
The Impact of Estrogens and Their Receptors on Immunity and Inflammation during Infection


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
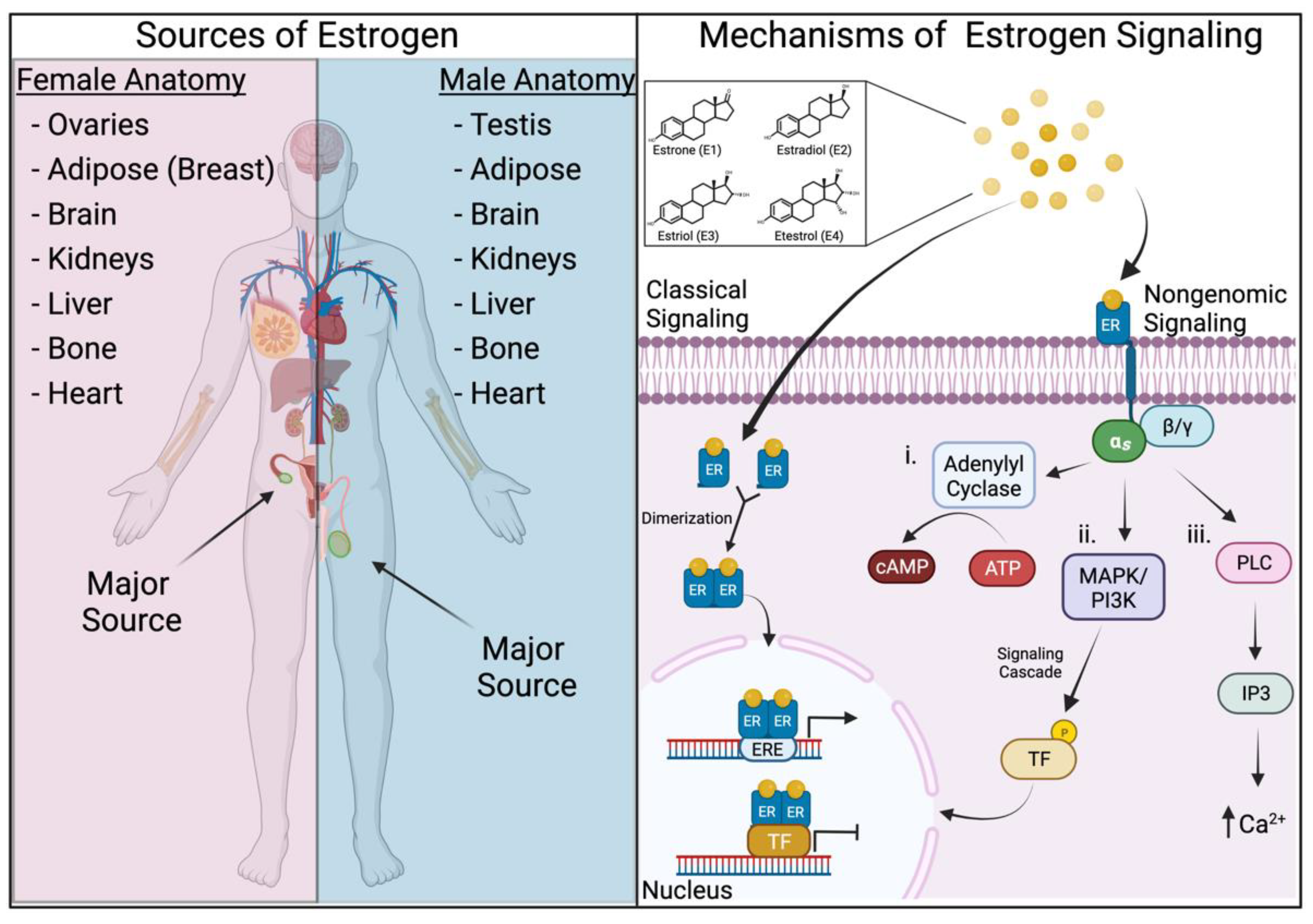
2. Estrogens and Their Mechanisms of Signaling
3. Influence of Estrogens during Viral Infection and Pathogenesis
3.1. Estrogens and Their Receptors Regulate Innate Immune Responses to Viral Infection
3.2. Estrogens and Their Receptors Modulate Viral Infection Severity
4. Influence of Estrogens during Bacterial Infection and Pathogenesis
4.1. Estrogens and Their Receptors Regulate Innate Immune Responses to Bacterial Infection
4.2. Estrogens and Their Receptors Modulate Bacterial Infection Severity
5. Influence of Estrogens during Eukaryotic Infection and Pathogenesis
5.1. Estrogens and Their Receptors Modulate Parasitic Infection Responses and Severity
5.2. Estrogens and Their Receptors Modulate Fungal Infection Responses and Severity
6. Effect of Estrogen on Recovery from Infection and Long-Term Immunity
6.1. Estrogens and Their Receptors Modulate Recovery after Infection
6.2. Estrogens and Their Receptors Regulate Adaptive Immune Responses to Infection
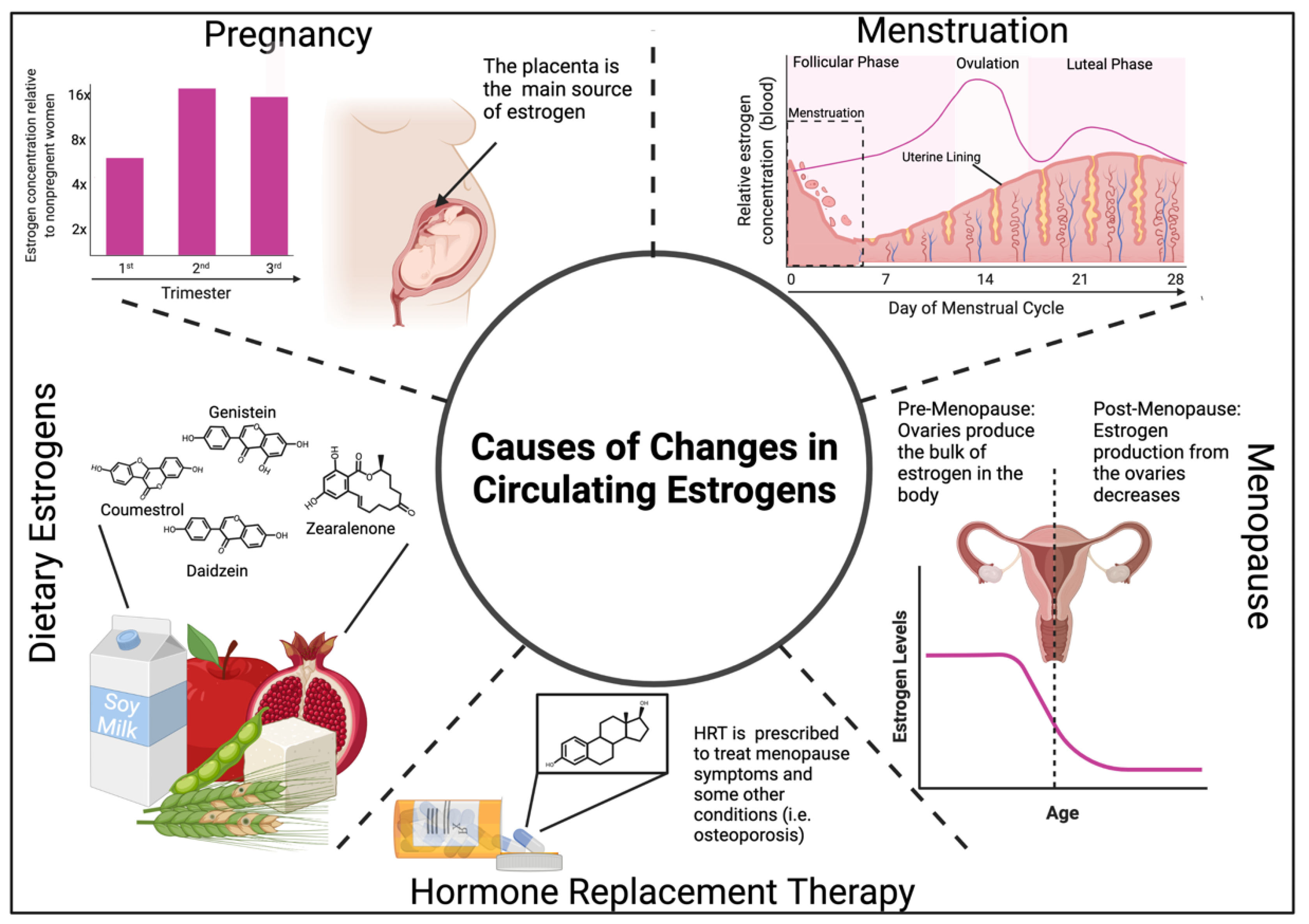
7. Changes in Estrogen Levels and Impacts on the Immune System
7.1. Estrogen Levels during Menstruation, Pregnancy, and Menopause Modulate Immunity
7.2. Exogenous Estrogens Modulate Immunity
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 3 December 2021).
- Murray, P.J.; Smale, S.T. Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways. Nat. Immunol. 2012, 13, 916–924. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Taylor, K.; Kimbrell, D.A. Host Immune Response and Differential Survival of the Sexes in Drosophila. Fly 2007, 1, 197–204. [Google Scholar] [CrossRef]
- Poulin, R. Sexual Size Dimorphism and Transition to Parasitism in Copepods. Evolution 1996, 50, 2520–2523. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.P.; Lorenzo, M.E.; Jian, W.; Klein, S.L. Elevated 17β-estradiol protects females from influenza a virus pathogenesis by suppressing inflammatory responses. PLoS Pathog. 2011, 7, 1002149. [Google Scholar] [CrossRef] [Green Version]
- Travi, B.L.; Osorio, Y.; Melby, P.C.; Chandrasekar, B.; Arteaga, L.; Saravia, N.G. Gender Is a Major Determinant of the Clinical Evolution and Immune Response in Hamsters Infected with Leishmania spp. Infect. Immun. 2002, 70, 2288–2296. [Google Scholar] [CrossRef] [Green Version]
- Merkel, S.M.; Alexander, S.; Zufall, E.; Oliver, J.D.; Huet-Hudson, Y.M. Essential Role for Estrogen in Protection against Vibrio vulnificus -Induced Endotoxic Shock. Infect. Immun. 2001, 69, 6119–6122. [Google Scholar] [CrossRef] [Green Version]
- DeRoo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Paterni, I.; Bertini, S.; Granchi, C.; Tuccinardi, T.; Macchia, M.; Martinelli, A.; Caligiuri, I.; Toffoli, G.; Rizzolio, F.; Carlson, K.E.; et al. Highly Selective Salicylketoxime-Based Estrogen Receptor β Agonists Display Antiproliferative Activities in a Glioma Model. J. Med. Chem. 2015, 58, 1184–1194. [Google Scholar] [CrossRef]
- Knowlton, A.A.; Lee, A.R. Estrogen and the cardiovascular system. Pharmacol. Ther. 2012, 135, 54–70. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, E.; Guignard, L.; Knaup Reymond, M.; Perreau, M.; Roth-Kleiner, M.; Calandra, T.; Roger, T. Estradiol and Progesterone Strongly Inhibit the Innate Immune Response of Mononuclear Cells in Newborns. Infect. Immun. 2011, 79, 2690–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Trevisi, L.; Bolego, C.; Cignarella, A. Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. Int. J. Mol. Sci. 2018, 19, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, A.T.; Goff, M.A.; Froggatt, H.M.; Lim, J.K.; Heaton, N.S. GPER1 is required to protect fetal health from maternal inflammation. Science 2021, 371, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; Drew, B.G.; Le, J.A.; Soleymani, T.; Daraei, P.; Sitz, D.; Mohammad, L.; Henstridge, D.C.; Febbraio, M.A.; Hewitt, S.C.; et al. Myeloid-specific estrogen receptor deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc. Natl. Acad. Sci. USA 2011, 108, 16457–16462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelekanou, V.; Kampa, M.; Kiagiadaki, F.; Deli, A.; Theodoropoulos, P.; Agrogiannis, G.; Patsouris, E.; Tsapis, A.; Castanas, E.; Notas, G. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPER1. J. Leukoc. Biol. 2015, 99, 333–347. [Google Scholar] [CrossRef]
- Toniolo, A.; Tedesco, S.; Cappellari, R.; Vegeto, E.; Maggi, A.; Avogaro, A.; Bolego, C.; Cignarella, A.; Fadini, G.P. Alternative Activation of Human Macrophages Is Rescued by Estrogen Treatment In Vitro and Impaired by Menopausal Status. J. Clin. Endocrinol. Metab. 2015, 100, E50–E58. [Google Scholar] [CrossRef] [Green Version]
