
The Cationic Amphiphilic Drug Hexamethylene Amiloride Eradicates Bulk Breast Cancer Cells and Therapy-Resistant Subpopulations with Similar Efficiencies


 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assays
2.3. Animal Therapeutic Studies
2.4. Cell Labeling, Flow Cytometry, and Sorting
2.5. Tumorsphere/Mammosphere Assays
2.6. Persister Cell Generation and Treatments
2.7. Ex Vivo Organoid Assay for Drug Cytotoxicity
2.8. Establishment of Orthotopic Xenograft Mouse Models
2.9. Organotypic Tumor Slice Preparation and Viability Analysis
2.10. Lung Analysis
2.11. Histology
2.12. Statistical, Data, and Image Analysis
3. Results
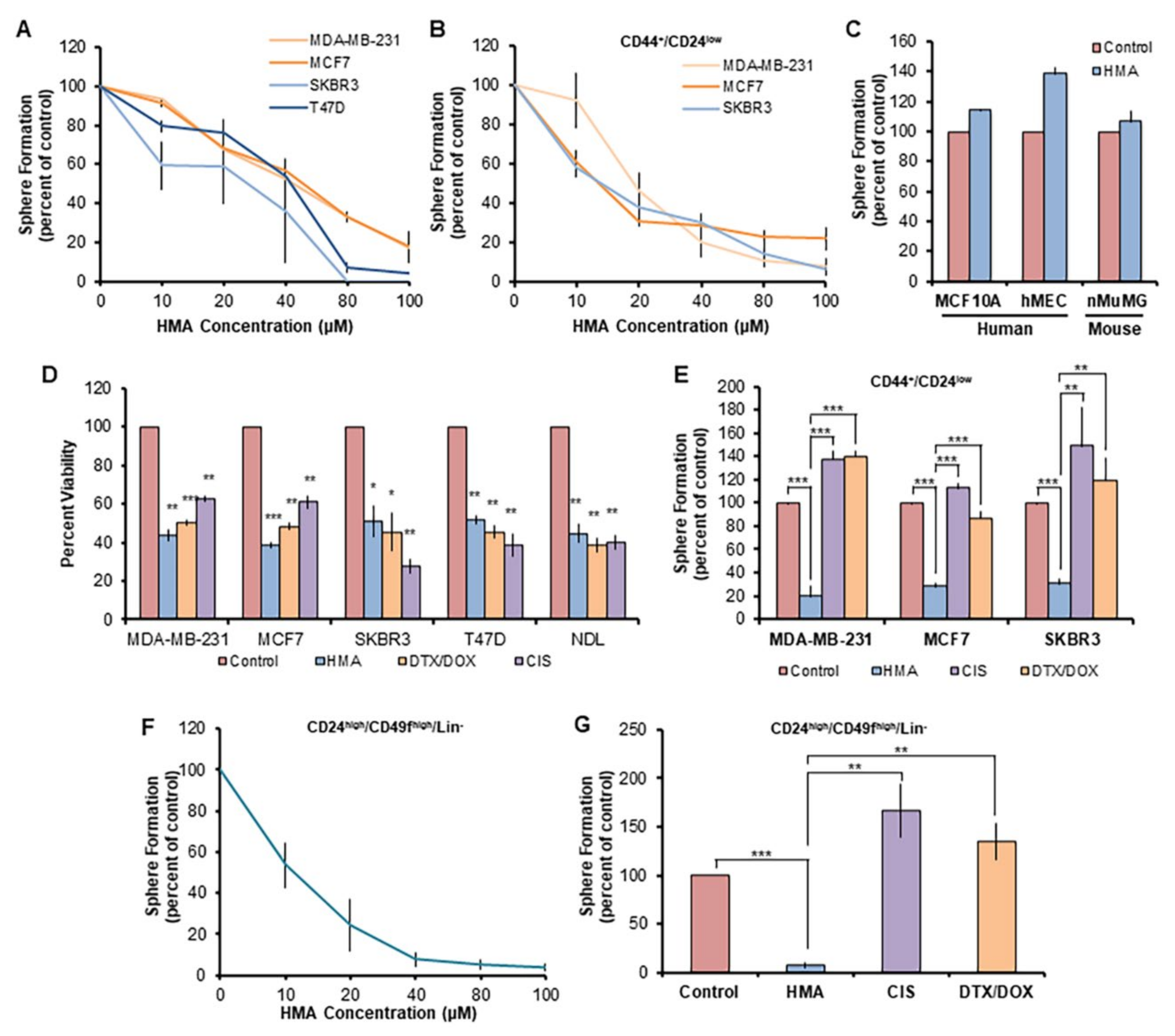
3.1. HMA Ablates Chemotherapy-Resistant BCSCs but Not Normal Mammary Stem-like Cells
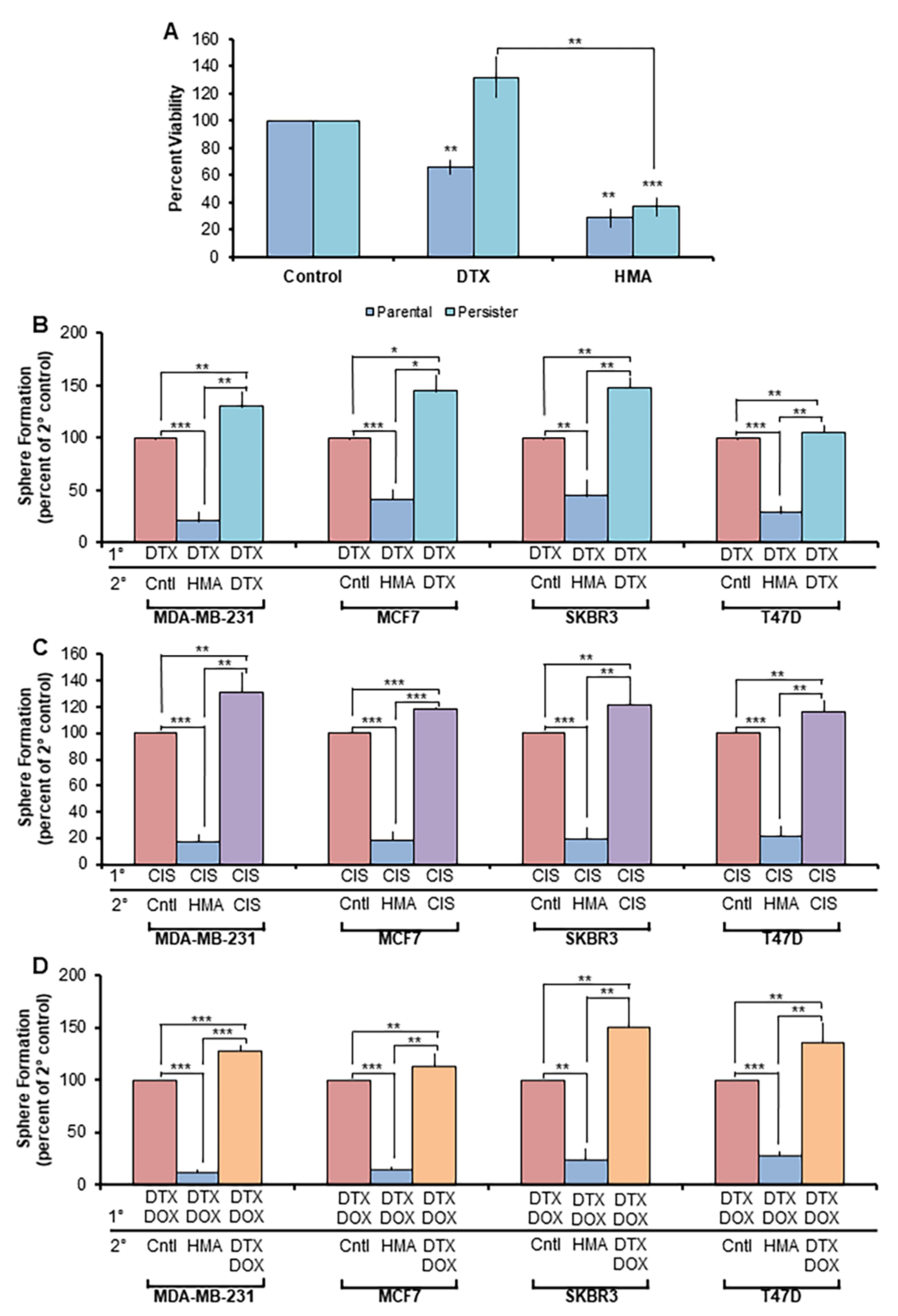
3.2. HMA Is Cytotoxic toward Tumor Cell Populations Insensitive to Conventional Chemotherapeutics
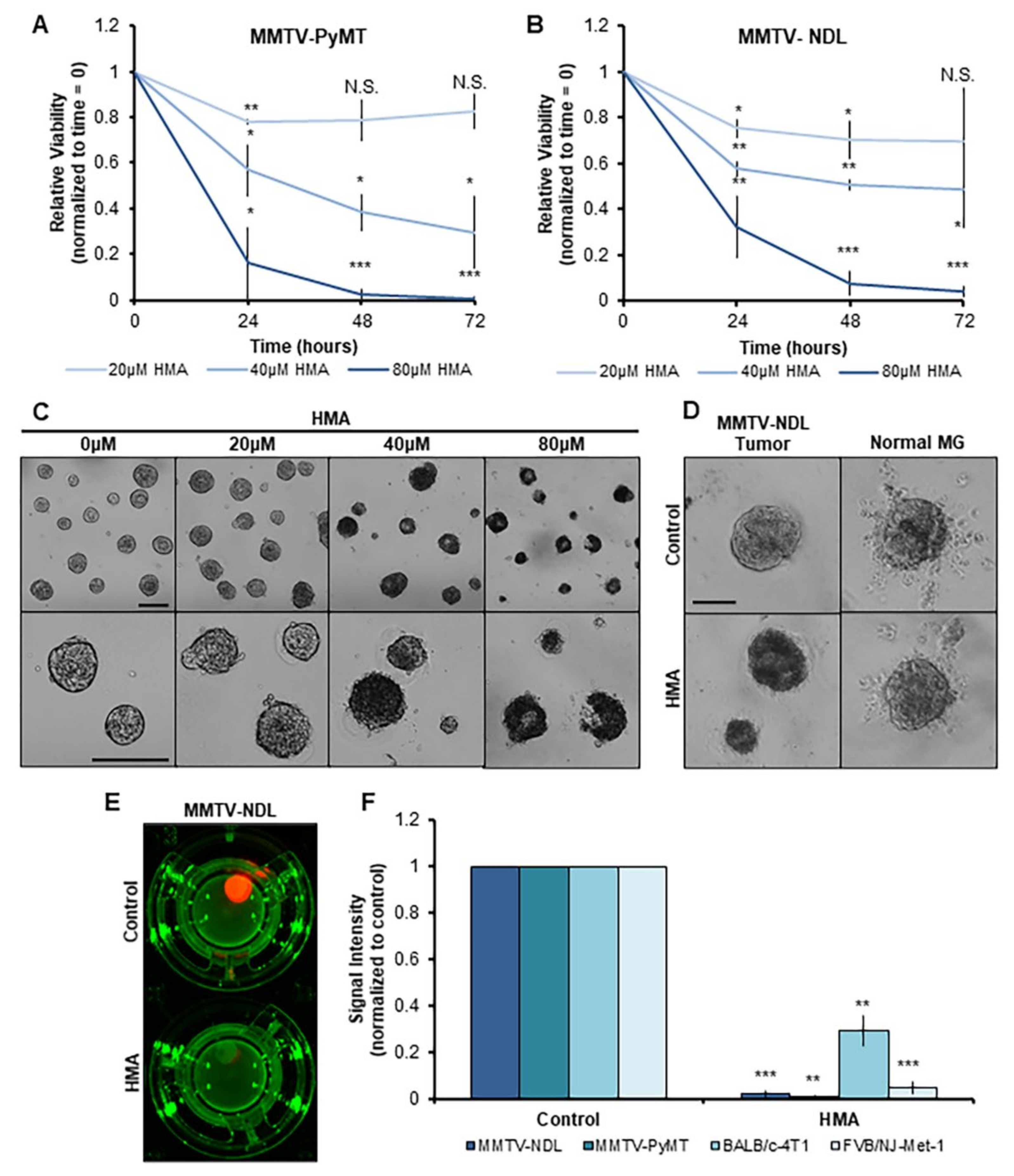
3.3. HMA Thwarts the Viability of Mouse Mammary Tumor Tissues Ex Vivo
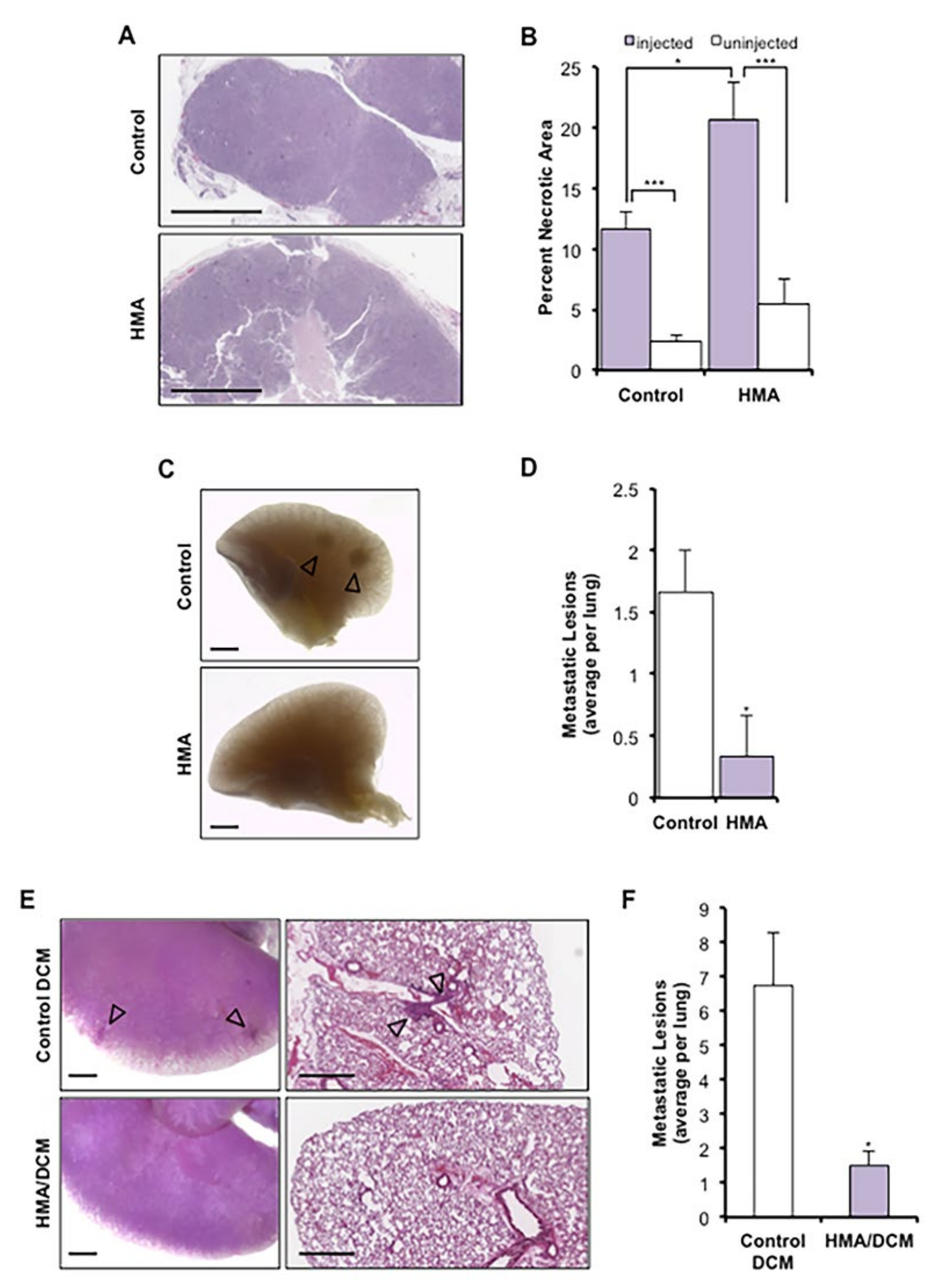
3.4. HMA Induces Necrosis In Vivo and Suppresses Metastasis
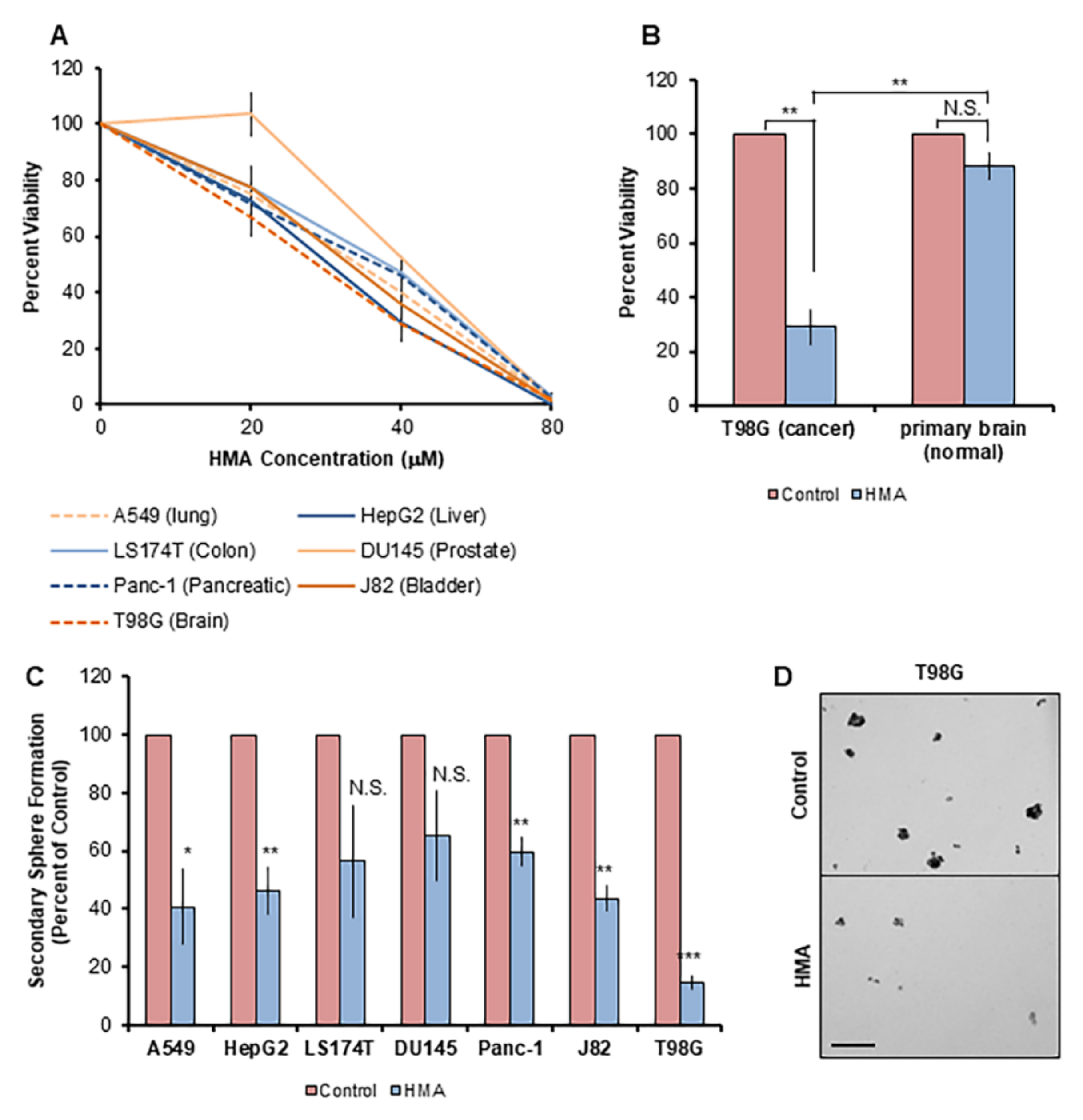
3.5. HMA Depletes Cancer Cells Derived from an Array of Human Tissue Types and Inhibits Outgrowth of Enriched Cancer Stem-like Populations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalerba, P.; Dylla, S.J.; Park, I.-K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.M.; Shaw, P.A.; Gedye, C.; Bernardini, M.Q.; Neel, B.G.; Ailles, L.E. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6468–6473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S.E. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Müller, V.; Stahmann, N.; Riethdorf, S.; Rau, T.; Zabel, T.; Goetz, A.; Jänicke, F.; Pantel, K. Circulating Tumor Cells in Breast Cancer: Correlation to Bone Marrow Micrometastases, Heterogeneous Response to Systemic Therapy and Low Proliferative Activity. Clin. Cancer Res. 2005, 11, 3678–3685. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Yao, H.; Liu, Q.; Chen, J.; Shi, J.; Su, F.; Song, E. Markers of Tumor-Initiating Cells Predict Chemoresistance in Breast Cancer. PLoS ONE 2010, 5, e15630. [Google Scholar] [CrossRef] [Green Version]
