
Morphological and Molecular Characterization of KRAS G12C-Mutated Lung Adenocarcinomas

 ,
,  , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Morphological Characterization
2.3. Immunohistochemistry
2.4. Next-Generation Sequencing
2.5. Statistical Analysis
3. Results
3.1. Demographic Characteristics
3.2. Morphology and Immunohistochemistry
3.3. Next-Generation Sequencing Analysis
3.4. KRAS G12C and STK11 Co-Mutated LUADs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- WHO. Thoracic Tumours, 5th ed.; Organisation Mondiale de la Santé Centre International de Recherche sur le Cancer, Ed.; World Health Organization Classification of Tumours; International Agency for Research on Cancer: Lyon, France, 2021; ISBN 978-92-832-4506-3. [Google Scholar]
- Ferrara, M.G.; Di Noia, V.; D’Argento, E.; Vita, E.; Damiano, P.; Cannella, A.; Ribelli, M.; Pilotto, S.; Milella, M.; Tortora, G.; et al. Oncogene-Addicted Non-Small-Cell Lung Cancer: Treatment Opportunities and Future Perspectives. Cancers 2020, 12, E1196. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. EGFR Mutations and Lung Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 49–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumbrink, H.L.; Heimsoeth, A.; Sos, M.L. The next Tier of EGFR Resistance Mutations in Lung Cancer. Oncogene 2021, 40, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J.; Wu, Y.-L.; Paz-Ares, L. Lung Cancer: Current Therapies and New Targeted Treatments. The Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Mack, P.C.; Banks, K.C.; Espenschied, C.R.; Burich, R.A.; Zill, O.A.; Lee, C.E.; Riess, J.W.; Mortimer, S.A.; Talasaz, A.; Lanman, R.B.; et al. Spectrum of Driver Mutations and Clinical Impact of Circulating Tumor DNA Analysis in Non–Small Cell Lung Cancer: Analysis of over 8000 Cases. Cancer 2020, 126, 3219–3228. [Google Scholar] [CrossRef]
- Riely, G.J.; Kris, M.G.; Rosenbaum, D.; Marks, J.; Li, A.; Chitale, D.A.; Nafa, K.; Riedel, E.R.; Hsu, M.; Pao, W.; et al. Frequency and Distinctive Spectrum of KRAS Mutations in Never Smokers with Lung Adenocarcinoma. Clin. Cancer Res. 2008, 14, 5731–5734. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-Occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [Green Version]
- Nagasaka, M.; Li, Y.; Sukari, A.; Ou, S.-H.I.; Al-Hallak, M.N.; Azmi, A.S. KRAS G12C Game of Thrones, Which Direct KRAS Inhibitor Will Claim the Iron Throne? Cancer Treat. Rev. 2020, 84, 101974. [Google Scholar] [CrossRef]
- Biernacka, A.; Tsongalis, P.D.; Peterson, J.D.; de Abreu, F.B.; Black, C.C.; Gutmann, E.J.; Liu, X.; Tafe, L.J.; Amos, C.I.; Tsongalis, G.J. The Potential Utility of Re-Mining Results of Somatic Mutation Testing: KRAS Status in Lung Adenocarcinoma. Cancer Genet. 2016, 209, 195–198. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Rybkin, I.I.; Spira, A.I.; Riely, G.J.; Papadopoulos, K.P.; Sabari, J.K.; Johnson, M.L.; Heist, R.S.; Bazhenova, L.; Barve, M.; et al. KRYSTAL-1: Activity and Safety of Adagrasib (MRTX849) in Advanced/Metastatic Non–Small-Cell Lung Cancer (NSCLC) Harboring KRAS G12C Mutation. Eur. J. Cancer 2020, 138, S1–S2. [Google Scholar] [CrossRef]
- Mirati Therapeutics. Mirati Therapeutics’ Adagrasib Receives Breakthrough Therapy Designation from u.s. Food and Drug Administration for Patients with Advanced Non-Small Cell Lung Cancer Harboring the KRAS G12C Mutation 2021. News release. Available online: http://www.prnewswire.com/news-releases/mirati-therapeutics-adagrasib-receives-breakthrough-therapy-designation-from-us-food-and-drug-administration-for-patients-with-advanced-non-small-cell-lung-cancer-harboring-the-kras-g12c-mutation-301319824.html (accessed on 24 June 2021).