- Blasko, E.; Haskell, C.A.; Leung, S.; Gualtieri, G.; Halks-Miller, M.; Mahmoudi, M.; Dennis, M.; Prossnitz, E.; Karpus, W.J.; Horuk, R. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J. Neuroimmunol. 2009, 214, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Xing, D.; Oparil, S.; Yu, H.; Gong, K.; Feng, W.; Black, J.; Chen, Y.F.; Nozell, S. Estrogen Modulates NFκB Signaling by Enhancing IκBα Levels and Blocking p65 Binding at the Promoters of Inflammatory Genes via Estrogen Receptor-β. PLoS ONE 2012, 7, e36890. [Google Scholar] [CrossRef]
- Dupuis, M.L.; Conti, F.; Maselli, A.; Pagano, M.T.; Ruggieri, A.; Anticoli, S.; Fragale, A.; Gabriele, L.; Gagliardi, M.C.; Sanchez, M.; et al. The Natural Agonist of Estrogen Receptor β Silibinin Plays an Immunosuppressive Role Representing a Potential Therapeutic Tool in Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1903. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, M.; Hatori, M.; Suzuki, T.; Miki, Y.; Darnel, A.D.; Tazawa, C.; Sawai, T.; Uzuki, M.; Tanaka, Y.; Kokubun, S.; et al. Sex steroid receptors in rheumatoid arthritis. Clin. Sci. 2004, 106, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Rider, V.; Li, X.; Peterson, G.; Dawson, J.; Kimler, B.F.; Abdou, N.I. Differential expression of estrogen receptors in women with systemic lupus erythematosus. J. Rheumatol. 2006, 33, 1093–1101. [Google Scholar] [PubMed]
- Fluhmann, C.F. Female Sex Hormones and Menstruation. Calif. West. Med. 1931, 35, 279–283. [Google Scholar]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakemore, J.; Naftolin, F. Aromatase: Contributions to Physiology and Disease in Women and Men. Physiology 2016, 31, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Wildman, R.P.; Colvin, A.; Powell, L.H.; Matthews, K.A.; Everson-Rose, S.; Hollenberg, S.; Johnston, J.M.; Sutton-Tyrrell, K. Associations of endogenous sex hormones with the vasculature in menopausal women: The Study of Women’s Health Across the Nation (SWAN). Menopause 2008, 15, 414–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Miki, Y.; Abe, K.; Hatori, M.; Hosaka, M.; Kariya, Y.; Kakuo, S.; Fujimura, T.; Hachiya, A.; Honma, S.; et al. Sex steroid synthesis in human skin in situ: The roles of aromatase and steroidogenic acute regulatory protein in the homeostasis of human skin. Mol. Cell. Endocrinol. 2012, 362, 19–28. [Google Scholar] [CrossRef]
- Cooke, P.S.; Nanjappa, M.K.; Ko, C.; Prins, G.S.; Hess, R. Estrogens in Male Physiology. Physiol. Rev. 2017, 97, 995–1043. [Google Scholar] [CrossRef]
- Nilsson, S.; Mäkelä, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of Estrogen Action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef]
- Lösel, R.; Wehling, M. Nongenomic actions of steroid hormones. Nat. Rev. Mol. Cell Biol. 2003, 4, 46–56. [Google Scholar] [CrossRef]
- Improta-Brears, T.; Whorton, A.R.; Codazzi, F.; York, J.D.; Meyer, T.; McDonnell, D.P. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc. Natl. Acad. Sci. USA 1999, 96, 4686–4691. [Google Scholar] [CrossRef] [Green Version]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosselman, S.; Polman, J.; Dijkema, R. ERβ: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996, 392, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjöld, M.; Gustafsson, J.-A. Human Estrogen Receptor β-Gene Structure, Chromosomal Localization, and Expression Pattern1. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sand, P.; Luckhaus, C.; Schlurmann, K.; Götz, M.; Deckert, J. Untangling the human estrogen receptor gene structure. J. Neural Transm. 2002, 109, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef]
- Pedrero, J.M.G.; Zuazua, P.; Martínez-Campa, C.; Lazo, P.S.; Ramos, S. The Naturally Occurring Variant of Estrogen Receptor (ER) ERΔE7 Suppresses Estrogen-Dependent Transcriptional Activation by Both Wild-Type ERα and ERβ. Endocrinology 2003, 144, 2967–2976. [Google Scholar] [CrossRef] [Green Version]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Stein, B.; Yang, M.X. Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-kappa B and C/EBP beta. Mol. Cell. Biol. 1995, 15, 4971–4979. [Google Scholar] [CrossRef] [Green Version]