- Stenning, S.P.; Parkinson, M.C.; Fisher, C.; Mead, G.M.; Cook, P.A.; Fossa, S.D.; Horwich, A.; Jones, W.G.; Newlands, E.S.; Oliver, R.T.; et al. Postchemotherapy residual masses in germ cell tumor patients: Content, clinical features, and prognosis. Medical Research Council Testicular Tumour Working Party. Cancer 1998, 83, 1409–1419. [Google Scholar] [CrossRef]
- Masui, K.; Gini, B.; Wykosky, J.; Zanca, C.; Mischel, P.S.; Furnari, F.; Cavenee, W.K. A tale of two approaches: Complementary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcinogenesis 2013, 34, 725–738. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A. Pathways to Breast Cancer Recurrence. ISRN Oncol. 2013, 2013, 290568. [Google Scholar] [CrossRef]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K., 3rd. Emerging mechanisms of drug-induced phospholipidosis. Biol. Chem. 2019, 401, 31–46. [Google Scholar] [CrossRef]
- Hu, M.; Carraway, K.L. Repurposing cationic amphiphilic drugs and derivatives to engage lysosomal cell death in cancer treatment. Front. Oncol. 2020, 10, 05361. [Google Scholar] [CrossRef]
- Ellegaard, A.-M.; Bach, P.; Jäättelä, M. Targeting Cancer Lysosomes with Good Old Cationic Amphiphilic Drugs. Rev. Physiol. Biochem. Pharmacol. 2020, 1–46. [Google Scholar] [CrossRef]
- Rowson-Hodel, A.R.; Berg, A.L.; Wald, J.H.; Hatakeyama, J.; VanderVorst, K.; Curiel, D.A. Hexamethylene amiloride engages a novel reactive oxygen species- and lysosome-dependent programmed necrotic mechanism to selectively target breast cancer cells. Cancer Lett. 2016, 375, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Kittaneh, M.; Montero, A.J.; Glück, S. Molecular Profiling for Breast Cancer: A Comprehensive Review. Biomark. Cancer 2013, 5, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.-S.; Koh, C.-G.; Li, H.-Y. Mitosis-targeted anti-cancer therapies: Where they stand. Cell Death Dis. 2012, 3, e411. [Google Scholar] [CrossRef] [Green Version]
