
Hormonal Therapy for Gynecological Cancers: How Far Has Science Progressed toward Clinical Applications?

 , ,
, ,  , ,
, ,  , ,
, ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Pathogenesis of Gynecological Cancers: Hormonal Landscape
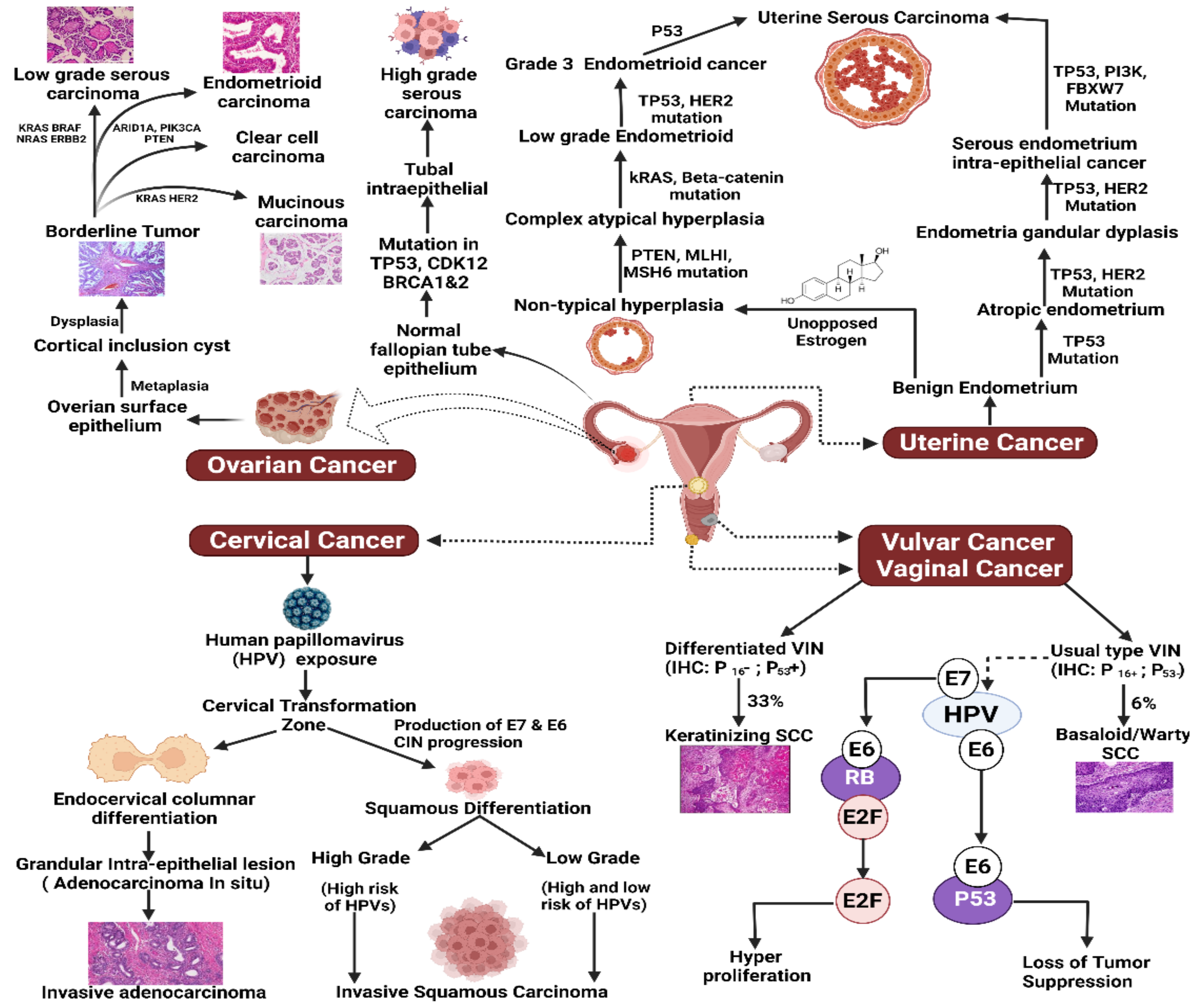
2.1. Cervical Cancer
2.2. Ovarian Cancer
2.3. Uterine and Endometrial Cancer
2.4. Vaginal Cancer
2.5. Vulvar Cancer
3. Hormonal Therapy for Gynecological Cancers
3.1. Hormonal Therapy for Cervical Cancer
3.2. Hormonal Therapy for Ovarian Cancer
3.3. Hormonal Therapy for Uterine Cancer
3.3.1. Progestins
3.3.2. GnRHa
3.3.3. Aromatase Inhibitors
3.3.4. SERMs
3.4. Hormonal Therapy for Vaginal Cancer
3.5. Hormonal Therapy for Vulvar Cancer
4. Risk Factors Associated with HRT in Gynecological Cancers
5. Concluding Remarks and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Salvo, G.; Odetto, D.; Pareja, R.; Frumovitz, M.; Ramirez, P.T. Revised 2018 International Federation of Gynecology and Obstetrics (FIGO) cervical cancer staging: A review of gaps and questions that remain. Int. J. Gynecol. Cancer 2020, 30, 873–878. [Google Scholar] [CrossRef]
- Ratner, E.S.; Foran, K.A.; Schwartz, P.E.; Minkin, M.J. Sexuality and intimacy after gynecological cancer. Maturitas 2010, 66, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Canavan, T.P.; Doshi, N.R. Cervical cancer. Am. Fam. Physician 2000, 61, 1369–1376. [Google Scholar]
- Ali, F.; Kuelker, R.; Wassie, B. Understanding cervical cancer in the context of developing countries. Ann. Trop. Med. Public Health 2012, 5, 3–15. [Google Scholar] [CrossRef]
- Chung, S.H. Targeting female hormone receptors as cervical cancer therapy. Trends Endocrinol. Metab. 2015, 26, 399–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tummala, M.K.; McGuire, W.P. Recurrent ovarian cancer. Clin. Adv. Hematol. Oncol. 2005, 3, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Eisenhauer, E.L.; Salani, R.; Copeland, L.J. Epithelial Ovarian Cancer. Clin. Gynecol. Oncol. Eighth Ed. 2012, 285–328. [Google Scholar] [CrossRef]
- Lukanova, A.; Lundin, E.; Toniolo, P.; Micheli, A.; Akhmedkhanov, A.; Rinaldi, S.; Muti, P.; Lenner, P.; Biessy, C.; Krogh, V.; et al. Circulating levels of insulin-like growth factor-I and risk of ovarian cancer. Int. J. Cancer 2002, 101, 549–554. [Google Scholar] [CrossRef]
- Nainu, F.; Masyita, A.; Bahar, M.A.; Raihan, M.; Prova, S.R.; Mitra, S.; Emran, T.B.; Simal- Gandara, J. Pharmaceutical Prospects of Bee Products: Special Focus on Anticancer and Antimicrobial Properties. Antibiotics 2021, 10, 822. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Ghoncheh, M.; Pakzad, R.; Hasanpour, H.; Salehiniya, H. Incidence and mortality of uterine cancer and relationship with Human Development Index in the world. Cukurova Med. J. 2017, 42, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Agrawal, G.; Mittal, M.; Mishra, P. Management of Vaginal Cancer. Rev. Recent Clin. Trials 2015, 10, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Kalliala, I.; Athanasiou, A.; Veroniki, A.A.; Salanti, G.; Efthimiou, O.; Raftis, N.; Bowden, S.; Paraskevaidi, M.; Aro, K.; Arbyn, M.; et al. Incidence and mortality from cervical cancer and other malignancies after treatment of cervical intraepithelial neoplasia: A systematic review and meta-analysis of the literature. Ann. Oncol. 