Guidelines Committee; American Association for the Study of Liver Diseases. Management of Hepatocellular Carcinoma. Hepatology 2005, 42, 1208–1236. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.C.; Farrell, A.T.; Madabushi, R.; Booth, B.; Chattopadhyay, S.; Sridhara, R.; Justice, R.; Pazdur, R. Sorafenib for the Treatment of Unresectable Hepatocellular Carcinoma. Oncologist 2009, 14, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, A.; Ho, M. Role of Sorafenib in the Treatment of Advanced Hepatocellular Carcinoma: An Update. Hepatol. Res. 2013, 43, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masao, O.; Ann-Lii, C.; Norihiro, K.; Masatoshi, K.; Min, L.J.; Jidong, J.; Ryosuke, T.; Kwang-Hyub, H. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: A 2017 update. Hepatol. Int. 2017, 11, 317–370. [Google Scholar] [CrossRef] [Green Version]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [Green Version]
- Heimbach, J.K.; Kulik, L.M.; Finn, R.S.; Sirlin, C.B.; Abecassis, M.M.; Roberts, L.R.; Zhu, A.X.; Hassan Murad, M.; Marrero, J.A. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018, 67, 358–380. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and Safety of Sorafenib in Patients in the Asia-Pacific Region with Advanced Hepatocellular Carcinoma: A phase III Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Cabral, L.K.D.; Tiribelli, C.; Sukowati, C.H.C. Sorafenib Resistance in Hepatocellular Carcinoma: The Relevance of Genetic Heterogeneity. Cancers 2020, 12, 1576. [Google Scholar] [CrossRef]
- Xia, S.; Pan, Y.; Liang, Y.; Xu, J.; Cai, X. The Microenvironmental and Metabolic Aspects of Sorafenib Resistance in Hepatocellular Carcinoma. E Bio. Med. 2020, 51, 102610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez-Blanco, C.; Fondevila, F.; García-Palomo, A.; González-Gallego, J.; Mauriz, J.L. Sorafenib Resistance in Hepatocarcinoma: Role of Hypoxia-Inducible Factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The Mechanisms of Sorafenib Resistance in Hepatocellular Carcinoma: Theoretical Basis and Therapeutic Aspects. Signal Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New Knowledge of the Mechanisms of Sorafenib Resistance in Liver Cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Du, D.; Liu, C.; Qin, M.; Zhang, X.; Xi, T.; Yuan, S.; Xiong, J. Metabolic Dysregulation and Emerging Therapeutical Targets for Hepatocellular Carcinoma. Acta Pharm. Sin. B 2021, 12, 558–580. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Q.; Yin, P.; Xing, W.; Wu, Z.; Chen, S.; Lu, X.; Zhang, Y.; Lin, X.; Xu, G. Serum Metabolomics Reveals the Deregulation of Fatty Acids Metabolism in Hepatocellular Carcinoma and Chronic Liver Diseases. Anal. Bioanal. Chem. 2012, 403, 203–213. [Google Scholar] [CrossRef]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging Roles and the Regulation of Aerobic Glycolysis in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, G.; Guo, F.; Li, Q.; Luo, H.; Shu, Y.; Shen, Y.; Gan, J.; Xu, L.; Yang, H. Mitochondrial UQCC3 Modulates Hypoxia Adaptation by Orchestrating OXPHOS and Glycolysis in Hepatocellular Carcinoma. Cell Rep. 2020, 33, 108340. [Google Scholar] [CrossRef]
- Lu, C.; Fang, S.; Weng, Q.; Lv, X.; Meng, M.; Zhu, J.; Zheng, L.; Hu, Y.; Gao, Y.; Wu, X.; et al. Integrated Analysis Reveals Critical Glycolytic Regulators in Hepatocellular Carcinoma. Cell Commun. Signal 2020, 18, 97. [Google Scholar] [CrossRef]
- De Matteis, S.; Ragusa, A.; Marisi, G.; De Domenico, S.; Casadei Gardini, A.; Bonafè, M.; Giudetti, A.M. Aberrant Metabolism in Hepatocellular Carcinoma Provides Diagnostic and Therapeutic Opportunities. Oxid. Med. Cell. Longev. 2018, 2018, 7512159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, J.; Wang, S.; Jiang, Q.; Xu, K. Identification and Validation of a Nine-Gene Amino Acid Metabolism-Related Risk Signature in HCC. Front. Cell Dev. Biol. 2021, 9, 731790. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and Glycine Metabolism in Cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Wang, S.; Zaal, E.A.; Wang, C.; Wu, H.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.; Lieftink, C.; et al. A Powerful Drug Combination Strategy Targeting Glutamine Addiction for the Treatment of Human Liver Cancer. eLife 2020, 9, e56749. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Choi, Y.K.; Park, S.Y.; Jang, S.Y.; Lee, J.Y.; Ham, H.J.; Kim, B.G.; Jeon, H.J.; Kim, J.H.; Kim, J.G.; et al. PPARδ Reprograms Glutamine Metabolism in Sorafenib-Resistant HCC. Mol. Cancer Res. 2017, 15, 1230–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Tan, X.; Luo, J.; Yao, H.; Si, Z.; Tong, J.S. The miR-30a-5p/CLCF1 Axis Regulates Sorafenib Resistance and Aerobic Glycolysis in Hepatocellular Carcinoma. Cell Death Dis. 2020, 11, 902. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; Keys, H.R.; Cantor, J.R.; Lewis, C.A.; Chan, S.H.; Kunchok, T.; Abu-Remaileh, M.; Freinkman, E.; Schweitzer, L.D.; Sabatini, D.M. Histidine Catabolism Is a Major Determinant of Methotrexate Sensitivity. Nature 2018, 559, 632–636. [Google Scholar] [CrossRef]