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Dafni, U.; Tsourti, Z.; Vervita, K.; Peters, S. Immune Checkpoint Inhibitors, Alone or in Combination with Chemotherapy, as First-Line Treatment for Advanced Non-Small Cell Lung Cancer. A Systematic Review and Network Meta-Analysis. Lung Cancer Amst. Neth. 2019, 134, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Svaton, M.; Fiala, O.; Pesek, M.; Bortlicek, Z.; Minarik, M.; Benesova, L.; Topolcan, O. The Prognostic Role of KRAS Mutation in Patients with Advanced NSCLC Treated with Second- or Third-Line Chemotherapy. Anticancer Res. 2016, 36, 1077–1082. [Google Scholar] [PubMed]
- Blair, H.A. Sotorasib: First Approval. Drugs 2021, 81, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Muzumdar, M.D.; Chen, P.-Y.; Dorans, K.J.; Chung, K.M.; Bhutkar, A.; Hong, E.; Noll, E.M.; Sprick, M.R.; Trumpp, A.; Jacks, T. Survival of Pancreatic Cancer Cells Lacking KRAS Function. Nat. Commun. 2017, 8, 1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Z.-F.; Ma, P.C. Emerging Insights of Tumor Heterogeneity and Drug Resistance Mechanisms in Lung Cancer Targeted Therapy. J. Hematol. Oncol.J Hematol Oncol 2019, 12, 134. [Google Scholar] [CrossRef] [Green Version]
- Gutiontov, S.I.; Turchan, W.T.; Spurr, L.F.; Rouhani, S.J.; Chervin, C.S.; Steinhardt, G.; Lager, A.M.; Wanjari, P.; Malik, R.; Connell, P.P.; et al. CDKN2A Loss-of-Function Predicts Immunotherapy Resistance in Non-Small Cell Lung Cancer. Sci. Rep. 2021, 11, 20059. [Google Scholar] [CrossRef]