2020, 31, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouwer, A.F.W.; Arts, H.J.; van der Velden, J.; de Hullu, J.A. Limiting the morbidity of inguinofemoral lymphadenectomy in vulvar cancer patients; a review. Expert Rev. Anticancer Ther. 2017, 17, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Guidozzi, F. Estrogen therapy in gynecological cancer survivors. Climacteric 2013, 16, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, P.; Asselin, E. Resistance to chemotherapy and hormone therapy in endometrial cancer. Endocr. Relat. Cancer 2009, 16, 363–380. [Google Scholar] [CrossRef]
- Ibeanu, O.A. Molecular pathogenesis of cervical cancer. Cancer Biol. Ther. 2011, 11, 295–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, U.; Khandia, R.; Singhal, S.; Puranik, N.; Tripathi, M.; Pateriya, A.K.; Khan, R.; Emran, T.B.; Dhama, K.; Munjal, A.; et al. Insight into Codon Utilization Pattern of Tumor Suppressor Gene EPB41L3 from Different Mammalian Species Indicates Dominant Role of Selection Force. Cancer 2021, 13, 2739. [Google Scholar] [CrossRef]
- Austin, R.M.; Zhao, C. Type 1 and type 2 cervical carcinomas: Some cervical cancers are more difficult to prevent with screening. Cytopathology 2012, 23, 6–12. [Google Scholar] [CrossRef]
- Bulk, S.; Visser, O.; Rozendaal, L.; Verheijen, R.H.M.; Meijer, C.J.L.M. Cervical cancer in the Netherlands 1989-1998: Decrease of squamous cell carcinoma in older women, increase of adenocarcinoma in younger women. Int. J. Cancer 2005, 113, 1005–1009. [Google Scholar] [CrossRef]
- Pater, A.; Bayatpour, M.; Pater, M.M. Oncogenic transformation by human papillomavirus type 16 deoxyribonucleic acid in the presence of progesterone or progestins from oral contraceptives. Am. J. Obstet. Gynecol. 1990, 162, 1099–1103. [Google Scholar] [CrossRef]
- Chan, W.K.; Klock, G.; Bernard, H.U. Progesterone and glucocorticoid response elements occur in the long control regions of several human papillomaviruses involved in anogenital neoplasia. J. Virol. 1989, 63, 3261–3269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.; Hotz, M.; Gissmann, L. Increased prevalence of human papillomaviruses in the lower genital tract of pregnant women. Int. J. Cancer 1987, 40, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Monsonego, J.; Magdelenat, H.; Catalan, F.; Coscas, Y.; Zerat, L.; Sastre, X. Estrogen and progesterone receptors in cervical human papillomavirus related lesions. Int. J. Cancer 1991, 48, 533–539. [Google Scholar] [CrossRef]
- Ibeanu, O.; Modesitt, S.C.; Ducie, J.; Von Gruenigen, V.; Agueh, M.; Fader, A.N. Hormone replacement therapy in gynecologic cancer survivors: Why not? Gynecol. Oncol. 2011, 122, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B.; Grodstein, F.; Hennekens, C.H.; Colditz, G.A.; Johnson, M.; Manson, J.A.E.; Rosner, B.; Stampfer, M.J. Age at natural menopause and risk of cardiovascular disease. Arch. Intern. Med. 1999, 159, 1061–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, B.K.; Heuch, I.; Kvåle, G. Age at natural menopause and all-cause mortality: A 37-year follow-up of 19,731 Norwegian women. Am. J. Epidemiol. 2003, 157, 923–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.H.; Franceschi, S.; Lambert, P.F. Estrogen and ERα: Culprits in cervical cancer? Trends Endocrinol. Metab. 2010, 21, 504–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, Y.A.; Son, J.; Mehta, F.F.; Demayo, F.J.; Lydon, J.P.; Chung, S.H. Progesterone signaling inhibits cervical carcinogenesis in mice. Am. J. Pathol. 2013, 183, 1679–1687. [Google Scholar] [CrossRef] [Green Version]