- Lee, M.; Jeon, J.Y.; Neugent, M.L.; Kim, J.W.; Yun, M. 18F-Fluorodeoxyglucose Uptake on Positron Emission Tomography/Computed Tomography Is Associated with Metastasis and Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma. Clin. Exp. Metastasis 2017, 34, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Lee, J.S.; Jo, H.; Park, Y.; Yun, M.; Song, Z.; Pyun, J.C.; Lee, M. Modified Parylene-N Films as Chemical Microenvironments for Differentiation and Spheroid Formation of Osteoblast Cells. Sci. Rep. 2020, 10, 15219. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Marinoni, I.; Irmler, M.; Psaras, T.; Honegger, J.B.; Beschorner, R.; Anastasov, N.; Beckers, J.; Theodoropoulou, M.; Roncaroli, F.; et al. Transcriptome Analysis of MENX-Associated Rat Pituitary Adenomas Identifies Novel Molecular Mechanisms Involved in the Pathogenesis of Human Pituitary Gonadotroph Adenomas. Acta Neuropathol. 2013, 126, 137–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalise, M.; Galluccio, M.; Console, L.; Pochini, L.; Indiveri, C. The Human SLC7A5 (LAT1): The Intriguing Histidine/Large Neutral Amino Acid Transporter and Its Relevance to Human Health. Front. Chem. 2018, 6, 243. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, M.E.; Brosnan, J.T. Histidine Metabolism and Function. J. Nutr. 2020, 150 (Suppl. S1), 2570S–2575S. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, R.; Tomi, M.; Akanuma, S.; Tabuchi, A.; Kubo, Y.; Tachikawa, M.; Hosoya, K. Up-Regulation of L-Type Amino Acid transporter 1 (LAT1) in Cultured Rat Retinal Capillary Endothelial Cells in Response to Glucose Deprivation. Drug Metab. Pharmacokinet. 2012, 27, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Polet, F.; Martherus, R.; Corbet, C.; Pinto, A.; Feron, O. Inhibition of Glucose Metabolism Prevents Glycosylation of the Glutamine Transporter ASCT2 and Promotes Compensatory LAT1 Upregulation in Leukemia Cells. Oncotarget 2016, 7, 46371–46383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clair, T.; Koh, E.; Ptaszynska, M.; Bandle, R.W.; Liotta, L.A.; Schiffmann, E.; Stracke, M.L. L-Histidine Inhibits Production of Lysophosphatidic Acid by the Tumor-Associated Cytokine, Autotaxin. Lipids Health Dis. 2005, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.L.; Wu, S.T.; Yin, M.C.; Chen, H.T.; Chen, H.C. Protective Effects from Carnosine and Histidine on Acetaminophen-Induced Liver Injury. J. Food Sci. 2009, 74, H259–H265. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, S.; Fuchs, B.C.; Tanabe, K.K. STAT3 Is a Key Transcriptional Regulator of Cancer Stem Cell Marker CD133 in HCC. Hepatobiliary Surg. Nutr. 2016, 5, 201–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, C.; Kim, B.H.; Yi, E.H.; Choi, K.J.; Kim, E.K.; Jeong, J.M.; Lee, J.H.; Jang, J.J.; Yoon, J.H.; Jeong, W.I.; et al. Signal Transducer and Activator of Transcription 3-Mediated CD133 Up-Regulation Contributes to Promotion of Hepatocellular Carcinoma. Hepatology 2015, 62, 1160–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid. Redox Signal 2017, 26, 462–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.Y.; Kim, E.H. Therapeutic Effects of Amino Acids in Liver Diseases: Current Studies and Future Perspectives. J. Cancer Prev. 2019, 24, 72–78. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.; Han, Y.; Kim, D.; Cho, S.; Kim, W.; Hwang, H.; Lee, H.W.; Han, D.H.; Kim, K.S.; Yun, M.; et al. Impact of Exogenous Treatment with Histidine on Hepatocellular Carcinoma Cells. Cancers 2022, 14, 1205. https://doi.org/10.3390/cancers14051205
Park Y, Han Y, Kim D, Cho S, Kim W, Hwang H, Lee HW, Han DH, Kim KS, Yun M, et al. Impact of Exogenous Treatment with Histidine on Hepatocellular Carcinoma Cells. Cancers. 2022; 14(5):1205. https://doi.org/10.3390/cancers14051205
Chicago/Turabian StylePark, Yusun, Yeonju Han, Dongwoo Kim, Sua Cho, WonJin Kim, Hyemin Hwang, Hye Won Lee, Dai Hoon Han, Kyung Sik Kim, Mijin Yun, and et al. 2022. "Impact of Exogenous Treatment with Histidine on Hepatocellular Carcinoma Cells" Cancers 14, no. 5: 1205. https://doi.org/10.3390/cancers14051205