
Precision Targets for Intercepting the Lethal Progression of Prostate Cancer: Potential Avenues for Personalized Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
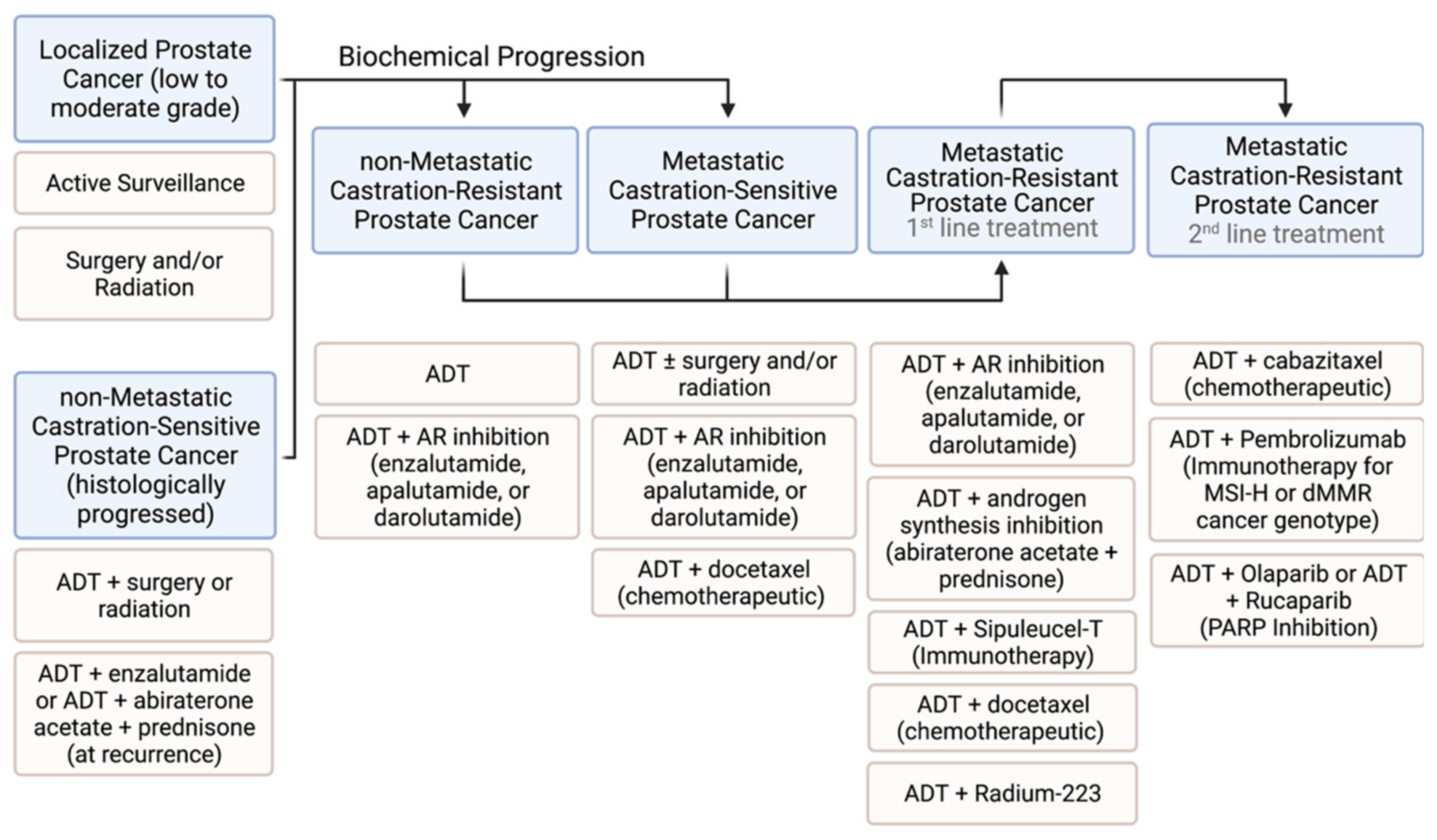
2. Current Prostate Cancer Therapies
2.1. Low-Risk and High-Risk Non-Metastatic Prostate Cancer
2.2. Targeting Metastatic Prostate Cancer
2.2.1. Hormone Therapy
2.2.2. Chemotherapy
2.2.3. PSMA-Targeted Radiation Therapy
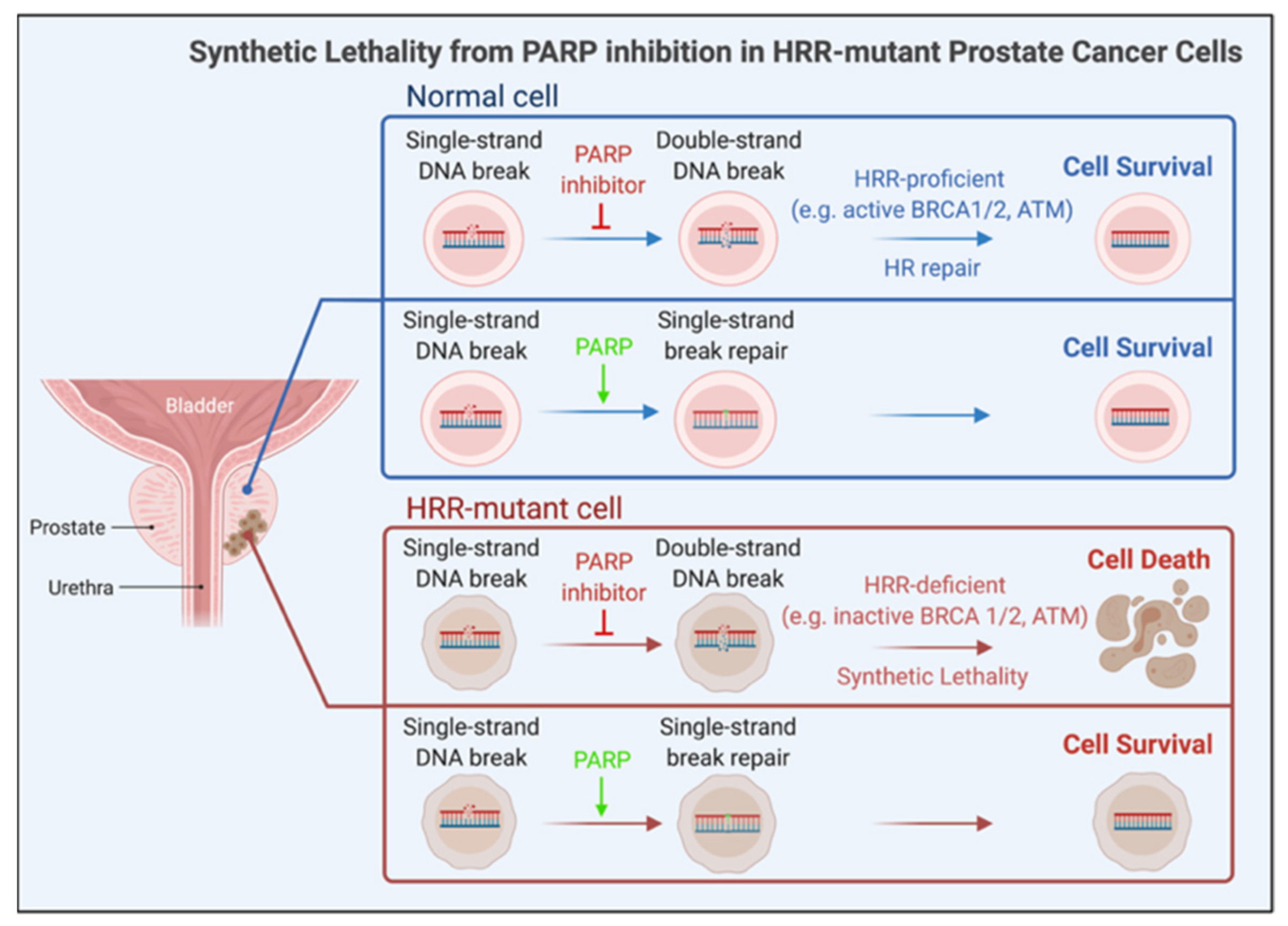
2.2.4. Genome-Targeted Precision Therapy
2.2.5. Immunotherapy
2.2.6. Summary of Approved Therapies
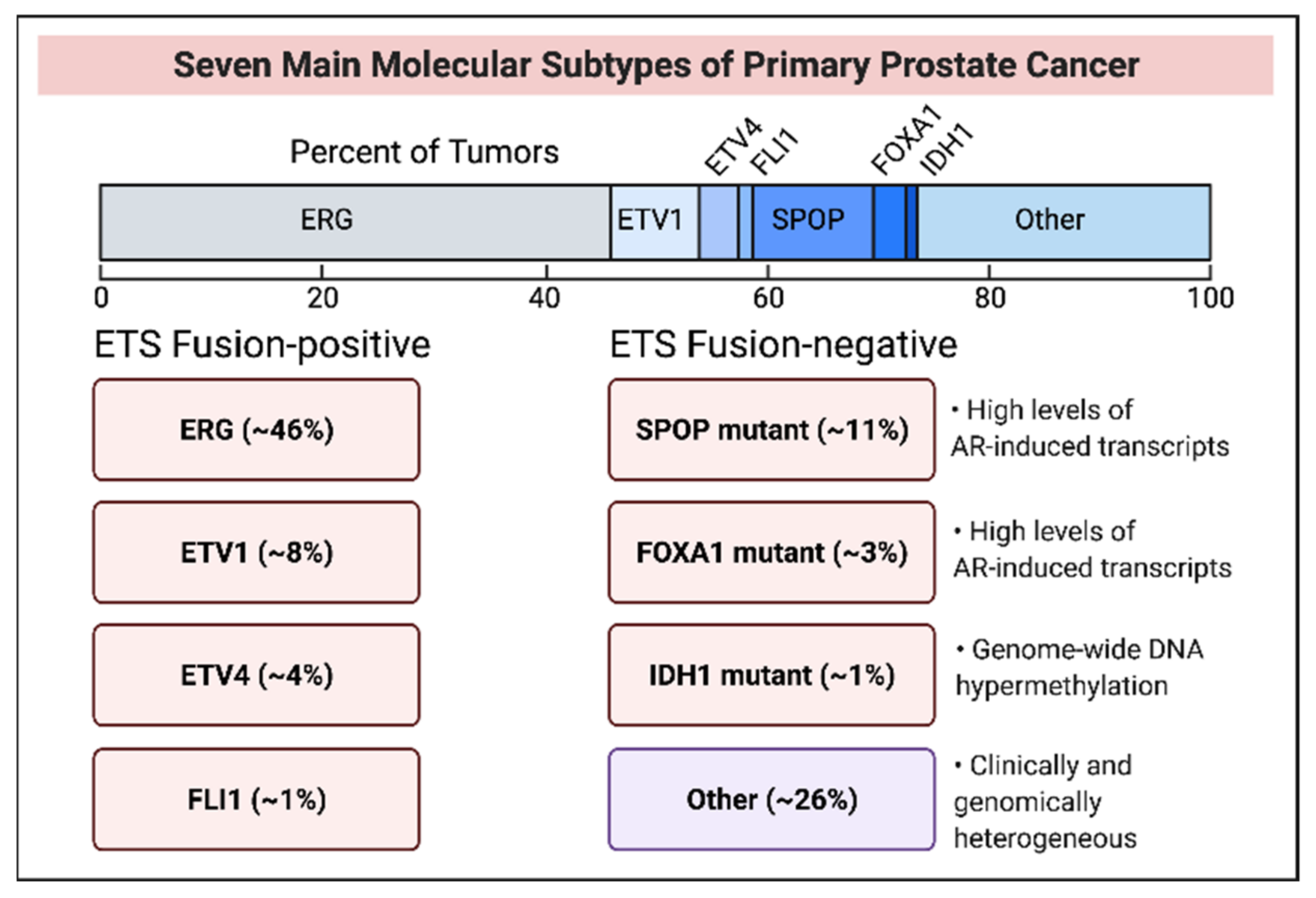
3. Molecular Subtypes of Early Stage and Late-Stage Prostate Cancer
3.1. ETS Fusion-Positive Subtypes
3.2. ETS Fusion-Negative Subtypes
4. Metastatic Castration-Resistant Prostate Cancer
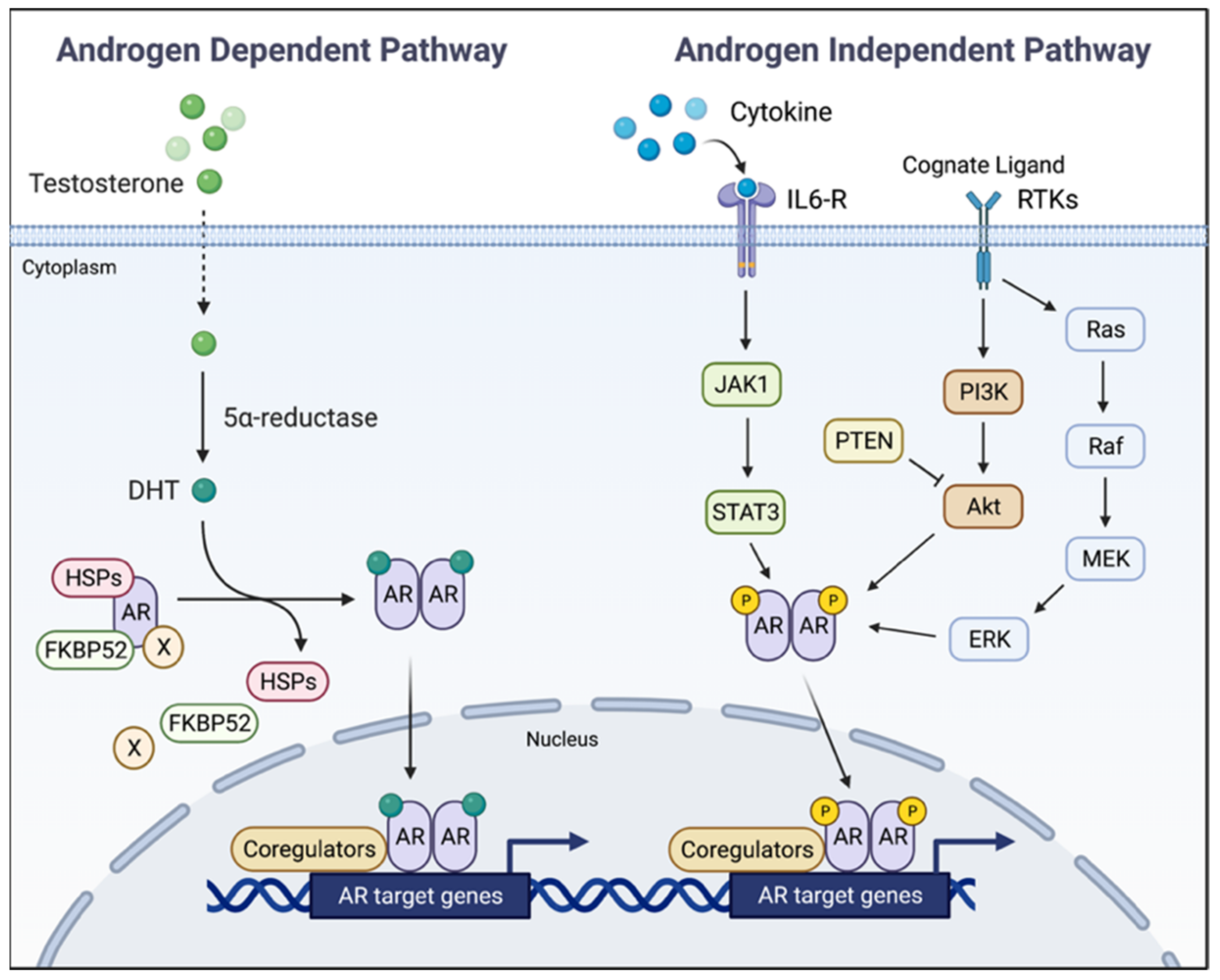
5. AR Ablation for Intercepting Advanced Prostate Cancer
6. Lineage Switch and mCRPC Progression
7. Concluding Perspectives and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Giona, S. The Epidemiology of Prostate Cancer. In Prostate Cancer; Bott, S.R.J., Ng, K.L., Eds.; Exon Publications: Brisbane, Australia, 2021; Chapter 1. [Google Scholar] [CrossRef]
- Nelson, P. Beyond the Androgen Receptor: Targeting Actionable Drivers of Prostate Cancer. JCO Precis. Oncol. 2017, 1, 1–3. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartor, O.; de Bono, J.S. Metastatic prostate cancer. N. Engl. J. Med. 2018, 378, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Mohler, J.L. A brief history of intracrine androgen metabolism by castration-recurrent prostate cancer. Am. J. Clin. Exp. Urol. 2018, 6, 101–106. [Google Scholar] [PubMed]
- Cunha, G.R.; Cao, M.; Franco, O.; Baskin, L.S. A comparison of prostatic development in xenografts of human fetal prostate and human female fetal proximal urethra grown in dihydrotestosterone-treated hosts. Differentiation 2020, 115, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, R.K.; Lavrovsky, Y.; Ahn, S.C.; Song, C.S.; Chatterjee, B.; Roy, A.K. Dynamics of Intracellular Movement and Nucleocytoplasmic Recycling of the Ligand-Activated Androgen Receptor in Living Cells. Mol. Endocrinol. 2000, 14, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Men’s Health 2019, 37, 288–295. [Google Scholar] [CrossRef]
- Brawley, S.; Mohan, R.; Nein, C.D. Localized Prostate Cancer: Treatment Options. Am. Fam. Physician 2018, 97, 798–805. [Google Scholar]
- Wilkins, L.J.; Tosoian, J.J.; Sundi, D.; Ross, A.E.; Grimberg, D.; Klein, E.A.; Chapin, B.F.; Nyame, Y.A. Surgical management of high-risk, localized prostate cancer. Nat. Rev. Urol. 2020, 17, 679–690. [Google Scholar] [CrossRef]
- Stattin, P.; Holmberg, E.; Johansson, J.-E.; Holmberg, L.; Adolfsson, J.; Hugosson, J.; National Prostate Cancer Register (NPCR) of Sweden. Outcomes in Localized Prostate Cancer: National Prostate Cancer Register of Sweden Follow-up Study. JNCI J. Natl. Cancer Inst. 2010, 102, 950–958. [Google Scholar] [CrossRef] [Green Version]