- Kaku, T.; Ogawa, S.; Kawano, Y.; Ohishi, Y.; Kobayashi, H.; Hirakawa, T.; Nakano, H. Histological classification of ovarian cancer. Med. Electron Microsc. 2003, 36, 9–17. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.M. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Boussios, S.; Zarkavelis, G.; Seraj, E.; Zerdes, I.; Tatsi, K.; Pentheroudakis, G. Non-epithelial ovarian cancer: Elucidating uncommon gynaecological malignancies. Anticancer Res. 2016, 36, 5031–5042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, I.M.; Davidson, B. Pathogenesis of ovarian cancer: Clues from selected overexpressed genes. Futur. Oncol. 2009, 5, 1641–1657. [Google Scholar] [CrossRef] [Green Version]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Charkhchi, P.; Cybulski, C.; Gronwald, J.; Wong, F.O.; Narod, S.A.; Akbari, M.R. Ca125 and ovarian cancer: A comprehensive review. Cancers 2020, 12, 3730. [Google Scholar] [CrossRef] [PubMed]
- Hough, C.D.; Sherman-Baust, C.A.; Pizer, E.S.; Montz, F.J.; Im, D.D.; Rosenshein, N.B.; Cho, K.R.; Riggins, G.J.; Morin, P.J. Large-scale serial analysis of gene expression reveals genes differentially expressed in ovarian cancer. Cancer Res. 2000, 60, 6281–6287. [Google Scholar] [PubMed]
- Kurman, R.J.; Shih, I.M. Pathogenesis of ovarian cancer: Lessons from morphology and molecular biology and their clinical implications. Int. J. Gynecol. Pathol. 2008, 27, 151–160. [Google Scholar] [CrossRef] [Green Version]
- McGrail, D.J.; Kieu, Q.M.N.; Dawson, M.R. The malignancy of metastatic ovarian cancer cells is increased on soft matrices through a mechanosensitive Rho-ROCK pathway. J. Cell Sci. 2014, 127, 2621–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, M.F.; Sahai, E. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 2009, 26, 273–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.S.; Yeung, T.L.; Yip, K.P.; Pradeep, S.; Balasubramanian, L.; Liu, J.; Wong, K.; Mangala, L.S.; Armaiz-Pena, G.N.; Lopez-Berestein, G.; et al. Calcium-dependent FAK/CREB/TNNC1 signalling mediates the effect of stromal MFAP5 on ovarian cancer metastatic potential. Nat Commun 2014, 5, 5092. [Google Scholar] [CrossRef]
- Yeung, T.L.; Leung, C.S.; Wong, K.K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-β Modulates ovarian cancer invasion by upregulating CAF-Derived versican in the tumor microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef] [Green Version]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Yeung, C.L.A.; Wong, S.T.C.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A review in the theme: Cell and molecular processes in cancer metastasis. Am. J. Physiol.-Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [Green Version]
- Chesang, J.J. Pathogenesis of ovarian cancer: Current perspectives. East Afr. Med. J. 2017, 94, 561–574. [Google Scholar]
