
Novel Repositioning Therapy for Drug-Resistant Glioblastoma: In Vivo Validation Study of Clindamycin Treatment Targeting the mTOR Pathway and Combination Therapy with Temozolomide

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Cell Viability Assays
2.4. Western Blotting
2.5. In Vitro mTOR Kinase Assays
2.6. Cell Cycle Analysis
2.7. Experimental Animals
2.8. Establishment of a Patient-Derived Xenograft Model
2.9. Histopathological Examination
2.10. Statistical Analyses
3. Results
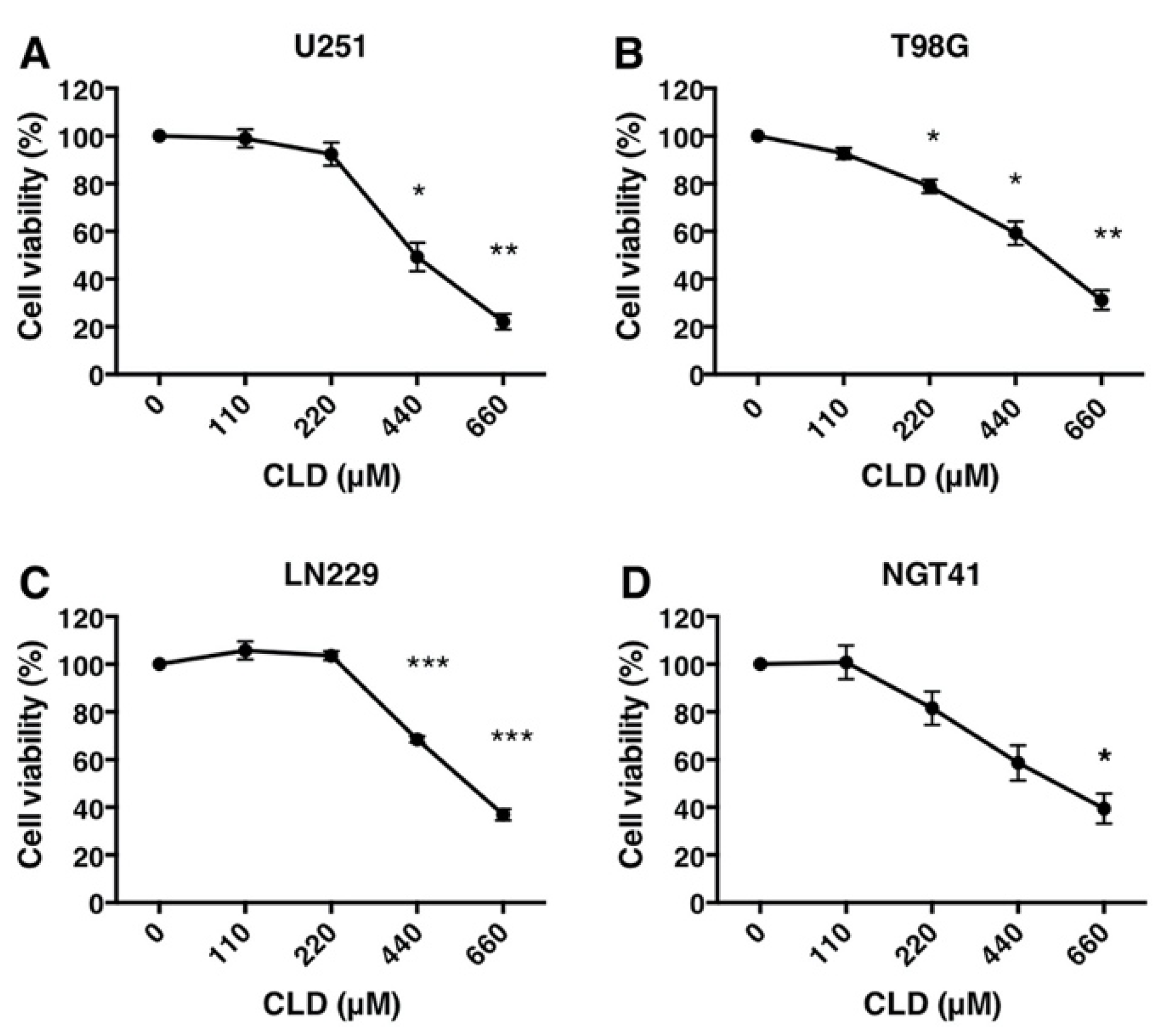
3.1. Antiproliferative Activity of Clindamycin in Cultured Glioblastoma Cells
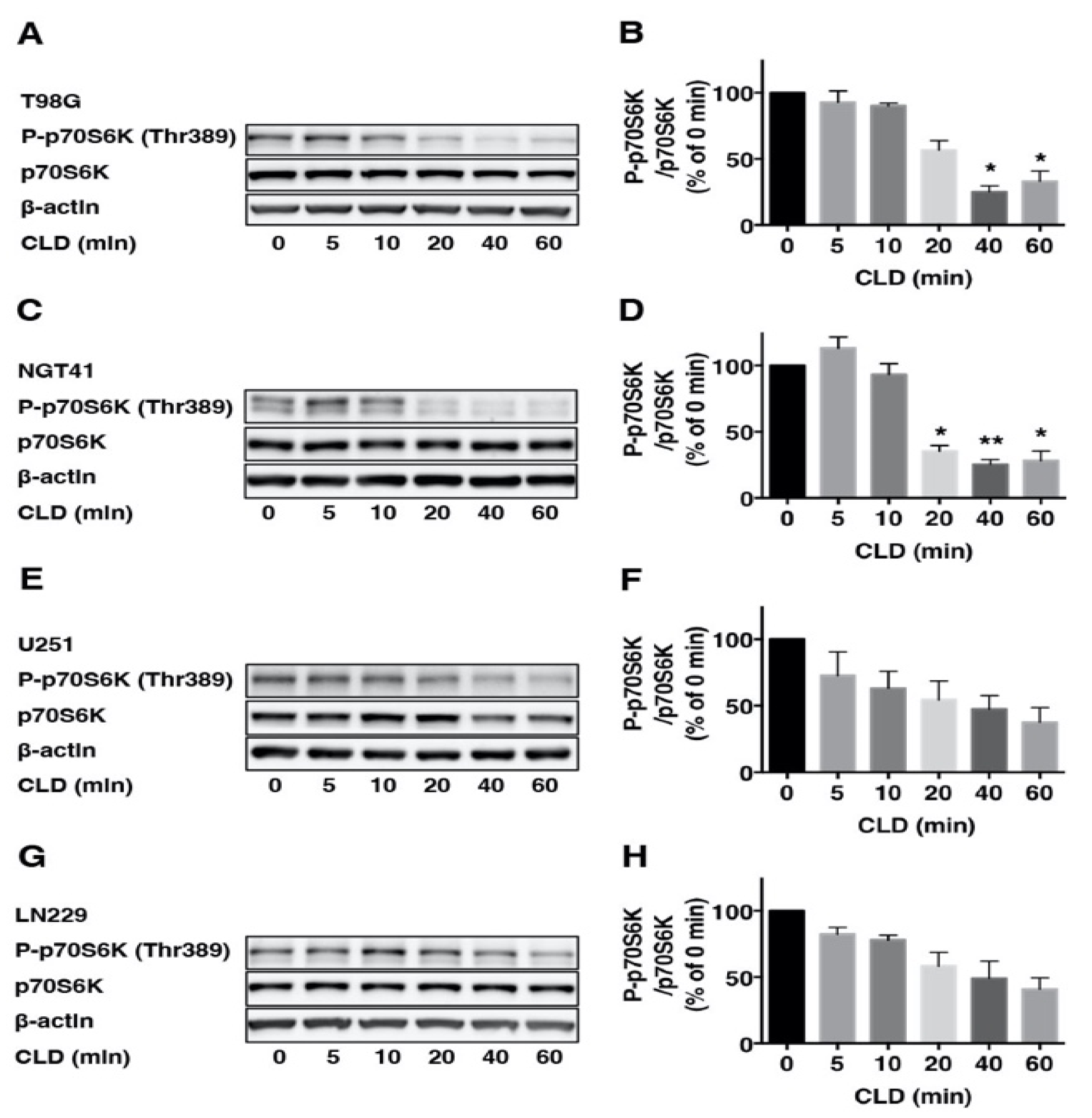
3.2. Effects of Clindamycin on mTOR Signaling in Glioblastoma Cell Lines
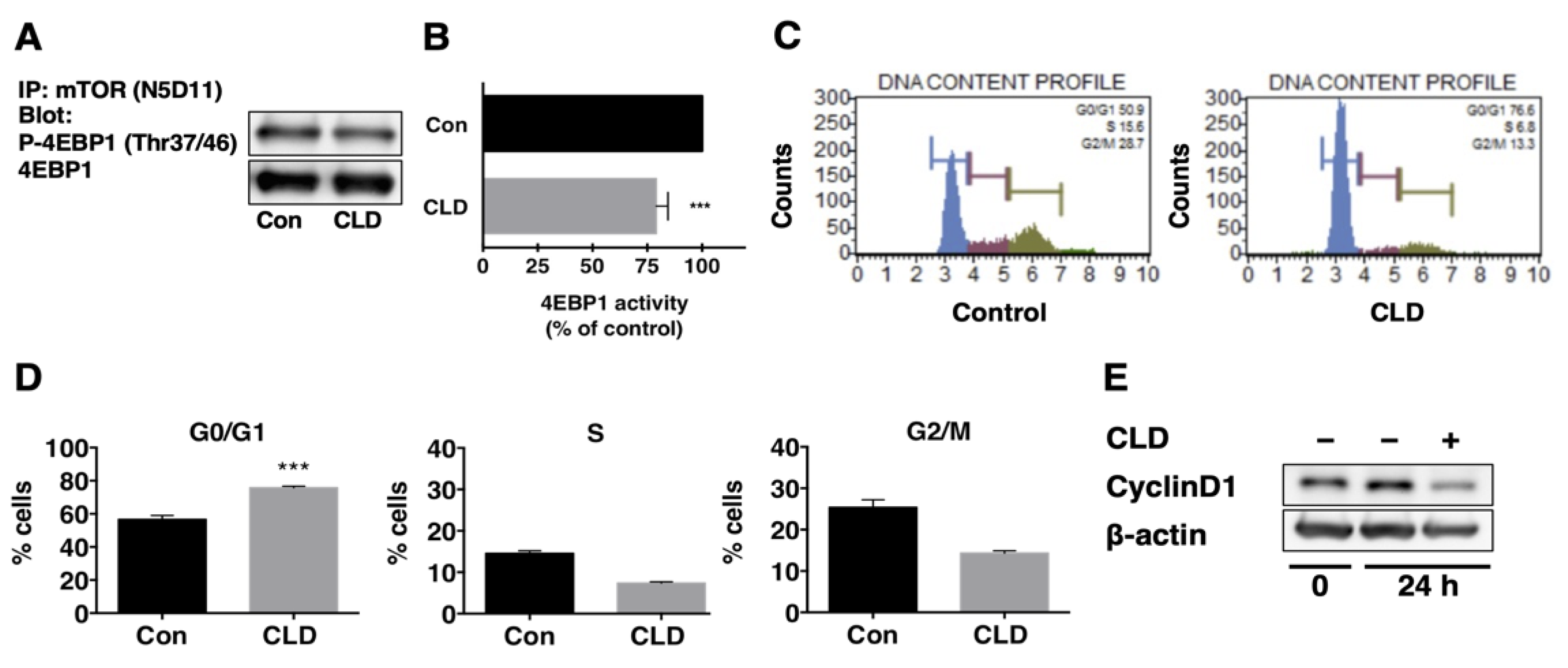
3.3. Direct Effect of Clindamycin on mTOR Kinase Activity
3.4. Effect of Clindamycin on Cell Cycle Progression in GBM Cells
3.5. Effects of Clindamycin and Temozolomide Combined Treatment on Glioma Cell Viability
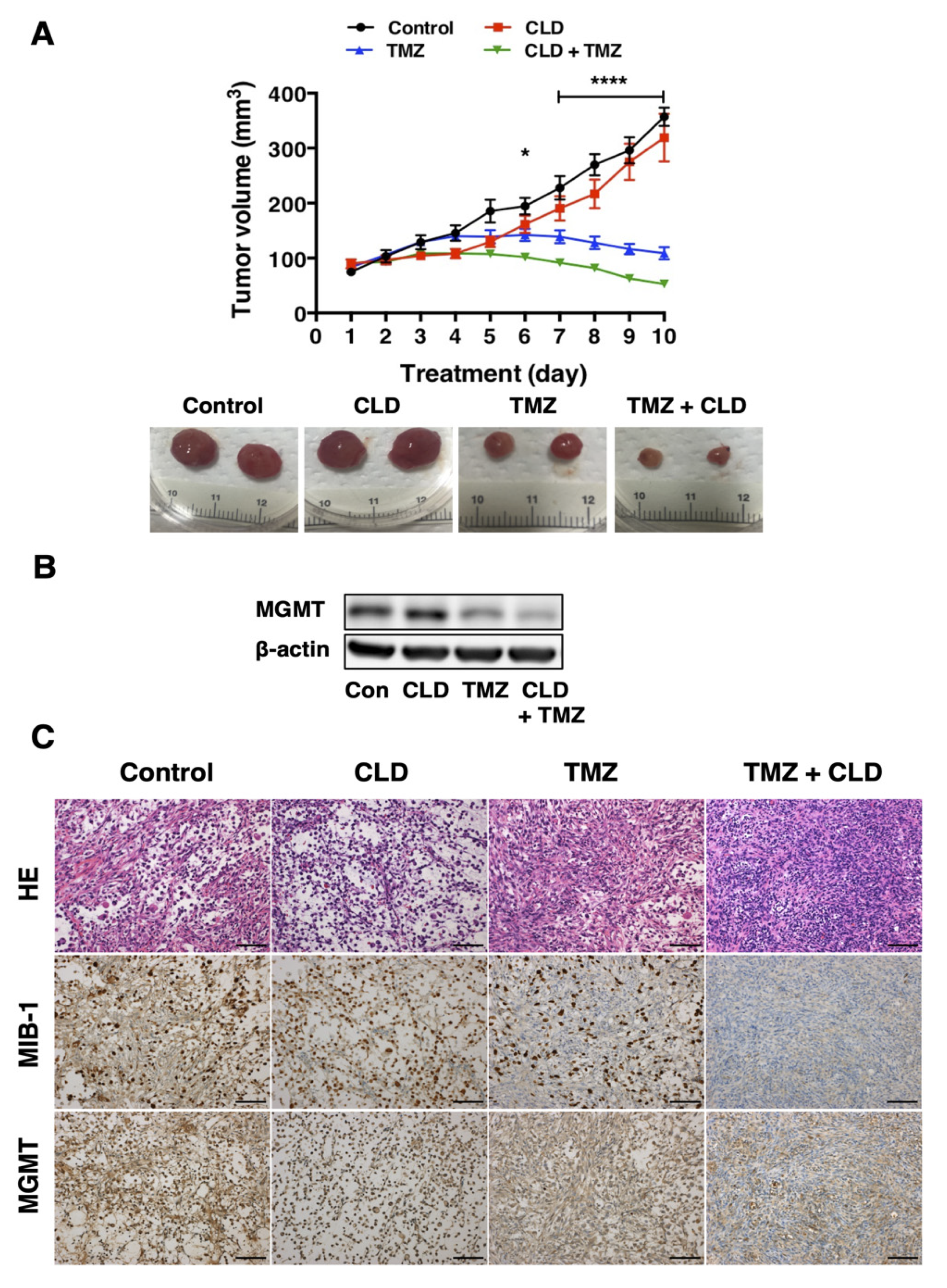
3.6. Synergistic Effects of Clindamycin and Temozolomide in a Subcutaneous Tumor Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; Nicolas, T.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, R.; Ji, X.D.; Harmon, R.C.; Lazar, C.S.; Gill, G.N.; Cavenee, W.K.; Huang, H.J. A mutant epidermal growth factor receptor common in human gliomaconfersenhancedtumorigenicity. Proc. Natl. Acad. Sci. USA 1994, 91, 7727–7731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanson, M.; Marie, Y.; Paris, S.; Idbaih, A.; Laffaire, J.; Ducray, F.; El Hallani, S.; Boisselier, B.; Mokhtari, K.; Hoang-Xuan, K.; et al. Isocitrate Dehydrogenase 1 Codon 132 Mutation Is an Important Prognostic Biomarker in Gliomas. J. Clin. Oncol. 2009, 27, 4150–4154. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.; Maiorka, P.; et al. Genetic Pathways to Glioblastoma. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Knobbe, C.B.; Reifenberger, G. Genetic Alterations and Aberrant Expression of Genes Related to the Phosphatidyl-Inositol-3′-Kinase/Protein Kinase B (Akt) Signal TransductionPathway in Glioblastomas. Brain Pathol. 2003, 13, 507–518. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Zarogoulidis, P.; Papanas, N.; Kioumis, I.; Chatzaki, E.; Maltezos, E.; Zarogoulidis, K. Macrolides: From in vitro anti-inflammatory and immunomodulatory properties to clinical practice in respiratory diseases. Eur. J. Clin. Pharmacol. 2012, 68, 479–503. [Google Scholar] [CrossRef] [PubMed]
- Kanoh, S.; Rubin, B. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin. Microbiol. Rev. 2010, 23, 590–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinkai, M.; Henke, M.O.; Rubin, B.K. Macrolide antibiotics as immunomodulatory medications: Proposed mechanisms of action. Pharmacol. Ther. 2008, 117, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, S.; Azuma, A.; Yamamoto, M.; Izumi, T.; Ando, M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am. J. Respir. Crit. Care Med. 1998, 157, 1829–1832. [Google Scholar] [CrossRef]
- Ratzinger, F.; Haslacher, H.; Poeppl, W.; Hoermann, G.; Kovarik, J.J.; Jutz, S.; Steinberger, P.; Burgmann, H.; Pickl, W.F.; Schmetterer, K.G. Azithromycin suppresses CD4(+) T-cell activation by direct modulation of mTOR activity. Sci. Rep. 2014, 4, 7438. [Google Scholar] [CrossRef]
- Akhavan, D.; Cloughesy, T.F.; Mischel, P.S. mTOR signaling in glioblastoma: Lessons learned from bench to bedside. Neuro Oncol. 2010, 12, 882–889. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Kanemaru, Y.; Natsumeda, M.; Okada, M.; Saito, R.; Kobayashi, D.; Eda, T.; Watanabe, J.; Saito, S.; Tsukamoto, Y.; Oishi, M.; et al. Dramatic response of BRAF V600E-mutant epithelioid glioblastoma to combination therapy with BRAF and MEK inhibitor: Establishment and xenograft of a cell line to predict clinical efficacy. Acta Neuropathol. Commun. 2019, 7, 119. [Google Scholar] [CrossRef]