- de Sá Moreira, E.; Robinson, D.; Hawthorne, S.; Zhao, L.; Hanson, M.; Kanas, G.; Turnure, M.; Davis, C.; Clark, O. Patterns of Care and Outcomes for Non-Metastatic Prostate Cancer in the United States: Results of the CancerMPact® Survey 2018. Cancer Manag Res. 2021, 13, 9127–9137. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Murphy, L.; Clarke, N.W.; Cross, W.; Jones, R.J.; Parker, C.C.; Gillessen, S.; Cook, A.; Brawley, C.; Amos, C.L.; et al. Abiraterone acetate and prednisolone with or without enzalutamide for high-risk non-metastatic prostate cancer: A meta-analysis of primary results from two randomised controlled phase 3 trials of the STAMPEDE platform protocol. Lancet 2021, 399, 447–460. [Google Scholar] [CrossRef]
- Mateo, J.; Fizazi, K.; Gillessen, S.; Heidenreich, A.; Perez-Lopez, R.; Oyen, W.J.G.; Shore, N.; Smith, M.; Sweeney, C.; Tombal, B.; et al. Managing Nonmetastatic Castration-resistant Prostate Cancer. Eur. Urol. 2019, 75, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.R. Progress in Nonmetastatic Prostate Cancer. N. Engl. J. Med. 2018, 378, 2531–2532. [Google Scholar] [CrossRef]
- Nguyen, P.L.; Huang, H.C.; Davicioni, E.; Sandler, H.M.; Shipley, W.E.; Efstathiou, J.A.; Simko, J.; Pollack, A.; Dicker, A.; Roach, M.; et al. Validation of a 22-Gene Genomic Classifier in the NRG Oncology/RTOG 9202, 9413 and 9902 Phase III Randomized Trials: A Biopsy-Based Individual Patient Meta-Analysis in High-Risk Prostate Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, S50. [Google Scholar] [CrossRef]
- Dawson, N.A.; Zibelman, M.; Lindsay, T.; Feldman, R.A.; Saul, M.; Gatalica, Z.; Korn, W.M.; Heath, E.I. An Emerging Landscape for Canonical and Actionable Molecular Alterations in Primary and Metastatic Prostate Cancer. Mol. Cancer Ther. 2020, 19, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Cancer Discovery-Research Watch, AR Enhancer Amplification Drives Castration-Resistant Prostate Cancer. Cancer Discov. 2018, 8, OF17. [CrossRef] [Green Version]
- Takeda, D.Y.; Spisa´k, S.; Seo, J.H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szállási, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.; Haradhvala, N.J.; Freeman, S.; Reed, S.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018, 174, 433–447.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769.e9, Erratum in Cell 2018, 175, 889. [Google Scholar] [CrossRef] [Green Version]
- van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef] [Green Version]
- de Bono, J.S.; Cook, N.; Yu, E.Y.; Lara, P.N.L.; Wang, J.S.; Yamasaki, Y.; Yamamiya, I.; Gao, P.; Calleja, E.M.; Rathkopf, D.E. First-in-Human Study of TAS3681, an Oral Androgen Receptor Antagonist with AR and AR Splice Variant Downregulation Activity, in Patients with mCRPC Refractory to Abiraterone And/or Enzalutamide and Chemotherapy. J. Clin. Oncol. 2021, 39, 5031. [Google Scholar] [CrossRef]