- McMurray, R.W.; Ndebele, K.; Hardy, K.J.; Jenkins, J.K. 17-beta-estradiol suppresses IL-2 and IL-2 receptor. Cytokine 2001, 14, 324–333. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Giraud, S.N.; Caron, C.M.; Pham-Dinh, D.; Kitabgi, P.; Nicot, A.B. Estradiol inhibits ongoing autoimmune neuroinflammation and NF B-dependent CCL2 expression in reactive astrocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 8416–8421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wira, C.R.; Fahey, J.V.; Ghosh, M.; Patel, M.V.; Hickey, D.K.; Ochiel, D.O. Sex Hormone Regulation of Innate Immunity in the Female Reproductive Tract: The Role of Epithelial Cells in Balancing Reproductive Potential with Protection against Sexually Transmitted Pathogens. Am. J. Reprod. Immunol. 2010, 63, 544–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvoro, A.; Tatomer, D.; Tee, M.-K.; Zogovic, T.; Harris, H.A.; Leitman, D.C. Selective Estrogen Receptor-β Agonists Repress Transcription of Proinflammatory Genes. J. Immunol. 2008, 180, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Dragin, N.; Nancy, P.; Villegas, J.; Roussin, R.; Le Panse, R.; Berrih-Aknin, S. Balance between Estrogens and Proinflammatory Cytokines Regulates Chemokine Production Involved in Thymic Germinal Center Formation. Sci. Rep. 2017, 7, 7970. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, M.; Tachibana, H.; Yamada, K. Effect of Estrogens on the Interferon-γ Producing Cell Population of Mouse Splenocytes. Biosci. Biotechnol. Biochem. 2006, 70, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- Ryabkova, V.A.; Churilov, L.P.; Shoenfeld, Y. Influenza infection, SARS, MERS and COVID-19: Cytokine storm—The common denominator and the lessons to be learned. Clin. Immunol. 2020, 223, 108652. [Google Scholar] [CrossRef]
- Vermillion, M.S.; Ursin, R.; Attreed, S.E.; Klein, S.L. Estriol Reduces Pulmonary Immune Cell Recruitment and Inflammation to Protect Female Mice from Severe Influenza. Endocrinology 2018, 159, 3306–3320. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.P.; Hall, O.J.; Nilles, T.L.; Bream, J.; Klein, S.L. 17 -Estradiol Protects Females against Influenza by Recruiting Neutrophils and Increasing Virus-Specific CD8 T Cell Responses in the Lungs. J. Virol. 2014, 88, 4711–4720. [Google Scholar] [CrossRef] [Green Version]
- Pazos, M.A.; Kraus, T.A.; Muñoz-Fontela, C.; Moran, T.M. Estrogen Mediates Innate and Adaptive Immune Alterations to Influenza Infection in Pregnant Mice. PLoS ONE 2012, 7, e40502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Channappanavar, R.; Fett, C.; Mack, M.; Ten Eyck, P.P.; Meyerholz, D.K.; Perlman, S. Sex-Based Differences in Susceptibility to Severe Acute Respiratory Syndrome Coronavirus Infection. J. Immunol. 2017, 198, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Seeland, U.; Coluzzi, F.; Simmaco, M.; Mura, C.; Bourne, P.E.; Heiland, M.; Preissner, R.; Preissner, S. Evidence for treatment with estradiol for women with SARS-CoV-2 infection. BMC Med. 2020, 18, 369. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Klein, S.L.; Levin, E.R. Estradiol, Progesterone, Immunomodulation, and COVID-19 Outcomes. Endocrinology 2020, 161, bqaa127. [Google Scholar] [CrossRef]
- Leong, H.-N.; Earnest, A.; Lim, H.-H.; Chin, C.-F.; Tan, C.S.H.; Puhaindran, M.E.; Tan, A.C.H.; Chen, M.I.C.; Leo, Y.-S. SARS in Singapore—Predictors of disease severity. Ann. Acad. Med. Singap. 2006, 35, 326–331. [Google Scholar]
- Alghamdi, I.; Hussain, I.; Alghamdi, M.; Almalki, S.; Alghamdi, M.; Elsheemy, M. The pattern of Middle East respiratory syndrome coronavirus in Saudi Arabia: A descriptive epidemiological analysis of data from the Saudi Ministry of Health. Int. J. Gen. Med. 2014, 7, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.M.; Bai, P.; He, W.; Wu, F.; Liu, X.F.; Han, D.M.; Liu, S.; Yang, J.K. Gender Differences in Patients With COVID-19: Focus on Severity and Mortality. Front. Public Health 2020, 8, 152. [Google Scholar] [CrossRef]
- Al-Raddadi, R.M.; Shabouni, O.I.; Alraddadi, Z.M.; Alzalabani, A.H.; Al-Asmari, A.M.; Ibrahim, A.; Almarashi, A.; Madani, T.A. Burden of Middle East respiratory syndrome coronavirus infection in Saudi Arabia. J. Infect. Public Health 2020, 13, 692–696. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Greten, F. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Tietze, K.; Dalpke, A.; Morath, S.; Mutters, R.; Heeg, K.; Nonnenmacher, C. Differences in innate immune responses upon stimulation with gram-positive and gram-negative bacteria. J. Periodontal Res. 2006, 41, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of Adaptor TRIF in the MyD88-Independent Toll-Like Receptor Signaling Pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, R.A.; Dorrington, M.G.; Dutta, B.; Krauss, K.S.; Martins, A.J.; Uderhardt, S.; Chan, W.; Tsang, J.S.; Torabi-Parizi, P.; Fraser, I.D.; et al. IFN-mediated negative feedback supports bacteria class-specific macrophage inflammatory responses. eLife 2019, 8, e46836. [Google Scholar] [CrossRef] [PubMed]