- Simpkins, F.; Garcia-Soto, A.; Slingerland, J. New insights on the role of hormonal therapy in ovarian cancer. Steroids 2013, 78, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Kavanagh, J.J.; Hu, W.; Liao, Q.; Fu, S. Hormonal therapy in ovarian cancer. Int. J. Gynecol. Cancer 2007, 17, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Eva, T.A.; Barua, N.; Chowdhury, M.M.; Yeasmin, S.; Rakib, A.; Islam, M.R.; Emran, T.B.; Simal-Gandara, J. Perspectives on signaling for biological- and processed food-related advanced glycation end-products and its role in cancer progression. Crit. Rev. Food Sci. Nutr. 2020, 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.S. Molecular carcinogenesis of endometrial cancer. Taiwan. J. Obstet. Gynecol. 2007, 46, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Sivridis, E.; Giatromanolaki, A. The pathogenesis of endometrial carcinomas at menopause: Facts and figures. J. Clin. Pathol. 2011, 64, 553–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgami, T.; Kato, K. Pathogenesis of endometrial cancer. Curr. Approaches Endometrial Cancer 2014, 2014, 18–32. [Google Scholar] [CrossRef]
- Kurra, V.; Krajewski, K.M.; Jagannathan, J.; Giardino, A.; Berlin, S.; Ramaiya, N. Typical and atypical metastatic sites of recurrent endometrial carcinoma. Cancer Imaging 2013, 13, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Thaker, P.H.; Sood, A.K. Molecular Oncology in Gynecologic Cancer. Compr. Gynecol. 2013, 2013, 623–633. [Google Scholar] [CrossRef]
- Colombo, N.; Preti, E.; Landoni, F.; Carinelli, S.; Colombo, A.; Marini, C.; Sessa, C. Endometrial cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24, vi33–vi38. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.S. The emerging role of speckle-type POZ protein (SPOP) in cancer development. Drug Discov. Today 2014, 19, 1498–1502. [Google Scholar] [CrossRef] [Green Version]
- Boyle, P.; Levin, B. International Agency for Research on Cancer, World Cancer Report; WHO: Geneva, Switzerland, 2014; p. 630. [Google Scholar]
- Colombo, N.; Preti, E.; Landoni, F.; Carinelli, S.; Colombo, A.; Marini, C.; Sessa, C. Endometrial cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2011, 22, vi35–vi39. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Dong, M.; Zhang, K.; Gao, C.; Guo, F.; Wang, Y.; Xue, F. Hormonal therapy in uterine sarcomas. Cancer Med. 2019, 8, 1339–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.J.; Chapman-Davis, E. Role of progesterone in endometrial cancer. Semin. Reprod. Med. 2010, 28, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Sherertz, T. Vaginal cancer. In Handbook of Evidence-Based Radiation Oncology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 693–705. [Google Scholar] [CrossRef]
- Slaughter, D.P.; Southwick, H.W.; Smejkal, W. “Field cancerization” in oral stratified squamous epithelium. Clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]
- Koyamatsu, Y.; Yokoyama, M.; Nakao, Y.; Fukuda, K.; Saito, T.; Matsukuma, K.; Iwasaka, T. A comparative analysis of human papillomavirus types 16 and 18 and expression of p53 gene and Ki-67 in cervical, vaginal, and vulvar carcinomas. Gynecol. Oncol. 2003, 90, 547–551. [Google Scholar] [CrossRef]
- Kouvaris, J.R.; Plataniotis, G.A.; Sykiotis, C.A.; Dapolla, V.J.; Vlahos, L.J. Dermal metastasis from vaginal squamous cell carcinoma. Br. J. Dermatol. 1999, 141, 579–580. [Google Scholar] [CrossRef]
- Forsberg, J.G. Estrogen, vaginal cancer, and vaginal development. Am. J. Obstet. Gynecol. 1972, 113, 83–87. [Google Scholar] [CrossRef]