- Xu, C.; Buczkowski, K.A.; Zhang, Y.; Asahina, H.; Beauchamp, E.M.; Terai, H.; Li, Y.Y.; Meyerson, M.; Wong, K.; Hammerman, P.S. NSCLC Driven by DDR2 Mutation Is Sensitive to Dasatinib and JQ1 Combination Therapy. Mol. Cancer Ther. 2015, 14, 2382–2389. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Yonesaka, K.; Teramura, T.; Takehara, T.; Kato, R.; Sakai, H.; Haratani, K.; Tanizaki, J.; Kawakami, H.; Hayashi, H.; et al. KRAS Inhibitor-Resistance in MET-Amplified KRAS G12C Non-Small Cell Lung Cancer Induced by RAS- and Non-RAS-Mediated Cell Signaling Mechanisms. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Bordi, P.; Tiseo, M.; Rofi, E.; Petrini, I.; Restante, G.; Danesi, R.; Del Re, M. Detection of ALK and KRAS Mutations in Circulating Tumor DNA of Patients With Advanced ALK-Positive NSCLC With Disease Progression During Crizotinib Treatment. Clin. Lung Cancer 2017, 18, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.; Gautschi, O.; Rothschild, S.; Mark, M.; Froesch, P.; Klingbiel, D.; Reichegger, H.; Jochum, W.; Diebold, J.; Früh, M. Clinical Outcome of ALK-Positive Non-Small Cell Lung Cancer (NSCLC) Patients with De Novo EGFR or KRAS Co-Mutations Receiving Tyrosine Kinase Inhibitors (TKIs). J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2017, 12, 681–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRASG12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Martín Martorell, P.; Huerta, M.; Compañ Quilis, A.; Abellán, R.; Seda, E.; Blesa, S.; Chaves, F.J.; Dualde Beltrán, D.; Roselló Keränen, S.; Franco, J.; et al. Coexistence of EGFR, KRAS, BRAF, and PIK3CA Mutations and ALK Rearrangement in a Comprehensive Cohort of 326 Consecutive Spanish Nonsquamous NSCLC Patients. Clin. Lung Cancer 2017, 18, e395–e402. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Murray, C.W.; Brady, J.J.; Tsai, M.K.; Li, C.; Winters, I.P.; Tang, R.; Andrejka, L.; Ma, R.K.; Kunder, C.A.; Chu, P.; et al. An LKB1-SIK Axis Suppresses Lung Tumor Growth and Controls Differentiation. Cancer Discov. 2019, 9, 1590–1605. [Google Scholar] [CrossRef] [Green Version]
- Wohlhieter, C.A.; Richards, A.L.; Uddin, F.; Hulton, C.H.; Quintanal-Villalonga, À.; Martin, A.; de Stanchina, E.; Bhanot, U.; Asher, M.; Shah, N.S.; et al. Concurrent Mutations in STK11 and KEAP1 Promote Ferroptosis Protection and SCD1 Dependence in Lung Cancer. Cell Rep. 2020, 33, 108444. [Google Scholar] [CrossRef]
- Pavan, A.; Bragadin, A.B.; Calvetti, L.; Ferro, A.; Zulato, E.; Attili, I.; Nardo, G.; Dal Maso, A.; Frega, S.; Menin, A.G.; et al. Role of next Generation Sequencing-Based Liquid Biopsy in Advanced Non-Small Cell Lung Cancer Patients Treated with Immune Checkpoint Inhibitors: Impact of STK11, KRAS and TP53 Mutations and Co-Mutations on Outcome. Transl. Lung Cancer Res. 2021, 10, 202–220. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Rizvi, H.; Plodkowski, A.J.; Hellmann, M.D.; Knezevic, A.; Heller, G.; Yu, H.A.; Ladanyi, M.; Kris, M.G.; Arcila, M.E.; et al. Treatment Outcomes and Clinical Characteristics of Patients with KRAS-G12C–Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 2209–2215. [Google Scholar] [CrossRef] [PubMed]
- Briere, D.M.; Li, S.; Calinisan, A.; Sudhakar, N.; Aranda, R.; Hargis, L.; Peng, D.H.; Deng, J.; Engstrom, L.D.; Hallin, J.; et al. The KRASG12C Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Sensitizes Tumors to Checkpoint Inhibitor Therapy. Mol. Cancer Ther. 2021, 20, 975–985. [Google Scholar] [CrossRef]