- Aoki, H.; Takada, Y.; Kondo, S.; Sawaya, R.; Aggarwal, B.B.; Kondo, Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: Role of Akt and extracellular signal-regulated kinase signaling pathways. Mol. Pharmacol. 2007, 72, 29–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiuma, T.; Hara, K.; Tsujishita, Y.; Kaneko, K.; Shii, K.; Yonezawa, K. Characterization of the Phosphoproteins and Protein Kinase Activity in mTOR Immunoprecipitates. Biochem. Biophys. Res. Commun. 1998, 252, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Kawamura, M.; Hara, K.; Yonezawa, K.; Nawa, H. Brain-derived neurotrophic factor enhances neuronal translation by activating multiple initiation processes: Comparison with the effects of insulin. J. Biol. Chem. 2001, 276, 42818–42825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsumeda, M.; Maitani, K.; Liu, Y.; Miyahara, H.; Kaur, H.; Chu, Q.; Zhang, H.; Kahlert, U.D.; Eberhart, C.G. Targeting Notch Signaling and Autophagy Increases Cytotoxicity in Glioblastoma Neurospheres. Brain Pathol. 2016, 26, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, Y.; Ohtsu, N.; Echizenya, S.; Otsuguro, S.; Ogura, R.; Natsumeda, M.; Isogawa, M.; Aoki, H.; Ichikawa, S.; Sakaitani, M.; et al. Chemical Screening Identifies EUrd as a Novel Inhibitor Against Temozolomide-Resistant Glioblastoma-Initiating Cells. Stem Cells 2016, 34, 2016–2025. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Kawagoe, Y.; Sato, Y.; Nozumi, M.; Ishikawa, Y.; Tamada, A.; Yamazaki, H.; Sekino, Y.; Kanemura, Y.; Shinmyo, Y.; et al. Phosphorylation of GAP-43 T172 is a molecular marker of growing axons in a wide range of mammals including primates. Mol. Brain 2021, 14, 66. [Google Scholar] [CrossRef]