- Atypical vaginal adenosis and cervical ectropion. Association with clear cell adenocarcinoma in diethylstilbestrol-exposed offspring. Cancer 1984, 54, 869–875. [Google Scholar] [CrossRef]
- Deli, T.; Orosz, M.; Jakab, A. Hormone Replacement Therapy in Cancer Survivors—Review of the Literature. Pathol. Oncol. Res. 2020, 26, 63–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Avoort, I.A.M.; Shirango, H.; Hoevenaars, B.M.; Grefte, J.M.M.; De Hullu, J.A.; De Wilde, P.C.M.; Bulten, J.; Melchers, W.J.G.; Massuger, L.F.A.G. Vulvar squamous cell carcinoma is a multifactorial disease following two separate and independent pathways. Int. J. Gynecol. Pathol. 2006, 25, 22–29. [Google Scholar] [CrossRef]
- Post, M.D. Differentiated vulvar intraepithelial neoplasia contains Tp53 mutations and is genetically linked to vulvar squamous cell carcinoma. Yearb. Pathol. Lab. Med. 2011, 2011, 85–86. [Google Scholar] [CrossRef]
- Mirghani, H.; Amen, F.; Tao, Y.; Deutsch, E.; Levy, A. Increased radiosensitivity of HPV-positive head and neck cancers: Molecular basis and therapeutic perspectives. Cancer Treat. Rev. 2015, 41, 844–852. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Melo Maia, B.; Fontes, A.M.; Lavorato-Rocha, A.M.; Rodrigues, I.S.A.; De Brot, L.; Baiocchi, G.; Stiepcich, M.M.; Soares, F.A.; Rocha, R.M. EGFR expression in vulvar cancer: Clinical implications and tumor heterogeneity. Hum. Pathol. 2014, 45, 917–925. [Google Scholar] [CrossRef]
- Woelber, L.; Hess, S.; Bohlken, H.; Tennstedt, P.; Eulenburg, C.; Simon, R.; Gieseking, F.; Jaenicke, F.; Mahner, S.; Choschzick, M. EGFR gene copy number increase in vulvar carcinomas is linked with poor clinical outcome. J. Clin. Pathol. 2012, 65, 133–139. [Google Scholar] [CrossRef]
- Hicklin, D.J.; Ellis, L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef]
- Dhakal, H.P.; Nesland, J.M.; Førsund, M.; Trope, C.G.; Holm, R. Primary Tumor Vascularity, HIF-1α and VEGF expression in vulvar squamous cell carcinomas: Their relationships with clinicopathological characteristics and prognostic impact. BMC Cancer 2013, 13, 506. [Google Scholar] [CrossRef] [Green Version]
- Obermair, A.; Kohlberger, P.; Bancher-Todesca, D.; Tempfer, C.; Sliutz, G.; Leodolter, S.; Reinthaller, A.; Kainz, C.; Breitenecker, G.; Gitsch, G. Influence of microvessel density and vascular permeability factor/vascular endothelial growth factor expression on prognosis in vulvar cancer. Gynecol. Oncol. 1996, 63, 204–209. [Google Scholar] [CrossRef]
- Bancher-Todesca, D.; Obermair, A.; Bilgi, S.; Kohlberger, P.; Kainz, C.; Breitenecker, G.; Leodolter, S.; Gitsch, G. Angiogenesis in vulvar intraepithelial neoplasia. Gynecol. Oncol. 1997, 64, 496–500. [Google Scholar] [CrossRef]
- Sherman, K.J.; Daling, J.R.; McKnight, B.; Chu, J. Hormonal factors in vulvar cancer: A case-control study. J. Reprod. Med. Obstet. Gynecol. 1994, 39, 857–861. [Google Scholar] [CrossRef]
- Mirkin, S.; Pinkerton, J.V.; Kagan, R.; Thompson, J.R.; Pan, K.; Pickar, J.H.; Komm, B.S.; Archer, D.F. Gynecologic Safety of Conjugated Estrogens Plus Bazedoxifene: Pooled Analysis of Five Phase 3 Trials. J. Women’s Health 2016, 25, 431–442. [Google Scholar] [CrossRef]