- Aits, S.; Jäättelä, M.; Nylandsted, J. Methods for the quantification of lysosomal membrane permeabilization: A hallmark of lysosomal cell death. Methods Cell Biol. 2015, 126, 261–285. [Google Scholar] [CrossRef]
- Ostenfeld, M.S.; Fehrenbacher, N.; Høyer-Hansen, M.; Thomsen, C.; Farkas, T.; Jäättelä, M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 2005, 65, 8975–8983. [Google Scholar] [CrossRef] [Green Version]
- Aits, S.; Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [Green Version]
- Fehrenbacher, N.; Gyrd-Hansen, M.; Poulsen, B.; Felbor, U.; Kallunki, T.; Boes, M.; Weber, E.; Leist, M.; Jäättelä, M. Sensitization to the Lysosomal Cell Death Pathway upon Immortalization and Transformation. Cancer Res. 2004, 64, 5301–5310. [Google Scholar] [CrossRef] [Green Version]
- Petersen, N.H.; Olsen, O.D.; Groth-Pedersen, L.; Ellegaard, A.M.; Bilgin, M.; Redmer, S.; Ostenfeld, M.S.; Ulanet, D.; Dovmark, T.H.; Lønborg, A.; et al. Transformation-Associated Changes in Sphingolipid Metabolism Sensitize Cells to Lysosomal Cell Death Induced by Inhibitors of Acid Sphingomyelinase. Cancer Cell 2013, 24, 379–393. [Google Scholar] [CrossRef] [Green Version]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Kallunki, T.; Olsen, O.D.; Jäättelä, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.K.; Shattuck, D.L.; Ingalla, E.Q.; Yen, L.; Borowsky, A.D.; Young, L.J.; Cardiff, R.D.; Carraway, K.L.; Sweeney, C. Suppression of the Negative Regulator LRIG1 Contributes to ErbB2 Overexpression in Breast Cancer. Cancer Res. 2008, 68, 8286–8294. [Google Scholar] [CrossRef] [Green Version]
- Borowsky, A.D.; Namba, R.; Young, L.J.T.; Hunter, K.W.; Hodgson, J.G.; Tepper, C.G.; McGoldrick, E.T.; Muller, W.J.; Cardiff, R.D.; Gregg, J.P. Syngeneic mouse mammary carcinoma cell lines: Two closely related cell lines with divergent metastatic behavior. Clin. Exp. Metastasis 2005, 22, 47–59. [Google Scholar] [CrossRef]