- Ogura, R.; Tsukamoto, Y.; Natsumeda, M.; Isogawa, M.; Aoki, H.; Kobayashi, T.; Yoshida, S.; Okamoto, K.; Takahashi, H.; Fujii, Y.; et al. Immunohistochemical profiles of IDH1, MGMT and P53: Practical significance for prognostication of patients with diffuse gliomas. Neuropathology 2015, 35, 324–335. [Google Scholar] [CrossRef]
- Abedalthagafi, M.; Bi, W.L.; Aizer, A.A.; Merrill, P.H.; Brewster, R.; Agarwalla, P.K.; Listewnik, M.L.; Dias-Santagata, D.; Thorner, A.R.; Van Hummelen, P.; et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro Oncol. 2016, 18, 649–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyahara, H.; Yadavilli, S.; Natsumeda, M.; Rubens, J.A.; Rodgers, L.; Kambhampati, M.; Taylor, I.C.; Kaur, H.; Asnaghi, L.; Eberhart, C.G.; et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett. 2017, 400, 110–116. [Google Scholar] [CrossRef]
- Gini, B.; Zanca, C.; Guo, D.; Matsutani, T.; Masui, K.; Ikegami, S.; Yang, H.; Nathanson, D.; Villa, G.R.; Shackelford, D.; et al. The mTOR kinase inhibitors, CC214-1 and CC214-2, preferentially block the growth of EGFRvIII-activated glioblastomas. Clin. Cancer Res. 2013, 19, 5722–5732. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Yonezawa, K.; Weng, Q.P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 1998, 273, 14484–14494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, N.; Nawa, H. mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, C.; Grummt, I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006, 25, 6384–6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005, 65, 3336–3346. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Liu, T.J.; Koul, D.; LaFortune, T.; Tiao, N.; Shen, R.J.; Maira, S.M.; Garcia-Echevrria, C.; Yung, W.K. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol. Cancer Ther. 2009, 8, 2204–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, G.; Sottero, T.; Yang, X.; Mueller, S.; James, C.D.; Weiss, W.A.; Polley, M.Y.; Ozawa, T.; Berger, M.S.; Aftab, D.T.; et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro Oncol. 2011, 13, 384–392. [Google Scholar] [CrossRef] [Green Version]