- Sawaya, G.F.; Grady, D.; Kerlikowske, K.; La Valleur, J.; Barnabei, V.M.; Bass, K.; Snyder, T.E.; Pickar, J.H.; Agarwal, S.K.; Mandelblatt, J. The positive predictive value of cervical smears in previously screened postmenopausal women: The heart and estrogen/progestin replacement study (HERS). Ann. Intern. Med. 2000, 133, 942–950. [Google Scholar] [CrossRef]
- Vargiu, V.; Amar, I.D.; Rosati, A.; Dinoi, G.; Turco, L.C.; Capozzi, V.A.; Scambia, G.; Villa, P. Hormone replacement therapy and cervical cancer: A systematic review of the literature. Climacteric 2021, 24, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Parazzini, F.; La Vecchia, C.; Negri, E.; Franceschi, S.; Moroni, S.; Chatenoud, L.; Bolis, G. Case-control study of oestrogen replacement therapy and risk of cervical cancer. Br. Med. J. 1997, 315, 85–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.H.; Lambert, P.F. Prevention and treatment of cervical cancer in mice using estrogen receptor antagonists. Proc. Natl. Acad. Sci. USA 2009, 106, 19467–19472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploch, E. Hormonal replacement therapy in patients after cervical cancer treatment. Gynecol. Oncol. 1987, 26, 169–177. [Google Scholar] [CrossRef]
- Roura, E.; Travier, N.; Waterboer, T.; de Sanjosé, S.; Xavier Bosch, F.; Pawlita, M.; Pala, V.; Weiderpass, E.; Margall, N.; Dillner, J.; et al. The influence of hormonal factors on the risk of developing cervical cancer and pre-cancer: Results from the EPIC cohort. PLoS ONE 2016, 11, 0147029. [Google Scholar] [CrossRef]
- Roig, L.M.V.; Lotfi, H.; Olcese, J.E.; Castro, G.L.; Ciocca, D.R. Effects of short-term tamoxifen administration in patients with invasive cervical carcinoma. Anticancer Res. 1993, 13, 2457–2463. [Google Scholar]
- Bigler, L.R.; Thigpen, J.T.; Blessing, J.A.; Fiorica, J.; Monk, B.J. Evaluation of tamoxifen in persistent or recurrent nonsquamous cell carcinoma of the cervix: A Gynecologic Oncology Group study. Int. J. Gynecol. Cancer 2004, 14, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Cervical cancer and hormonal contraceptives: Collaborative reanalysis of individual data for 16 573 women with cervical cancer and 35 509 women without cervical cancer from 24 epidemiological studies. Lancet 2007, 370, 1609–1621. [CrossRef]
- Shields, T.S.; Falk, R.T.; Herrero, R.; Schiffman, M.; Weiss, N.S.; Bratti, C.; Rodriguez, A.C.; Sherman, M.E.; Burk, R.D.; Hildesheim, A. A case-control study of endogenous hormones and cervical cancer. Br. J. Cancer 2004, 90, 146–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauh, L.A.; Pannone, A.F.; Cantrell, L.A. Hormone replacement therapy after treatment for cervical cancer: Are we adhering to standard of care? Gynecol. Oncol. 2017, 147, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Grenman, S.; Shapira, A.; Carey, T.E. In vitro response of cervical cancer cell lines CaSki, HeLa, and ME-180 to the antiestrogen tamoxifen. Gynecol. Oncol. 1988, 30, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Everhov, A.H.; Nyberg, T.; Bergmark, K.; Citarella, A.; Rådestad, A.F.; Hirschberg, A.L.; Smedby, K.E. Hormone therapy after uterine cervical cancer treatment: A Swedish population-based study. Menopause 2015, 22, 633–639. [Google Scholar] [CrossRef]
- Iversen, L.; Fielding, S.; Lidegaard, Ø.; Hannaford, P.C. Contemporary hormonal contraception and cervical cancer in women of reproductive age. Int. J. Cancer 2021, 149, 769–777. [Google Scholar] [CrossRef]