- Cho, R.W.; Wang, X.; Diehn, M.; Shedden, K.; Chen, G.Y.; Sherlock, G.; Gurney, A.; Lewicki, J.; Clarke, M.F. Isolation and Molecular Characterization of Cancer Stem Cells in MMTV-Wnt-1 Murine Breast Tumors. Stem Cells 2008, 26, 364–371. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N. Association of reactive oxygen species levels and radi-oresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Siegel, P.M.; Ryan, E.D.; Cardiff, R.D.; Muller, W.J. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: Implications for human breast cancer. EMBO J. 1999, 18, 2149–2164. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiao, K.; Luo, J.; Xiao, W.; Lee, J.S.; Gonik, A.M.; Kato, J.; Dong, T.A.; Lam, K.S. Well-defined, reversible disulfide cross-linked micelles for on-demand paclitaxel delivery. Biomaterials 2011, 32, 6633–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.Y.-F.; Lin, Y.; Bratman, S.V.; Feng, W.; Kuo, A.H.; Scheeren, F.A. Neuregulin autocrine signaling promotes self-renewal of breast tumor-initiating cells by triggering HER2/HER3 activation. Cancer Res. 2014, 74, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.C.; Deng, T.; Lehal, R.S.; Kim, J.; Zacksenhaus, E. Identification of tumorsphere- and tumor-initiating cells in HER2/Neu-induced mammary tumors. Cancer Res. 2007, 67, 8671–8681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A. Drug-tolerant persister cancer cells are vul-nerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Ngoc, K.-V.; Cheung, K.J.; Brenot, A.; Shamir, E.R.; Gray, R.S.; Hines, W.C. ECM microenvironment regulates collec-tive migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, E2595–E2604. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Roberto, J.B.; Knupp, A.; Kenerson, H.L.; Truong, C.D.; Yuen, S.Y.; Brempelis, K.J.; Tuefferd, M.; Chen, A.; Horton, H.; et al. Precision-cut human liver slice cultures as an immunological platform. J. Immunol. Methods 2018, 455, 71–79. [Google Scholar] [CrossRef]