- Karaghiosoff, M.; Steinborn, R.; Kovarik, P.; Kriegshäuser, G.; Baccarini, M.; Donabauer, B.; Reichart, U.; Kolbe, T.; Bogdan, C.; Leanderson, T.; et al. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat. Immunol. 2003, 4, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Notas, G.; Panagiotopoulos, A.; Vamvoukaki, R.; Kalyvianaki, K.; Kiagiadaki, F.; Deli, A.; Kampa, M.; Castanas, E. ERα36–GPER1 Collaboration Inhibits TLR4/NFκB-Induced Pro-Inflammatory Activity in Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 7603. [Google Scholar] [CrossRef]
- Rettew, J.A.; McCall, S.H.; Marriott, I. GPR30/GPER-1 mediates rapid decreases in TLR4 expression on murine macrophages. Mol. Cell. Endocrinol. 2010, 328, 87–92. [Google Scholar] [CrossRef]
- Rettew, J.A.; Huet, Y.; Marriott, I. Estrogens Augment Cell Surface TLR4 Expression on Murine Macrophages and Regulate Sepsis Susceptibility in Vivo. Endocrinology 2009, 150, 3877–3884. [Google Scholar] [CrossRef] [Green Version]
- Hotchkiss, R.R.; Moldawer, L.L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.I.; Vincent, J.-L. Sepsis and septic shock. Nat. Rev. Dis. Prim. 2016, 2, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Opal, S.M. Endotoxin desynchronizes biological clocks. Crit. Care Med. 2010, 38, 977–978. [Google Scholar] [CrossRef]
- Alexandraki, I.; Palacio, C. Gram-negative versus Gram-positive bacteremia: What is more alarmin(g)? Crit. Care 2010, 14, 161. [Google Scholar] [CrossRef]
- Vandereyken, M.M.; Singh, P.; Wathieu, C.P.; Jacques, S.; Zurashvilli, T.; Dejager, L.; Amand, M.; Musumeci, L.; Singh, M.; Moutschen, M.P.; et al. Dual-Specificity Phosphatase 3 Deletion Protects Female, but Not Male, Mice from Endotoxemia-Induced and Polymicrobial-Induced Septic Shock. J. Immunol. 2017, 199, 2515–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Qu, J.; Xia, X.; Pan, Y.; Liu, X.; Liang, H.; Dou, H.; Hou, Y. 17β-Estradiol promotes LC3B-associated phagocytosis in trained immunity of female mice against sepsis. Int. J. Biol. Sci. 2021, 17, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Guo, Y.; Jiang, M.; Hu, B.; Li, Z.; Fan, J.; Deng, M.; Billiar, T.R.; Kucera, H.R.; Gaikwad, N.W.; et al. Oestrogen sulfotransferase ablation sensitizes mice to sepsis. Nat. Commun. 2015, 6, 7979. [Google Scholar] [CrossRef] [Green Version]
- Sakr, Y.; Elia, C.; Mascia, L.; Barberis, B.; Cardellino, S.; Livigni, S.; Fiore, G.; Filippini, C.; Ranieri, V. The influence of gender on the epidemiology of and outcome from severe sepsis. Crit. Care 2013, 17, R50. [Google Scholar] [CrossRef] [Green Version]
- Oberholzer, A.; Keel, M.; Zellweger, R.; Steckholzer, U.; Trentz, O.; Ertel, W. Incidence of Septic Complications and Multiple Organ Failure in Severely Injured Patients Is Sex Specific. J. Trauma Inj. Infect. Crit. Care 2000, 48, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.V.; Wright, J.A.; Shen, L.; Smith, J.M.; Ghosh, M.; Rossoll, R.M.; Wira, C.R. Estradiol selectively regulates innate immune function by polarized human uterine epithelial cells in culture. Mucosal Immunol. 2008, 1, 317–325. [Google Scholar] [CrossRef]
- Crane-Godreau, M.A.; Wira, C.R. Effects of Estradiol on Lipopolysaccharide and Pam 3 Cys Stimulation of CCL20/Macrophage Inflammatory Protein 3 Alpha and Tumor Necrosis Factor Alpha Production by Uterine Epithelial Cells in Culture. Infect. Immun. 2005, 73, 4231–4237. [Google Scholar] [CrossRef] [Green Version]
- Kowsar, R.; Hambruch, N.; Liu, J.; Shimizu, T.; Pfarrer, C.; Miyamoto, A. Regulation of Innate Immune Function in Bovine Oviduct Epithelial Cells in Culture: The Homeostatic Role of Epithelial Cells in Balancing Th1/Th2 Response. J. Reprod. Dev. 2013, 59, 470–478. [Google Scholar] [CrossRef] [Green Version]
- Von Aulock, S.; Deininger, S.; Draing, C.; Gueinzius, K.; Dehus, O.; Hermann, C. Gender Difference in Cytokine Secretion on Immune Stimulation with LPS and LTA. J. Interf. Cytokine Res. 2006, 26, 887–892. [Google Scholar] [CrossRef]
- Phillips, M.A.; Burrows, J.N.; Manyando, C.; Van Huijsduijnen, R.H.; Van Voorhis, W.C.; Wells, T.N.C. Malaria. Nat. Rev. Dis. Prim. 2017, 3, 17050. [Google Scholar] [CrossRef]
- Landgraf, B.; Kollaritsch, H.; Wiedermann, G.; Wernsdorfer, W.H. Parasite density of Plasmodium falciparum malaria in Ghanaian schoolchildren: Evidence for influence of sex hormones? Trans. R. Soc. Trop. Med. Hyg. 1994, 88, 73–74. [Google Scholar] [CrossRef]
- Briggs, J.; Teyssier, N.; Nankabirwa, J.I.; Rek, J.; Jagannathan, P.; Arinaitwe, E.; Bousema, T.; Drakeley, C.; Murray, M.; Crawford, E.; et al. Sex-based differences in clearance of chronic Plasmodium falciparum infection. eLife 2020, 9, e59872. [Google Scholar] [CrossRef] [PubMed]