- Köhler, J.; Jänne, P.A. If Virchow and Ehrlich Had Dreamt Together: What the Future Holds for KRAS-Mutant Lung Cancer. Int. J. Mol. Sci. 2021, 22, 3025. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Variable/Molecular Subtype | Total Cases | KRAS G12C-STK11 Co-Mutated LUADs | KRAS G12C-nonstk11 Mutated LUADs | p-Value |
|---|---|---|---|---|
| (n = 202 Cases) | (n = 51 Cases) | (n = 151 Cases) | ||
| Histology | 0.3263 | |||
| acinar | 60 (29.7%) | 20 (39.3%) | 40 (26.5%) | |
| solid | 36 (17.8) | 6 (11.8%) | 30 (19.9%) | |
| lepidic | 15 (7.5%) | 4 (7.8%) | 11 (7.3%) | |
| mucinous | 7 (3.4%) | 2 (3.9%) | 5 (3.3%) | |
| poorly differentiated | 8 (3.9%) | 0 (0%) | 8 (5.3%) | |
| material of cytologic value | 26 (12.9%) | 8 (15.7%) | 18 (11.9%) | |
| NOS * | 50 (24.8%) | 11 (21.5%) | 39 (25.8%) | |
| Tumor cellularity | 0.8789 | |||
| >50% | 40 (19.8%) | 9 (17.6%) | 31 (20.6%) | |
| 25–50% | 55 (27.2%) | 14 (27.5%) | 41 (27.2%) | |
| 15–25% | 83 (41.1%) | 23 (45.1%) | 60 (39.7%) | |
| 5–15% | 24 (11.9%) | 5 (9.8%) | 19 (12.5%) | |
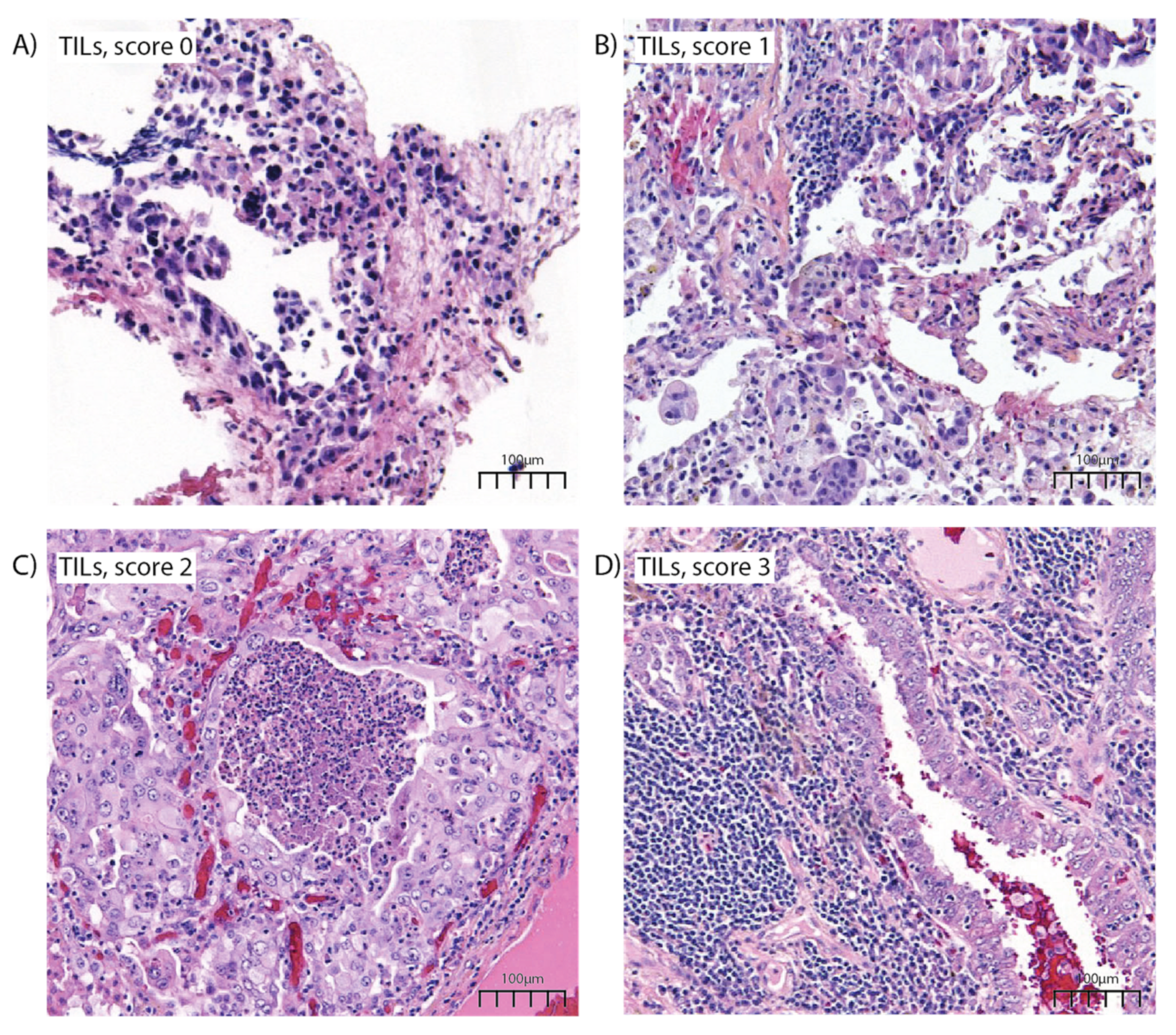
| TILs | 0.921 | |||
| 3+ | 76 (37.7%) | 20 (39.2%) | 56 (37.1%) | |
| 2+ | 58 (28.7%) | 14 (27.5%) | 44 (29.1%) | |
| 1+ | 37 (18.3%) | 8 (15.7%) | 29 (19.2%) | |
| 0+ | 31 (15.3%) | 9 (17.6%) | 22 (14.6%) | |
| Intratumor necrosis | 0.1222 | |||
| Present | 66 (32.3%) | 12 (23.5%) | 54 (35.8%) | |
| Absent | 136 (67.7%) | 39 (76.5%) | 97 (64.2%) | |
| Nuclear atypia | 0.7206 | |||
| Present | 144 (71.2%) | 35 (68.6%) | 109 (72.2%) | |