- Fizazi, K.; Maldonado, X.; Foulon, S.; Roubaud, G.; McDermott, R.S.; Flechon, A.; Tombal, B.F.; Supiot, S.; Berthold, D.R.; Ronchin, P.; et al. A phase 3 trial with a 2x2 factorial design of abiraterone acetate plus prednisone and/or local radiotherapy in men with de novo metastatic castration-sensitive prostate cancer (mCSPC): First results of PEACE-1. J. Clin. Oncol. 2021, 39, 5000. [Google Scholar] [CrossRef]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef] [Green Version]
- Ravery, V.; Fizazi, K.; Oudard, S.; Drouet, L.; Eymard, J.-C.; Culine, S.; Gravis, G.; Hennequin, C.; Zerbib, M. The use of estramustine phosphate in the modern management of advanced prostate cancer. Br. J. Urol. 2011, 108, 1782–1786. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Le Maitre, A.; Hudes, G.; Berry, W.R.; Kelly, W.K.; Eymard, J.-C.; Logothetis, C.J.; Pignon, J.-P.; Michiels, S. Meta-analysis of Estramustine in Prostate Cancer (MECaP) Trialists’ Collaborative Group. Addition of estramustine to chemotherapy and survival of patients with castration-refractory prostate cancer: A meta-analysis of individual patient data. Lancet Oncol. 2007, 8, 994–1000. [Google Scholar] [CrossRef]
- Saylor, P.J.; Rumble, R.B.; Michalski, J.M. Bone Health and Bone-Targeted Therapies for Prostate Cancer: American Society of Clinical Oncology Endorsement Summary of a Cancer Care Ontario Guideline. JCO Oncol. Pract. 2020, 16, 389–393. [Google Scholar] [CrossRef]
- Paller, C.J.; Carducci, M.A.; Philips, G.K. Management of bone metastases in refractory prostate cancer—Role of denosumab. Clin. Interv. Aging 2012, 7, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Hegemann, M.; Bedke, J.; Stenzl, A.; Todenhöfer, T. Denosumab treatment in the management of patients with advanced prostate cancer: Clinical evidence and experience. Ther. Adv. Urol. 2017, 9, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, A.K.; Bhoopathi, P.; Talukdar, S.; Shen, X.-N.; Emdad, L.; Das, S.K.; Sarkar, D.; Fisher, P.B. Recombinant MDA-7/IL24 Suppresses Prostate Cancer Bone Metastasis through Downregulation of the Akt/Mcl-1 Pathway. Mol. Cancer Ther. 2018, 17, 1951–1960. [Google Scholar] [CrossRef] [Green Version]
- Bolomsky, A.; Vogler, M.; Köse, M.C.; Heckman, C.A.; Ehx, G.; Ludwig, H.; Caers, J. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J. Hematol. Oncol. 2020, 13, 1–19. [Google Scholar] [CrossRef]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): A multicentre, randomised, double-blind, phase 3 trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.S.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- De Giorgi, U.; Sansovini, M.; Severi, S.; Nicolini, S.; Monti, M.; Gurioli, G.; Foca, F.; Casadei, C.; Conteduca, V.; Celli, M.; et al. Circulating androgen receptor gene amplification and resistance to 177Lu-PSMA-617 in metastatic castration-resistant prostate cancer: Results of a Phase 2 trial. Br. J. Cancer 2021, 125, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Sun, M.; Sartor, A.O.; Thomas, C.; Singh, S.; Bissassar, M.; Fernandez, E.; Niaz, M.J.; Ho, B.; Vallabhajosula, S.; et al. Phase I study of 225Ac-J591 for men with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2021, 39, 5015. [Google Scholar] [CrossRef]