- King, T.; Lamb, T. Interferon-γ: The Jekyll and Hyde of Malaria. PLOS Pathog. 2015, 11, e1005118. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.; Easterbrook, J.D.; Lalime, E.N.; Klein, S.L. Estrogen and progesterone affect responses to malaria infection in female C57BL/6 mice. Gend. Med. 2008, 5, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cernetich, A.; Garver, L.S.; Jedlicka, A.E.; Klein, P.; Kumar, N.; Scott, A.L.; Klein, S.L. Involvement of Gonadal Steroids and Gamma Interferon in Sex Differences in Response to Blood-Stage Malaria Infection. Infect. Immun. 2006, 74, 3190–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blessmann, J.; Thi, H.D.; Van Linh, P.; Tannich, E.; Buss, H.; Nu, P.A.T.; Muller-Myhsok, B. Epidemiology of amebiasis in a region of high incidence of amebic liver abscess in central Vietnam. Am. J. Trop. Med. Hyg. 2002, 66, 578–583. [Google Scholar] [CrossRef] [Green Version]
- Lotter, H.; Helk, E.; Bernin, H.; Jacobs, T.; Prehn, C.; Adamski, J.; González-Roldán, N.; Holst, O.; Tannich, E. Testosterone Increases Susceptibility to Amebic Liver Abscess in Mice and Mediates Inhibition of IFNγ Secretion in Natural Killer T Cells. PLoS ONE 2013, 8, e55694. [Google Scholar] [CrossRef] [Green Version]
- Liesenfeld, O.; Nguyen, T.A.; Pharke, C.; Suzuki, Y. Importance of Gender and Sex Hormones in Regulation of Susceptibility of the Small Intestine to Peroral Infection with Toxoplasma gondii Tissue Cysts. J. Parasitol. 2001, 87, 1491. [Google Scholar] [CrossRef]
- Walker, W.; Roberts, C.W.; Ferguson, D.J.; Jebbari, H.; Alexander, J. Innate immunity to Toxoplasma gondii is influenced by gender and is associated with differences in interleukin-12 and gamma interferon production. Infect. Immun. 1997, 65, 1119–1121. [Google Scholar] [CrossRef] [Green Version]
- Sobel, J.D.; Faro, S.; Force, R.W.; Foxman, B.; Ledger, W.; Nyirjesy, P.R.; Reed, B.D.; Summers, P.R. Vulvovaginal candidiasis: Epidemiologic, diagnostic, and therapeutic considerations. Am. J. Obstet. Gynecol. 1998, 178, 203–211. [Google Scholar] [CrossRef]
- Cotch, M.F.; Hillier, S.L.; Gibbs, R.S.; Eschenbach, D.A. Epidemiology and outcomes associated with moderate to heavy Candida colonization during pregnancy. Vaginal Infections and Prematurity Study Group. Am. J. Obstet. Gynecol. 1998, 178, 374–380. [Google Scholar] [CrossRef]
- Spinillo, A.; Capuzzo, E.; Nicola, S.; Baltaro, F.; Ferrari, A.; Monaco, A. The impact of oral contraception on vulvovaginal candidiasis. Contraception 1995, 51, 293–297. [Google Scholar] [CrossRef]
- Spinillo, A.; Bernuzzi, A.M.; Cevini, C.; Gulminetti, R.; Luzi, S.; De Santolo, A. The relationship of bacterial vaginosis, candida and trichomonas infection to symptomatic vaginitis in postmenopausal women attending a vaginitis clinic. Maturitas 1997, 27, 253–260. [Google Scholar] [CrossRef]
- Kinsman, O.; Pitblado, K.; Coulson, C. Effect of Mammalian Steroid Hormones and Luteinizing Hormone on the Germination of Candida albicans and Implications for Vaginal Candidosis. Mycoses 1988, 31, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Skowronski, R.; Feldman, D. Characterization of an Estrogen-Binding Protein in the Yeast Candida albicans. Endocrinology 1989, 124, 1965–1972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Essmann, M.; Burt, E.T.; Larsen, B. Estrogen Effects on Candida albicans: A Potential Virulence-Regulating Mechanism. J. Infect. Dis. 2000, 181, 1441–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Yeater, K.M.; Hoyer, L.L. Cellular and Molecular Biology of Candida albicans Estrogen Response. Eukaryot. Cell 2006, 5, 180–191. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.E.; Restrepo, A.; Stevens, D.A. Inhibition by estrogens of conidium-to-yeast conversion in the fungus Paracoccidioides brasiliensis. Infect. Immun. 1988, 56, 711–713. [Google Scholar] [CrossRef] [Green Version]
- Bellissimo-Rodrigues, F.; Bollela, V.; Fonseca, B.; Martinez, R. Endemic paracoccidioidomycosis: Relationship between clinical presentation and patients’ demographic features. Med. Mycol. 2013, 51, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Aristizábal, B.H.; Clemons, K.V.; Cock, A.M.; Restrepo, A.; Stevens, D.A. ExperimentalParacoccidioides brasiliensisinfection in mice: Influence of the hormonal status of the host on tissue responses. Med. Mycol. 2002, 40, 169–178. [Google Scholar] [CrossRef]
- Pinzan, C.F.; Ruas, L.P.; Casabona-Fortunato, A.S.; Carvalho, F.C.; Roque-Barreira, M.-C. Immunological Basis for the Gender Differences in Murine Paracoccidioides brasiliensis Infection. PLoS ONE 2010, 5, e10757. [Google Scholar] [CrossRef] [Green Version]