- Hridy, A.U.; Shabnaz, S.; Asaduzzaman, M.; Shahriar, M.; Bhuiyan, M.A.; Islam, M.S.; Hossen, S.M.M.; Emran, T.B. Genetic variations of RAD51 and XRCC2 genes increase the risk of colorectal cancer in Bangladeshi population. Asian Pac. J. Cancer Prev. 2020, 21, 1445–1451. [Google Scholar] [CrossRef]
- Danforth, K.N.; Tworoger, S.S.; Hecht, J.L.; Rosner, B.A.; Colditz, G.A.; Hankinson, S.E. A prospective study of postmenopausal hormone use and ovarian cancer risk. Br. J. Cancer 2007, 96, 151–156. [Google Scholar] [CrossRef]
- Puett, D.; Angelova, K.; da Costa, M.R.; Warrenfeltz, S.W.; Fanelli, F. The luteinizing hormone receptor: Insights into structure-function relationships and hormone-receptor-mediated changes in gene expression in ovarian cancer cells. Mol. Cell. Endocrinol. 2010, 329, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.; Simera, I. Tamoxifen for relapse of ovarian cancer. In Cochrane Database of Systematic Reviews; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2001. [Google Scholar] [CrossRef]
- Langdon, S.P.; Herrington, C.S.; Hollis, R.L.; Gourley, C. Estrogen signaling and its potential as a target for therapy in ovarian cancer. Cancers 2020, 12, 1647. [Google Scholar] [CrossRef] [PubMed]
- Symer, M.M.; Wong, N.Z.; Abelson, J.S.; Milsom, J.W.; Yeo, H.L. Hormone Replacement Therapy and Colorectal Cancer Incidence and Mortality in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Clin. Colorectal Cancer 2018, 17, e281–e288. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, J.J.; Roberts, W.; Townsend, P.; Hewitt, S. Leuprolide acetate in the treatment of refractory or persistent epithelial ovarian cancer. J. Clin. Oncol. 1989, 7, 115–118. [Google Scholar] [CrossRef]
- Bowman, A.; Gabra, H.; Langdon, S.P.; Lessells, A.; Stewart, M.; Young, A.; Smyth, J.F. CA125 Response Is Associated with Estrogen Receptor Expression in a Phase II Trial of Letrozole in Ovarian Cancer. Am. Assoc. Cancer Res. 2002, 6, 2233–2239. [Google Scholar]
- Papadimitriou, C.A.; Markaki, S.; Siapkaras, J.; Vlachos, G.; Efstathiou, E.; Grimani, I.; Hamilos, G.; Zorzou, M.; Dimopoulos, M.-A. Hormonal Therapy with Letrozole for Relapsed Epithelial Ovarian Cancer. Oncology 2004, 66, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Veenhof, C.H.N.; van der Burg, M.E.L.; Nooy, M.; Aalders, J.G.; Pecorelli, S.; Oliveira, C.F.; Rotmensz, N.; Vermorken, J.B. Phase II study of high-dose megestrol acetate in patients with advanced ovarian carcinoma. Eur. J. Cancer 1994, 30, 697–698. [Google Scholar] [CrossRef]
- Post, M.D. Hormone-receptor expression and ovarian cancer survival: An Ovarian Tumor Tissue Analysis consortium study. Yearb. Pathol. Lab. Med. 2014, 2014, 113–115. [Google Scholar] [CrossRef]
- Colon-Otero, G.; Weroha, S.J.; Foster, N.R.; Haluska, P.; Hou, X.; Wahner-Hendrickson, A.E.; Jatoi, A.; Block, M.S.; Dinh, T.A.; Robertson, M.W.; et al. Phase 2 trial of everolimus and letrozole in relapsed estrogen receptor-positive high-grade ovarian cancers. Gynecol. Oncol. 2017, 146, 64–68. [Google Scholar] [CrossRef]
- Decruze, S.B.; Green, J.A. Hormone therapy in advanced and recurrent endometrial cancer: A systematic review. Int. J. Gynecol. Cancer 2007, 17, 964–978. [Google Scholar] [CrossRef]