- Sartor, O.; Coleman, R.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; et al. Effect of radium-223 dichloride on symptomatic skeletal events in patients with castration-resistant prostate cancer and bone metastases: Results from a phase 3, double-blind, randomised trial. Lancet Oncol. 2014, 15, 738–746. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 2017, PO.17.00029. [Google Scholar] [CrossRef]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual Roles of PARP-1 Promote Cancer Growth and Progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, B.; Gui, F.; Takai, T.; Feng, C.; Bai, X.; Fazli, L.; Dong, X.; Liu, S.; Zhang, X.; Zhang, W.; et al. Selective targeting of PARP-2 inhibits androgen receptor signaling and prostate cancer growth through disruption of FOXA1 function. Proc. Natl. Acad. Sci. USA 2019, 116, 14573–14582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef] [Green Version]
- Pezaro, C. PARP inhibitor combinations in prostate cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835919897537. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor–induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, eaam7479. [Google Scholar] [CrossRef] [Green Version]
- Fay, E.K.; Graff, J.N. Immunotherapy in Prostate Cancer. Cancers 2020, 12, 1752. [Google Scholar] [CrossRef]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Ravindranathan, D.; Russler, G.A.; Yantorni, L.; Drusbosky, L.M.; Bilen, M.A. Detection of Microsatellite Instability via Circulating Tumor DNA and Response to Immunotherapy in Metastatic Castration-Resistant Prostate Cancer: A Case Series. Case Rep. Oncol. 2021, 14, 190–196. [Google Scholar] [CrossRef]
- De Almeida, D.V.P.; Fong, L.; Rettig, M.B.; Autio, K.A. Immune Checkpoint Blockade for Prostate Cancer: Niche Role or Next Breakthrough? Am. Soc. Clin. Oncol. Educ. Book 2020, e89–e106. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.-R.; Lee, J.H.; Ponnazhagan, S. Revisiting Immunotherapy: A Focus on Prostate Cancer. Cancer Res. 2020, 80, 1615–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graff, J.N.; Alumkal, J.J.; Thompson, R.; Moran, A.; Thomas, G.V.; Wood, M.A.; Drake, C.G.; Slottke, R.; Beer, T.M. Pembroluzimab (Pembro) plus enzalutamide (Enz) in metastatic castration resistant prostate cancer (mCRPC): Extended follow up. J. Clin. Oncol. 2018, 36, 5047. [Google Scholar] [CrossRef]
- Yu, E.; Piulats, J.; Gravis, G.; Fong, P.; Todenhöfer, T.; Laguerre, B.; Arranz, J.; Oudard, S.; Massard, C.; Stoeckle, M.; et al. 73P Association between homologous recombination repair mutations and response to pembrolizumab (pembro) plus olaparib (ola) in metastatic castration-resistant prostate cancer (mCRPC): KEYNOTE-365 Cohort A biomarker analysis. Ann. Oncol. 2021, 32, S387. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782.e14. [Google Scholar] [CrossRef] [Green Version]