- Gebremariam, T.; Alkhazraji, S.; Alqarihi, A.; Wiederhold, N.; Najvar, L.; Patterson, T.; Filler, S.; Ibrahim, A. Evaluation of Sex Differences in Murine Diabetic Ketoacidosis and Neutropenic Models of Invasive Mucormycosis. J. Fungi 2021, 7, 313. [Google Scholar] [CrossRef] [PubMed]
- Vannella, K.M.; Wynn, T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017, 199, 2191–2193. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, J.B.; Taintor, R.R.; Vavrin, Z.; Rachlin, E.M. Nitric oxide: A cytotoxic activated macrophage effector molecule. Biochem. Biophys. Res. Commun. 1988, 157, 87–94. [Google Scholar] [CrossRef]
- Mills, C.D. Macrophage Arginine Metabolism to Ornithine/Urea or Nitric Oxide/Citrulline: A Life or Death Issue. Crit. Rev. Immunol. 2001, 21, 399–425. [Google Scholar] [CrossRef]
- Keselman, A.; Fang, X.; White, P.B.; Heller, N.M. Estrogen Signaling Contributes to Sex Differences in Macrophage Polarization during Asthma. J. Immunol. 2017, 199, 1573–1583. [Google Scholar] [CrossRef]
- Campbell, L.; Emmerson, E.; Williams, H.; Saville, C.R.; Krust, A.; Chambon, P.; Mace, K.A.; Hardman, M.J. Estrogen Receptor-Alpha Promotes Alternative Macrophage Activation during Cutaneous Repair. J. Investig. Dermatol. 2014, 134, 2447–2457. [Google Scholar] [CrossRef] [Green Version]
- Bolego, C.; Cignarella, A.; Staels, B.; Chinetti-Gbaguidi, G. Macrophage Function and Polarization in Cardiovascular Disease: A role of estrogen signaling? Arter. Thromb. Vasc. Biol. 2013, 33, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Lu, Y.; Xu, Y.; Xu, L.; Zheng, W.; Wu, Y.; Li, L.; Shen, P. Estrogen Represses Hepatocellular Carcinoma (HCC) Growth via Inhibiting Alternative Activation of Tumor-associated Macrophages (TAMs). J. Biol. Chem. 2012, 287, 40140–40149. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.L. Estrogen, a double-edged sword: Modulation of TH1- and TH2-mediated inflammations by differential regulation of TH1/TH2 cytokine production. Curr. Drug Target Inflamm. Allergy 2004, 3, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Marzi, M.; Vigano, A.; Trabattoni, D.; Villa, M.L.; Salvaggio, A.; Clerici, E.; Clerici, M. Characterization of type 1 and type 2 cytokine production profile in physiologic and pathologic human pregnancy. Clin. Exp. Immunol. 1996, 106, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Horng, H.-C.; Chang, W.-H.; Yeh, C.-C.; Huang, B.-S.; Chang, C.-P.; Chen, Y.-J.; Tsui, K.-H.; Wang, P.-H. Estrogen Effects on Wound Healing. Int. J. Mol. Sci. 2017, 18, 2325. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Greenwell-Wild, T.; Horan, M.A.; Wahl, S.M.; Ferguson, M.W. Topical Estrogen Accelerates Cutaneous Wound Healing in Aged Humans Associated with an Altered Inflammatory Response. Am. J. Pathol. 1999, 155, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Zhu, B.T. Unique effect of the pregnancy hormone estriol on antigen-induced production of specific antibodies in female BALB/c mice. Steroids 2008, 73, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.-S.; Chang, Y.-L.; Chao, A.; Wu, T.-S.; Yang, L.-Y.; Lian, R.; Huang, Y.-C. Seropositivity of influenza A H1NI in mothers and infants following maternal vaccination with trivalent seasonal influenza vaccine after the 2009 pandemic. Taiwan. J. Obstet. Gynecol. 2017, 56, 37–40. [Google Scholar] [CrossRef]
- Potluri, T.; Fink, A.L.; Sylvia, K.E.; Dhakal, S.; Vermillion, M.S.; Steeg, L.V.; Deshpande, S.; Narasimhan, H.; Klein, S.L. Age-associated changes in the impact of sex steroids on influenza vaccine responses in males and females. NPJ Vaccines 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Yockey, L.J.; Jurado, K.A.; Arora, N.; Millet, A.; Rakib, T.; Milano, K.M.; Hastings, A.K.; Fikrig, E.; Kong, Y.; Horvath, T.L.; et al. Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol. 2018, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Pennock, N.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naïve to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Phiel, K.L.; Henderson, R.A.; Adelman, S.J.; Elloso, M.M. Differential estrogen receptor gene expression in human peripheral blood mononuclear cell populations. Immunol. Lett. 2005, 97, 107–113. [Google Scholar] [CrossRef]
- Laffont, S.; Rouquié, N.; Azar, P.; Seillet, C.; Plumas, J.; Aspord, C.; Guéry, J.-C. X-Chromosome Complement and Estrogen Receptor Signaling Independently Contribute to the Enhanced TLR7-Mediated IFN-α Production of Plasmacytoid Dendritic Cells from Women. J. Immunol. 2014, 193, 5444–5452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staples, J.E.; Gasiewicz, T.A.; Fiore, N.C.; Lubahn, D.B.; Korach, K.S.; Silverstone, A.E. Estrogen receptor alpha is necessary in thymic development and estradiol-induced thymic alterations. J. Immunol. 1999, 163, 4168–4174. [Google Scholar] [PubMed]