- Rowson-Hodel, A.; Wald, J.; Hatakeyama, J.; O’Neal, W.; Stonebraker, J.; Vandervorst, K.; Saldana, M.; Borowsky, A.; Sweeney, C.; Carraway, K. Membrane Mucin Muc4 promotes blood cell association with tumor cells and mediates efficient metastasis in a mouse model of breast cancer. Oncogene 2018, 37, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokdang, N.; Hatakeyama, J.; Wald, J.H.; Simion, C.; Tellez, J.D.; Chang, D.Z.; Swamynathan, M.M.; Chen, M.; Murphy, W.J.; Iii, K.L.C.; et al. LRIG1 opposes epithelial-to-mesenchymal transition and inhibits invasion of basal-like breast cancer cells. Oncogene 2016, 35, 2932–2947. [Google Scholar] [CrossRef]
- Rowson-Hodel, A.R.; Manjarin, R.; Trott, J.F.; Cardiff, R.D.; Borowsky, A.D.; Hovey, R.C. Neoplastic transformation of porcine mammary epithelial cells in vitro and tumor formation in vivo. BMC Cancer 2015, 15, 562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillmore, C.M.; Kuperwasser, C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.-H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast Cancer Cell Lines Contain Functional Cancer Stem Cells with Metastatic Capacity and a Distinct Molecular Signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Diehn, M.; Cho, R.W.; Clarke, M.F. Therapeutic Implications of the Cancer Stem Cell Hypothesis. Semin. Radiat. Oncol. 2009, 19, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Guy, C.T.; Webster, M.A.; Schaller, M.; Parsons, T.J.; Cardiff, R.D.; Muller, W.J. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10578–10582. [Google Scholar] [CrossRef] [Green Version]
- Ingalla, E.Q.; Miller, J.K.; Wald, J.H.; Workman, H.C.; Kaur, R.P.; Yen, L.; Fry, W.H.D.; Borowsky, A.D.; Young, L.J.T.; Sweeney, C.; et al. Post-transcriptional Mechanisms Contribute to the Suppression of the ErbB3 Negative Regulator Protein Nrdp1 in Mammary Tumors. J. Biol. Chem. 2010, 285, 28691–28697. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.-H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to Malignancy in the Polyoma Middle T Oncoprotein Mouse Breast Cancer Model Provides a Reliable Model for Human Diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef] [Green Version]