- Powers, E.; Karachaliou, G.S.; Kao, C.; Harrison, M.R.; Hoimes, C.J.; George, D.J.; Armstrong, A.J.; Zhang, T. Novel therapies are changing treatment paradigms in metastatic prostate cancer. J. Hematol. Oncol. 2020, 13, 144. [Google Scholar] [CrossRef]
- Dulos, J.; Carven, G.J.; van Boxtel, S.J.; Evers, S.; Driessen-Engels, L.J.A.; Hobo, W.; Gorecka, M.A.; de Haan, A.F.J.; Mulders, P.; Punt, C.J.A.; et al. PD-1 Blockade Augments Th1 and Th17 and Suppresses Th2 Responses in Peripheral Blood From Patients With Prostate and Advanced Melanoma Cancer. J. Immunother. 2012, 35, 169–178. [Google Scholar] [CrossRef]
- Peng, W.; Liu, C.; Xu, C.; Lou, Y.; Chen, J.; Yang, Y.; Yagita, H.; Overwijk, W.W.; Lizée, G.; Radvanyi, L.; et al. PD-1 Blockade Enhances T-cell Migration to Tumors by Elevating IFN-γ Inducible Chemokines. Cancer Res. 2012, 72, 5209–5218. [Google Scholar] [CrossRef] [Green Version]
- Ni, L.; Lu, J. Interferon gamma in cancer immunotherapy. Cancer Med. 2018, 7, 4509–4516. [Google Scholar] [CrossRef]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, M.; Solimando, A.G.; Kalogirou, C.; Marquardt, A.; Frank, T.; Sokolakis, I.; Hatzichristodoulou, G.; Kneitz, S.; Bargou, R.; Kübler, H.; et al. miR-221-3p Regulates VEGFR2 Expression in High-Risk Prostate Cancer and Represents an Escape Mechanism from Sunitinib In Vitro. J. Clin. Med. 2020, 9, 670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelson, D.; Regan, M.M.; Oh, W.; Kaufman, D.S.; Olivier, K.; Michaelson, S.Z.; Spicer, B.; Gurski, C.; Kantoff, P.; Smith, M.R. Phase II study of sunitinib in men with advanced prostate cancer. Ann. Oncol. 2009, 20, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Halabi, S.; Healy, P.; Lee, W.R.; Koontz, B.F.; Moul, J.W.; Mundy, K.; Creel, P.; Wood, S.; Davis, K.; et al. A phase 2 multimodality trial of docetaxel/prednisone with sunitinib followed by salvage radiation therapy in men with PSA recurrent prostate cancer after radical prostatectomy. Prostate Cancer Prostatic Dis. 2016, 19, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Kneitz, B.; Krebs, M.; Kalogirou, C.; Schubert, M.; Joniau, S.; Van Poppel, H.; Lerut, E.; Kneitz, S.; Scholz, C.-J.; Ströbel, P.; et al. Survival in Patients with High-Risk Prostate Cancer Is Predicted by miR-221, Which Regulates Proliferation, Apoptosis, and Invasion of Prostate Cancer Cells by Inhibiting IRF2 and SOCS3. Cancer Res. 2014, 74, 2591–2603. [Google Scholar] [CrossRef] [Green Version]
- Perner, S.; Rupp, N.J.; Braun, M.; Rubin, M.; Moch, H.; Dietel, M.; Wernert, N.; Jung, K.; Stephan, C.; Kristiansen, G. Loss of SLC45A3 protein (prostein) expression in prostate cancer is associated withSLC45A3-ERGgene rearrangement and an unfavorable clinical course. Int. J. Cancer 2013, 132, 807–812. [Google Scholar] [CrossRef]