- Lambert, K.C.; Curran, E.M.; Judy, B.M.; Milligan, G.N.; Lubahn, D.B.; Estes, D.M. Estrogen Receptor α (ERα) Deficiency in Macrophages Results in Increased Stimulation of CD4+T Cells while 17β-Estradiol Acts through ERα to Increase IL-4 and GATA-3 Expression in CD4+T Cells Independent of Antigen Presentation. J. Immunol. 2005, 175, 5716–5723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javadian, A.; Salehi, E.; Bidad, K.; Sahraian, M.A.; Izad, M. Effect of Estrogen on Th1, Th2 and Th17 Cytokines Production by Proteolipid Protein and PHA Activated Peripheral Blood Mononuclear Cells Isolated from Multiple Sclerosis Patients. Arch. Med. Res. 2014, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Zittermann, A.; Schwarz, I.; Scheld, K.; Sudhop, T.; Berthold, H.K.; Von Bergmann, K.; Van Der Ven, H.; Stehle, P. Physiologic Fluctuations of Serum Estradiol Levels Influence Biochemical Markers of Bone Resorption in Young Women. J. Clin. Endocrinol. Metab. 2000, 85, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Puder, J.J.; Blum, C.A.; Mueller, B.; De Geyter, C.; Dye, L.; Keller, U. Menstrual cycle symptoms are associated with changes in low-grade inflammation. Eur. J. Clin. Investig. 2006, 36, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Gold, E.B.; Wells, C.; Rasor, M.O. The Association of Inflammation with Premenstrual Symptoms. J. Women’s Health 2016, 25, 865–874. [Google Scholar] [CrossRef]
- Abbassi-Ghanavati, M.; Greer, L.G.; Cunningham, F.G. Pregnancy and Laboratory Studies. Obstet. Gynecol. 2009, 114, 1326–1331. [Google Scholar] [CrossRef]
- Wegmann, T.G.; Lin, H.; Guilbert, L.; Mosmann, T.R. Bidirectional cytokine interactions in the maternal-fetal relationship: Is successful pregnancy a TH2 phenomenon? Immunol. Today 1993, 14, 353–356. [Google Scholar] [CrossRef]
- Polanczyk, M.J.; Hopke, C.; Huan, J.; Vandenbark, A.A.; Offner, H. Enhanced FoxP3 expression and Treg cell function in pregnant and estrogen-treated mice. J. Neuroimmunol. 2005, 170, 85–92. [Google Scholar] [CrossRef]
- Nelson, J.L.; Østensen, M. Pregnancy and rheumatoid arthritis. Rheum. Dis. Clin. N. Am. 1997, 23, 195–212. [Google Scholar] [CrossRef]
- Bränn, E.; Edvinsson, A.; Punga, A.R.; Sundström-Poromaa, I.; Skalkidou, A. Inflammatory and anti-inflammatory markers in plasma: From late pregnancy to early postpartum. Sci. Rep. 2019, 9, 1863. [Google Scholar] [CrossRef] [PubMed]
- Richardson, H.; Ho, V.; Pasquet, R.; Singh, R.J.; Goetz, M.P.; Tu, D.; Goss, P.E.; Ingle, J.N. Baseline estrogen levels in postmenopausal women participating in the MAP.3 breast cancer chemoprevention trial. Menopause 2020, 27, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Shieh, A.; Epeldegui, M.; Karlamangla, A.S.; Greendale, G.A. Gut permeability, inflammation, and bone density across the menopause transition. JCI Insight 2020, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triebner, K.; Johannessen, A.; Puggini, L.; Benediktsdóttir, B.; Bertelsen, R.; Bifulco, E.; Dharmage, S.; Dratva, J.; Franklin, K.A.; Gíslason, T.; et al. Menopause as a predictor of new-onset asthma: A longitudinal Northern European population study. J. Allergy Clin. Immunol. 2016, 137, 50–57.e6. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, N.M.; Morris, P.G.; Zhou, X.K.; Gucalp, A.; Giri, D.; Harbus, M.D.; Falcone, D.J.; Krasne, M.; Vahdat, L.T.; Subbaramaiah, K.; et al. Menopause Is a Determinant of Breast Adipose Inflammation. Cancer Prev. Res. 2015, 8, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Rocca, W.A.; Rocca, L.G.; Smith, C.Y.; Grossardt, B.R.; Faubion, S.S.; Shuster, L.T.; Kirkland, J.L.; Lebrasseur, N.K.; Schafer, M.J.; Mielke, M.M.; et al. Loss of Ovarian Hormones and Accelerated Somatic and Mental Aging. Physiology 2018, 33, 374–383. [Google Scholar] [CrossRef]
- Rocca, W.A.; Gazzuola-Rocca, L.; Smith, C.Y.; Grossardt, B.; Faubion, S.S.; Shuster, L.T.; Kirkland, J.L.; Stewart, E.; Miller, V.M. Accelerated Accumulation of Multimorbidity After Bilateral Oophorectomy: A Population-Based Cohort Study. Mayo Clin. Proc. 2016, 91, 1577–1589. [Google Scholar] [CrossRef]
- Doran, M.F.; Crowson, C.S.; O’Fallon, W.M.; Gabriel, S.E. The effect of oral contraceptives and estrogen re-placement therapy on the risk of rheumatoid arthritis: A population based study. J. Rheumatol. 2004, 31, 207–213. [Google Scholar]
- Hazes, J.M.W.; Dijkmans, B.A.C.; Vandenbroucke, J.P.; De Vries, R.R.P.; Cats, A. Oral Contraceptives and Rheumatoid Arthritis; Further Evidence for a Protective Effect Independent of Duration of Pill use. Br. J. Rheumatol. 1989, XXVIII, 34. [Google Scholar] [CrossRef]