- von Achenbach, C.; Weller, M.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Fabbro, D.; Reifenberger, G.; Szabo, E. Synergistic growth inhibition mediated by dual PI3K/mTOR pathway targeting and genetic or direct pharmacological AKT inhibition in human glioblastoma models. J. Neurochem. 2020, 153, 510–524. [Google Scholar] [CrossRef]
- Pegg, A.E. Repair of O6-alkylguanine by alkyltransferases. Mutat. Res. 2000, 462, 83–100. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, T.; Natsume, A.; Mizusawa, J.; Katayama, H.; Fukuda, H.; Sumi, M.; Nishikawa, R.; Narita, Y.; Muragaki, Y.; Maruyama, T.; et al. JCOG0911 INTEGRA study: A randomized screening phase II trial of interferonbeta plus temozolomide in comparison with temozolomide alone for newly diagnosed glioblastoma. J. Neurooncol. 2018, 138, 627–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spriggs, K.A.; Bushell, M.; Willis, A.E. Translational regulation of gene expression during conditions of cell stress. Mol. Cell 2010, 40, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Sievert, A.J.; Lang, S.S.; Boucher, K.L.; Madsen, P.J.; Slaunwhite, E.; Choudhari, N.; Kellet, M.; Storm, P.B.; Resnick, A.C. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc. Natl. Acad. Sci. USA 2013, 110, 8750. [Google Scholar] [CrossRef] [Green Version]
- Arnold, A.; Yuan, M.; Price, A.; Harris, L.; Eberhart, C.G.; Raabe, E.H. Synergistic activity of mTORC1/2 kinase and MEK inhibitors suppresses pediatric low-grade glioma tumorigenicity and vascularity. Neuro Oncol. 2020, 22, 563–574. [Google Scholar] [CrossRef]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Imaoka, M.; Uzuka, Y.; Noguchi, Y.; Watanabe, K.; Matsumoto, K. Basic and clinical studies on clindamycin for intravenous injection. Jpn. J. Antibiot. 1977, 30, 51–58. [Google Scholar]