- Sivakumar, R.; Chan, M.; Shin, J.S.; Nishida-Aoki, N.; Kenerson, H.L.; Elemento, O.; Beltran, H.; Yeung, R.; Gujral, T.S. Organotypic tumor slice cultures provide a versatile platform for immune-oncology and drug discovery. OncoImmunology 2019, 8, e1670019. [Google Scholar] [CrossRef]
- Lee, C.; Tannock, I. Pharmacokinetic studies of amiloride and its analogs using reversed-phase high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1996, 685, 151–157. [Google Scholar] [CrossRef]
- Luo, J.; Tannock, I. Inhibition of the regulation of intracellular pH: Potential of 5-(N,N-hexamethylene) amiloride in tumour-selective therapy. Br. J. Cancer 1994, 70, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Bianco, J.; Bastiancich, C.; Jankovski, A.; des Rieux, A.; Préat, V.; Danhier, F. On glioblastoma and the search for a cure: Where do we stand? Cell. Mol. Life Sci. 2017, 74, 2451–2466. [Google Scholar] [CrossRef]
- Lee, C.-H.; Yu, C.-C.; Wang, B.-Y.; Chang, W.-W. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget 2016, 7, 1215–1226. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Valtorta, S.; Dico, A.L.; Raccagni, I.; Gaglio, D.; Belloli, S.; Politi, L.S.; Martelli, C.; Diceglie, C.; Bonanomi, M.; Ercoli, G.; et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget 2017, 8, 113090–113104. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Zhang, Y.; Yu, X.; Yang, J.; LeBrun, D.G.; Chen, C.; Yao, Q.; Li, M. Overcoming drug resistance in pancreatic cancer. Expert Opin. Ther. Targets 2011, 15, 817–828. [Google Scholar] [CrossRef] [Green Version]
- Lundholm, L.; Hååg, P.; Zong, D.; Juntti, T.; Mörk, B.; Lewensohn, R.; Viktorsson, K. Resistance to DNA-damaging treatment in non-small cell lung cancer tumor-initiating cells involves reduced DNA-PK/ATM activation and diminished cell cycle arrest. Cell Death Dis. 2013, 4, e478. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Cives-Losada, C.; Asensio, M.; Lozano, E.; Briz, O.; Macias, R.I.R. Mechanisms of Anticancer Drug Resistance in Hepatoblastoma. Cancers 2019, 11, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghe, T.V.