- Mao, N.; Zhang, Z.; Lee, Y.S.; Choi, D.; Rivera, A.A.; Li, D.; Lee, C.; Haywood, S.; Chen, X.; Chang, Q.; et al. Defining the therapeutic selective dependencies for distinct subtypes of PI3K pathway-altered prostate cancers. Nat. Commun. 2021, 12, 5053. [Google Scholar] [CrossRef]
- Wee, S.; Wiederschain, D.; Maira, S.-M.; Loo, A.; Miller, C.; Debeaumont, R.; Stegmeier, F.; Yao, Y.-M.; Lengauer, C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. USA 2008, 105, 13057–13062. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, S.; Wongvipat, J.; Trigwell, C.B.; Hancox, U.; Carver, B.S.; Rodrik-Outmezguine, V.; Will, M.; Yellen, P.; de Stanchina, E.; Baselga, J.; et al. Feedback Suppression of PI3Kα Signaling in PTEN-Mutated Tumors Is Relieved by Selective Inhibition of PI3Kβ. Cancer Cell 2014, 27, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Kaffenberger, S.D.; Barbieri, C. Molecular subtyping of prostate cancer. Curr. Opin. Urol. 2016, 26, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Geng, C.; He, B.; Xu, L.; Barbieri, C.; Eedunuri, V.K.; Chew, S.A.; Zimmermann, M.; Bond, R.; Shou, J.; Li, C.; et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc. Natl. Acad. Sci. USA 2013, 110, 6997–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boysen, G.; Barbieri, C.E.; Prandi, D.; Blattner, M.; Chae, S.-S.; Dahija, A.; Nataraj, S.; Huang, D.; Marotz, C.; Xu, L.; et al. SPOP mutation leads to genomic instability in prostate cancer. eLife 2015, 4, e09207. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Yu, J.; Varambally, S.; Mehra, R.; Perner, S.; Demichelis, F.; Helgeson, B.E.; Laxman, B.; Morris, D.S.; et al. The role of SPINK1 in ETS rearrangement-negative prostate cancers. Cancer Cell 2008, 13, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Ku, S.-Y.; Gleave, M.; Beltran, H. Towards precision oncology in advanced prostate cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Sumanasuriya, S.; De Bono, J.S. Treatment of Advanced Prostate Cancer-A Review of Current Therapies and Future Promise. Cold Spring Harb. Perspect. Med. 2018, 8, a030635. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Ludkovski, O.; DeGrace, D.; Williams, J.L.; Evans, A.; Sircar, K.; Bismar, T.A.; Nuin, P.; Squire, J.A. PTEN genomic deletions that characterize aggressive prostate cancer originate close to segmental duplications. Genes Chromosom. Cancer 2011, 51, 149–160. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Mirkheshti, N.; Park, S.; Jiang, S.; Cropper, J.; Werner, S.L.; Song, C.S.; Chatterjee, B. Dual targeting of androgen receptor and mTORC1 by salinomycin in prostate cancer. Oncotarget 2016, 7, 62240–62254. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-Y.; Yu, S.-N.; Lee, S.-Y.; Chun, S.-S.; Choi, Y.-L.; Park, Y.-M.; Song, C.S.; Chatterjee, B.; Ahn, S.-C. Salinomycin-induced apoptosis of human prostate cancer cells due to accumulated reactive oxygen species and mitochondrial membrane depolarization. Biochem. Biophys. Res. Commun. 