- Hirata, N.; Hiramatsu, K.; Kishi, K.; Yamasaki, T.; Ichimiya, T.; Nasu, M. Pretreatment of mice with clindamycin improves survival of endotoxic shock by modulating the release of inflammatory cytokines. Antimicrob. Agents Chemother. 2001, 45, 2638–2642. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, J.; Siu, I.M.; Thomale, U.W.; Jallo, G.I. The effects of temozolomide delivered by prolonged intracerebral microinfusion against the rat brainstem GBM allograft model. Childs Nerv. Syst. 2012, 28, 707–713. [Google Scholar] [CrossRef]
- Saito, R.; Sonoda, Y.; Kumabe, T.; Nagamatsu, K.; Watanabe, M.; Tominaga, T. Regression of recurrent glioblastoma infiltrating the brainstem after convection-enhanced delivery of nimustine hydrochloride. J. Neurosurg. Pediatr. 2011, 7, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Ozawa, T.; Gryaznov, S.M.; Bollen, A.W.; Lamborn, K.R.; Frey, W.H., II; Deen, D.F. New therapeutic approach for brain tumors: Intranasal delivery of telomerase inhibitor GRN163. Neuro Oncol. 2008, 10, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, N.; Liu, S.; He, X.; Drummond, D.C.; Noble, C.O.; Goldman, S.; Mueller, S.; Bankiewicz, K.; Gupta, N.; Hashizume, R. New therapeutic approaches for brainstem tumors: A comparison of delivery routes using nanoliposomal irinotecan in an animal model. J. Neurooncol. 2018, 136, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.J.; Bisht, S.; Bar, E.E.; Maitra, A.; Eberhart, C.G. A polymeric nanoparticle formulation of curcumin inhibits growth, clonogenicity and stem-like fraction in malignant brain tumors. Cancer Biol. Ther. 2011, 11, 464–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, K.C.; Chu, P.C.; Wang, H.Y.; Huang, C.Y.; Chen, P.Y.; Tsai, H.C.; Lu, Y.J.; Lee, P.Y.; Tseng, I.C.; Feng, L.Y.; et