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010, 17, 922–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunk, U.T.; Ericsson, J.L. Cytochemical evidence for the leakage of acid phosphatase through ultrastructurally intact lyso-somal membranes. Histochem. J. 1972, 4, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Joyce, J.A. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 2007, 6, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D—Many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef] [Green Version]
- Gocheva, V.; Wang, H.-W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L. IL-4 induces cathepsin protease activity in tu-mor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [Green Version]
- Kirkegaard, T.; Jäättelä, M. Lysosomal involvement in cell death and cancer. Biochim. Biophys. Acta 2009, 1793, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Pišlar, A.; Nanut, M.P.; Kos, J. Lysosomal cysteine peptidases—Molecules signaling tumor cell death and survival. Semin. Cancer Biol. 2015, 35, 168–179. [Google Scholar] [CrossRef]
- Davidson, S.M.; Vander Heiden, M.G. Critical Functions of the Lysosome in Cancer Biology. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 481–507. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [Green Version]
- Le Joncour, V.; Filppu, P.; Hyvönen, M.; Holopainen, M.; Turunen, S.P.; Sihto, H. Vulnerability of invasive glioblastoma cells to lysosomal membrane destabilization. EMBO Mol. Med. 2019, 11, e9034. [Google Scholar] [CrossRef]
- Das, S.; Dielschneider, R.; Chanas-LaRue, A.; Johnston, J.B.; Gibson, S.B. Antimalarial drugs trigger lysosome-mediated cell death in chronic lymphocytic leukemia (CLL) cells. Leuk. Res. 2018, 70, 79–86. [Google Scholar] [CrossRef]