2011, 413, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Zafeiriou, Z.; Bianchini, D.; Chandler, R.; Rescigno, P.; Yuan, W.; Carreira, S.; Barrero, M.; Petremolo, A.; Miranda, S.; Riisnaes, R.; et al. Genomic Analysis of Three Metastatic Prostate Cancer Patients with Exceptional Responses to Carboplatin Indicating Different Types of DNA Repair Deficiency. Eur. Urol. 2018, 75, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.H.; Pritchard, C.C.; Boyd, T.; Nelson, P.S.; Montgomery, B. Biallelic Inactivation of BRCA2 in Platinum-sensitive Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2015, 69, 992–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Lee, Y.; Koo, K. Current Status and Future Perspectives of Androgen Receptor Inhibition Therapy for Prostate Cancer: A Comprehensive Review. Biomolecules 2021, 11, 492. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, J.L.; Neklesa, T.K.; Cox, C.S.; Roth, A.G.; Buckley, D.L.; Tae, H.S.; Sundberg, T.B.; Stagg, D.; Hines, J.; McDonnell, D.P.; et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew. Chem. Int. Ed. 2015, 54, 9659–9662. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Tian, J.; Chou, F.; Lin, C.; Xing, E.Z.; Zuo, L.; Niu, Y.; Yeh, S.; Chang, C. Targeting the androgen receptor (AR) with AR degradation enhancer ASC-J9® led to increase docetaxel sensitivity via suppressing the p21 expression. Cancer Lett. 2018, 444, 35–44. [Google Scholar] [CrossRef]
- Wang, R.; Lin, W.; Lin, C.; Li, L.; Sun, Y.; Chang, C. ASC-J9® suppresses castration resistant prostate cancer progression via degrading the enzalutamide-induced androgen receptor mutant AR-F876L. Cancer Lett. 2016, 379, 154–160. [Google Scholar] [CrossRef]
- Omlin, A.; Jones, R.; Van Der Noll, R.; Satoh, T.; Niwakawa, M.; Smith, S.A.; Graham, J.; Ong, M.; Finkelman, R.D.; Schellens, J.H.M.; et al. AZD3514, an oral selective androgen receptor down-regulator in patients with castration-resistant prostate cancer—Results of two parallel first-in-human phase I studies. Investig. New Drugs 2015, 33, 679–690. [Google Scholar] [CrossRef]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.; Jin, M.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K.; et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. [Google Scholar] [CrossRef]
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.; et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 2020, 22, 111–119. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Gao, X.; Vogelzang, N.J.; Garfield, M.H.; Taylor, I.; Moore, M.D.; Peck, R.A.; Burris, H.A. First-in-human phase I study of ARV-110, an androgen receptor (AR) PROTAC degrader in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) following enzalutamide (ENZ) and/or abiraterone (ABI). J. Clin. Oncol. 2020, 38, 3500. [Google Scholar] [CrossRef]