- Khalili, H.; Higuchi, L.M.; Ananthakrishnan, A.N.; Richter, J.M.; Feskanich, D.; Fuchs, C.S.; Chan, A.T. Oral contraceptives, reproductive factors and risk of inflammatory bowel disease. Gut 2013, 62, 1153–1159. [Google Scholar] [CrossRef] [Green Version]
- Divani, A.A.; Luo, X.; Datta, Y.H.; Flaherty, J.D.; Panoskaltsis-Mortari, A. Effect of Oral and Vaginal Hormonal Contraceptives on Inflammatory Blood Biomarkers. Mediat. Inflamm. 2015, 2015, 1–8. [Google Scholar] [CrossRef]
- Hellwig, K.; Chen, L.H.; Stancyzk, F.Z.; Langer-Gould, A.M. Oral Contraceptives and Multiple Sclerosis/Clinically Isolated Syndrome Susceptibility. PLoS ONE 2016, 11, e0149094. [Google Scholar] [CrossRef]
- Miller, A.P.; Chen, Y.-F.; Xing, N.; Feng, W.; Oparil, S. Hormone Replacement Therapy and Inflammation. Hypertension 2003, 42, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiadou, P.; Sbarouni, E. Chapter 3 Effect of Hormone Replacement Therapy on Inflammatory Biomarkers. Adv. Clin. Chem. 2009, 47, 59–93. [Google Scholar] [CrossRef] [PubMed]
- Forsblad-D’Elia, H.; Larsen, A.; Mattsson, L.-A.; Waltbrand, E.; Kvist, G.; Mellström, D.; Saxne, T.; Ohlsson, C.; Nordborg, E.; Carlsten, H. Influence of hormone replacement therapy on disease progression and bone mineral density in rheumatoid arthritis. J. Rheumatol. 2003, 30, 1456–1463. [Google Scholar]
- Deli, T.; Orosz, M.; Jakab, A. Hormone Replacement Therapy in Cancer Survivors—Review of the Literature. Pathol. Oncol. Res. 2020, 26, 63–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, J.; Russo, I.H. The role of estrogen in the initiation of breast cancer. J. Steroid Biochem. Mol. Biol. 2006, 102, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Curran, E.M.; Judy, B.M.; Newton, L.G.; Lubahn, D.B.; Rottinghaus, G.E.; Macdonald, R.S.; Franklin, C.; Estes, D.M. Dietary soy phytoestrogens and ERalpha signalling modulate interferon gamma production in response to bacterial infection. Clin. Exp. Immunol. 2004, 135, 219–225. [Google Scholar] [CrossRef]
- Saleh, H.A.; Yousef, M.H.; Abdelnaser, A. The Anti-Inflammatory Properties of Phytochemicals and Their Effects on Epigenetic Mechanisms Involved in TLR4/NF-κB-Mediated Inflammation. Front. Immunol. 2021, 12, 606069. [Google Scholar] [CrossRef]
- Jefferson, W.N.; Williams, C.J. Circulating levels of genistein in the neonate, apart from dose and route, predict future adverse female reproductive outcomes. Reprod. Toxicol. 2011, 31, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.-S.; Pan, T.-M. Beneficial effects of phytoestrogens and their metabolites produced by intestinal microflora on bone health. Appl. Microbiol. Biotechnol. 2013, 97, 1489–1500. [Google Scholar] [CrossRef]
- Yellayi, S.; Zakroczymski, M.A.; Selvaraj, V.; Valli, V.E.; Ghanta, V.; Helferich, W.G.; Cooke, P.S. The phytoestrogen genistein suppresses cell-mediated immunity in mice. J. Endocrinol. 2003, 176, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Bulgaru, C.V.; Marin, D.E.; Pistol, G.C.; Taranu, I. Zearalenone and the Immune Response. Toxins 2021, 13, 248. [Google Scholar] [CrossRef] [PubMed]
- Pistol, G.C.; Gras, M.A.; Marin, D.E.; Israel-Roming, F.; Stancu, M.; Taranu, I. Natural feed contaminant zearalenone decreases the expressions of important pro- and anti-inflammatory mediators and mitogen-activated protein kinase/NF-κB signalling molecules in pigs. Br. J. Nutr. 2014, 111, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salah-Abbès, J.B.; Abbès, S.; Houas, Z.; Abdel-Wahhab, M.A.; Oueslati, R. Zearalenone induces immunotoxicity in mice: Possible protective effects of radish extract (Raphanus sativus). J. Pharm. Pharmacol. 2008, 60, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Ben Salah-Abbès, J.; Belgacem, H.; Ezzdini, K.; Abdel-Wahhab, M.A.; Abbès, S. Zearalenone nephrotoxicity: DNA fragmentation, apoptotic gene expression and oxidative stress protected by Lactobacillus plantarum MON03. Toxicon 2020, 175, 28–35. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harding, A.T.; Heaton, N.S. The Impact of Estrogens and Their Receptors on Immunity and Inflammation during Infection. Cancers 2022, 14, 909. https://doi.org/10.3390/cancers14040909
Harding AT, Heaton NS. The Impact of Estrogens and Their Receptors on Immunity and Inflammation during Infection. Cancers. 2022; 14(4):909. https://doi.org/10.3390/cancers14040909
Chicago/Turabian StyleHarding, Alfred T., and Nicholas S. Heaton. 2022. "The Impact of Estrogens and Their Receptors on Immunity and Inflammation during Infection" Cancers 14, no. 4: 909. https://doi.org/10.3390/cancers14040909