| Absent | 58 (28.8%) | 16 (31.4%) | 42 (27.8%) | |
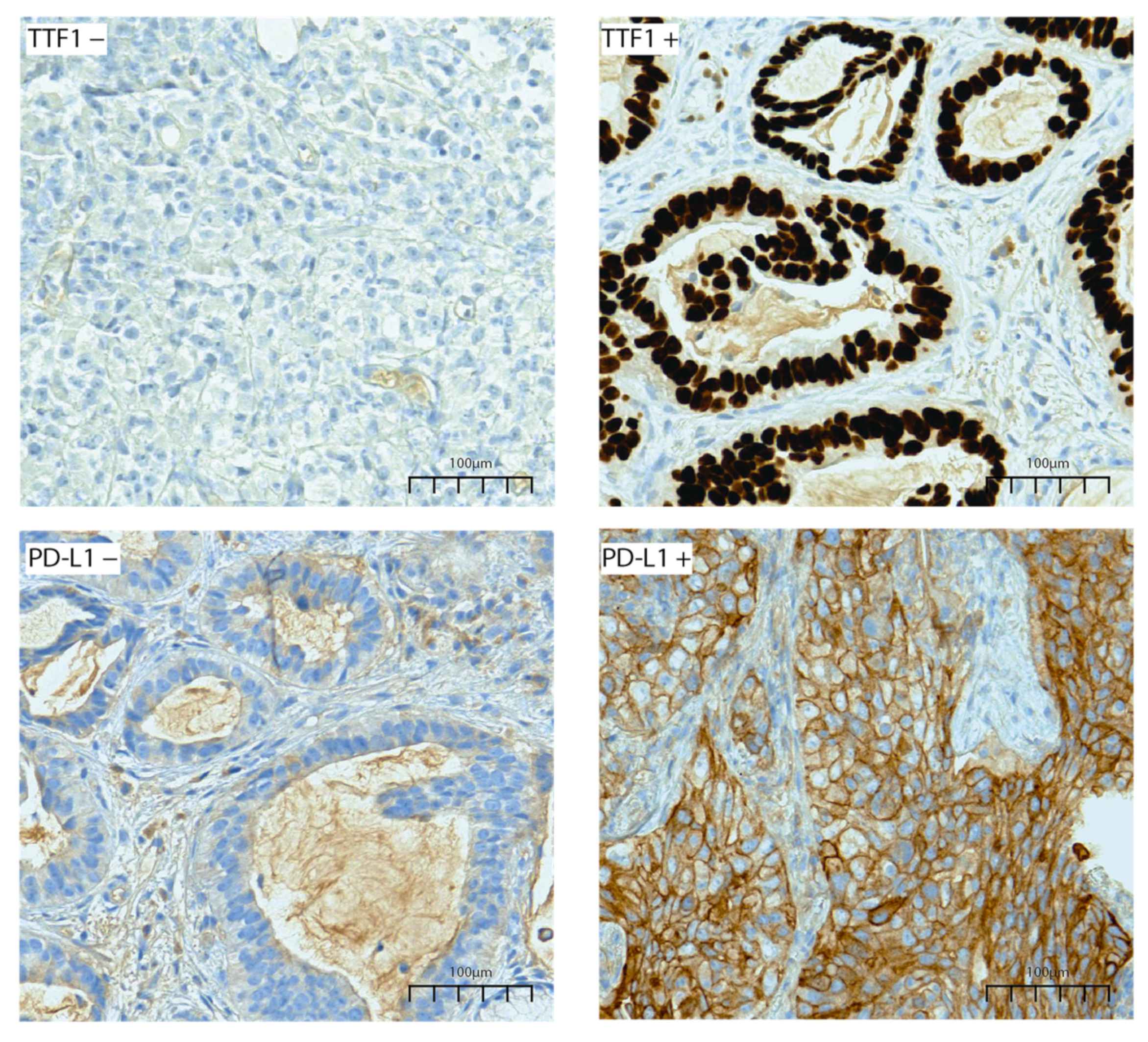
| TTF1 IHC | 0.0092 | |||
| Positive | 159 (78.7%) | 33 (64.7%) | 126 (83.4%) | |
| Negative | 43 (21.3%) | 18 (35.3%) | 25 (16.6%) | |
| PD-L1 IHC | <0.0001 | |||
| Positive | 89 (44.1%) | 10 (19.6%) | 79 (52.3%) | |
| Negative | 113 (55.9%) | 41 (80.4%) | 72 (47.7%) |
| Mutated Gene | Cases, n (%) | Mutated Gene | Cases, n (%) |
|---|---|---|---|
| ALK | 4 (1.9%) | IDH2 | 1 (0.5%) |
| BRAF | 1 (0.5%) | KIT | 2 (1%) |
| CDK2NA | 15 (7.5%) | KRAS * | 4 (2%) |
| CTNNB1 | 7 (2.5%) | MAPK | 2 (1%) |
| DDR2 | 9 (4.5%) | MET | 4(2%) |
| FGFR2 | 1 (0.5%) | NRAS | 2 (1%) |
| FGFR3 | 3 (1.5%) | PIK3CA | 15 (7.4%) |
| H3F3A | 2 (1%) | PTEN | 5 (2.4%) |
| HIST1H3B | 4 (2%) | STK11 | 51 (25.2%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirlog, R.; Piton, N.; Lamy, A.; Guisier, F.; Berindan-Neagoe, I.; Sabourin, J.-C.; Marguet, F. Morphological and Molecular Characterization of KRAS G12C-Mutated Lung Adenocarcinomas. Cancers 2022, 14, 1030. https://doi.org/10.3390/cancers14041030
Pirlog R, Piton N, Lamy A, Guisier F, Berindan-Neagoe I, Sabourin J-C, Marguet F. Morphological and Molecular Characterization of KRAS G12C-Mutated Lung Adenocarcinomas. Cancers. 2022; 14(4):1030. https://doi.org/10.3390/cancers14041030
Chicago/Turabian StylePirlog, Radu, Nicolas Piton, Aude Lamy, Florian Guisier, Ioana Berindan-Neagoe, Jean-Christophe Sabourin, and Florent Marguet. 2022. "Morphological and Molecular Characterization of KRAS G12C-Mutated Lung Adenocarcinomas" Cancers 14, no. 4: 1030. https://doi.org/10.3390/cancers14041030