- Chen, L.; Han, L.; Mao, S.; Xu, P.; Xu, X.; Zhao, R.; Wu, Z.; Zhong, K.; Yu, G.; Wang, X. Discovery of A031 as effective proteolysis targeting chimera (PROTAC) androgen receptor (AR) degrader for the treatment of prostate cancer. Eur. J. Med. Chem. 2021, 216, 113307. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; McLaughlin, B.; Lu, D.; Louw, J.; Danila, D.C.; Dugan, L.; Johnson, A.; Heller, G.; et al. Nuclear-specific AR-V7 Protein Localization is Necessary to Guide Treatment Selection in Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2017, 71, 874–882. [Google Scholar] [CrossRef] [Green Version]
- Ghildiyal, R.; Sawant, M.; Renganathan, A.; Mahajan, K.; Kim, E.H.; Luo, J.; Dang, H.X.; Maher, C.A.; Feng, F.Y.; Mahajan, N.P. Loss of Long Noncoding RNA NXTAR in Prostate Cancer Augments Androgen Receptor Expression and Enzalutamide Resistance. Cancer Res. 2021, 82, 155–168. [Google Scholar] [CrossRef]
- Kelly, K.; Balk, S.P. Reprogramming to resist. Science 2017, 355, 29–30. [Google Scholar] [CrossRef]
- Beltran, H.; Demichelis, F. Therapy considerations in neuroendocrine prostate cancer: What next? Endocrine-Related Cancer 2021, 28, T67–T78. [Google Scholar] [CrossRef]
- Labrecque, M.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; Gil Da Costa, R.M.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 Oncogenic Activity in Castration-Resistant Prostate Cancer Cells Is Polycomb-Independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [Green Version]
- Ko, S.; Ahn, J.; Song, C.S.; Kim, S.; Knapczyk-Stwora, K.; Chatterjee, B. Lysine Methylation and Functional Modulation of Androgen Receptor by Set9 Methyltransferase. Mol. Endocrinol. 2011, 25, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Peng, S.; Pilié, P.G.; Geng, C.; Park, S.; Manyam, G.C.; Lu, Y.; Yang, G.; Tang, Z.; Kondraganti, S.; et al. PARP and CDK4/6 Inhibitor Combination Therapy Induces Apoptosis and Suppresses Neuroendocrine Differentiation in Prostate Cancer. Mol. Cancer Ther. 2021, 20, 1680–1691. [Google Scholar] [CrossRef] [PubMed]
- Pernigoni, N.; Zagato, E.; Calcinotto, A.; Troiani, M.; Mestre, R.P.; Calì, B.; Attanasio, G.; Troisi, J.; Minini, M.; Mosole, S.; et al. Commensal bacteria promote endocrine resistance in prostate cancer through androgen biosynthesis. Science 2021, 374, 216–224. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christenson, M.; Song, C.-S.; Liu, Y.-G.; Chatterjee, B. Precision Targets for Intercepting the Lethal Progression of Prostate Cancer: Potential Avenues for Personalized Therapy. Cancers 2022, 14, 892. https://doi.org/10.3390/cancers14040892
Christenson M, Song C-S, Liu Y-G, Chatterjee B. Precision Targets for Intercepting the Lethal Progression of Prostate Cancer: Potential Avenues for Personalized Therapy. Cancers. 2022; 14(4):892. https://doi.org/10.3390/cancers14040892
Chicago/Turabian StyleChristenson, Max, Chung-Seog Song, Ya-Guang Liu, and Bandana Chatterjee. 2022. "Precision Targets for Intercepting the Lethal Progression of Prostate Cancer: Potential Avenues for Personalized Therapy" Cancers 14, no. 4: 892. https://doi.org/10.3390/cancers14040892