al. Focused ultrasound-induced blood-brain barrier opening to enhance temozolomide delivery for glioblastoma treatment: A preclinical study. PLoS ONE 2013, 8, e58995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Dong, Y.; Furuta, T.; Sabit, H.; Kitabayashi, T.; Jiapaer, S.; Kobayashi, M.; Ino, Y.; Todo, T.; Teng, L.; Hirao, A.; et al. Identification of antipsychotic drug fluspirilene as a potential anti-glioma stem cell drug. Oncotarget 2017, 8, 111728–111741. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef]
- Pfab, C.; Schnobrich, L.; Eldnasoury, S.; Gessner, A.; El-Najjar, N. Repurposing of Antimicrobial Agents for Cancer Therapy: What Do We Know? Cancers 2021, 13, 3193. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eda, T.; Okada, M.; Ogura, R.; Tsukamoto, Y.; Kanemaru, Y.; Watanabe, J.; On, J.; Aoki, H.; Oishi, M.; Takei, N.; et al. Novel Repositioning Therapy for Drug-Resistant Glioblastoma: In Vivo Validation Study of Clindamycin Treatment Targeting the mTOR Pathway and Combination Therapy with Temozolomide. Cancers 2022, 14, 770. https://doi.org/10.3390/cancers14030770
Eda T, Okada M, Ogura R, Tsukamoto Y, Kanemaru Y, Watanabe J, On J, Aoki H, Oishi M, Takei N, et al. Novel Repositioning Therapy for Drug-Resistant Glioblastoma: In Vivo Validation Study of Clindamycin Treatment Targeting the mTOR Pathway and Combination Therapy with Temozolomide. Cancers. 2022; 14(3):770. https://doi.org/10.3390/cancers14030770
Chicago/Turabian StyleEda, Takeyoshi, Masayasu Okada, Ryosuke Ogura, Yoshihiro Tsukamoto, Yu Kanemaru, Jun Watanabe, Jotaro On, Hiroshi Aoki, Makoto Oishi, Nobuyuki Takei, and et al. 2022. "Novel Repositioning Therapy for Drug-Resistant Glioblastoma: In Vivo Validation Study of Clindamycin Treatment Targeting the mTOR Pathway and Combination Therapy with Temozolomide" Cancers 14, no. 3: 770. https://doi.org/10.3390/cancers14030770