- Verdoodt, F.; Dehlendorff, C.; Jäättelä, M.; Strauss, R.; Pottegård, A.; Hallas, J.; Friis, S.; Kjaer, S.K. Antihistamines and Ovarian Cancer Survival: Nationwide Cohort Study and in Vitro Cell Viability Assay. JNCI J. Natl. Cancer Inst. 2020, 112, 964–967. [Google Scholar] [CrossRef]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.-Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Marquardt, S.; Solanki, M.; Spitschak, A.; Vera, J.; Pützer, B.M. Emerging functional markers for cancer stem cell-based therapies: Understanding signaling networks for targeting metastasis. Semin. Cancer Biol. 2018, 53, 90–109. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Jensen, S.S.; Aaberg-Jessen, C.; Christensen, K.G.; Kristensen, B. Expression of the lysosomal-associated membrane protein-1 (LAMP-1) in astrocytomas. Int. J. Clin. Exp. Pathol. 2013, 6, 1294–1305. [Google Scholar]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef] [Green Version]
- Du Rietz, H.; Hedlund, H.; Wilhelmson, S.; Nordenfelt, P.; Wittrup, A. Imaging small molecule-induced endosomal escape of siRNA. Nat. Commun. 2020, 11, 1809. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berg, A.L.; Rowson-Hodel, A.; Hu, M.; Keeling, M.; Wu, H.; VanderVorst, K.; Chen, J.J.; Hatakeyama, J.; Jilek, J.; Dreyer, C.A.; et al. The Cationic Amphiphilic Drug Hexamethylene Amiloride Eradicates Bulk Breast Cancer Cells and Therapy-Resistant Subpopulations with Similar Efficiencies. Cancers 2022, 14, 949. https://doi.org/10.3390/cancers14040949
Berg AL, Rowson-Hodel A, Hu M, Keeling M, Wu H, VanderVorst K, Chen JJ, Hatakeyama J, Jilek J, Dreyer CA, et al. The Cationic Amphiphilic Drug Hexamethylene Amiloride Eradicates Bulk Breast Cancer Cells and Therapy-Resistant Subpopulations with Similar Efficiencies. Cancers. 2022; 14(4):949. https://doi.org/10.3390/cancers14040949
Chicago/Turabian StyleBerg, Anastasia L., Ashley Rowson-Hodel, Michelle Hu, Michael Keeling, Hao Wu, Kacey VanderVorst, Jenny J. Chen, Jason Hatakeyama, Joseph Jilek, Courtney A. Dreyer, and et al. 2022. "The Cationic Amphiphilic Drug Hexamethylene Amiloride Eradicates Bulk Breast Cancer Cells and Therapy-Resistant Subpopulations with Similar Efficiencies" Cancers 14, no. 4: 949. https://doi.org/10.3390/cancers14040949