- Ghafarnegad, M.; Arjmand, N.; Khazaeipour, Z. Pregnancy rate of gonadotrophin therapy and laparoscopic ovarian electrocautery in polycystic ovary syndrome resistant to clomiphene citrate: A comparative study. Tehran Univ. Med. J. 2010, 67, 712–717. [Google Scholar]
- Kirilovas, D.; Schedvins, K.; Naessén, T.; Von Schoultz, B.; Carlström, K. Conversion of circulating estrone sulfate to 17β-estradiol by ovarian tumor tissue: A possible mechanism behind elevated circulating concentrations of 17β-estradiol in postmenopausal women with ovarian tumors. Gynecol. Endocrinol. 2007, 23, 25–28. [Google Scholar] [CrossRef]
- Carter, J.; Pather, S. An overview of uterine cancer and its management. Expert Rev. Anticancer Ther. 2006, 6, 33–41. [Google Scholar] [CrossRef]
- Seo, S.y.; Shin, J.Y.; Ji, Y. Il Metastatic uterine cancer looking as cervical fibroid in recurrent breast cancer woman: A case report. Obstet. Gynecol. Sci. 2017, 60, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Banno, K.; Kisu, I.; Yanokura, M.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; Nomura, H.; Susumu, N.; et al. Progestin therapy for endometrial cancer: The potential of fourth-generation progestin (Review). Int. J. Oncol. 2012, 40, 1755–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, H.; Choi, Y.S.; Song, I.C.; Yun, H.J.; Jo, D.Y.; Kim, S.; Lee, H.J. Long-term treatment of residual or recurrent low-grade endometrial stromal sarcoma with aromatase inhibitors: A report of two cases and a review of the literature. Oncol. Lett. 2015, 10, 3310–3314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiderpass, E.; Adami, H.O.; Baron, J.A.; Magnusson, C.; Bergström, R.; Lindgren, A.; Correia, N.; Persson, I. Risk of endometrial cancer following estrogen replacement with and without progestins. J. Natl. Cancer Inst. 1999, 91, 1131–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pike, M.C.; Peters, R.K.; Cozen, W.; Probst-Hensch, N.M.; Felix, J.C.; Wan, P.C.; Mack, T.M. Estrogen-Progestin Replacement Therapy and Endometrial Cancer. Obstet. Gynecol. Surv. 1998, 53, 285–287. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.S.; Belland, L. Contemporary management of uterine fibroids: Focus on emerging medical treatments. Curr. Med. Res. Opin. 2015, 31, 1–12. [Google Scholar] [CrossRef]
- Reich, O.; Nogales, F.F.; Regauer, S. Gonadotropin-releasing hormone receptor expression in endometrial stromal sarcomas: An immunohistochemical study. Mod. Pathol. 2005, 18, 573–576. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ding, J.X.; Tao, X.; Lu, Z.Y.; Wang, J.J.; Feng, W.W.; Hua, K.Q. Goserelin can inhibit ovarian cancer proliferation and simultaneously protect ovarian function from cisplatin: An in vitro and in vivo study. J. Chemother. 2013, 25, 96–103. [Google Scholar] [CrossRef]
- Morgan, K.; Stewart, A.J.; Miller, N.; Mullen, P.; Muir, M.; Dodds, M.; Medda, F.; Harrison, D.; Langdon, S.; Millar, R.P. Gonadotropin-releasing hormone receptor levels and cell context affect tumor cell responses to agonist in vitro and in vivo. Cancer Res. 2008, 68, 6331–6340. [Google Scholar] [CrossRef] [Green Version]
- Emons, G.; Gründker, C. The role of gonadotropin-releasing hormone (GnRH) in endometrial cancer. Cells 2021, 10, 292. [Google Scholar] [CrossRef]
- Chlebowski, R.T.; Schottinger, J.E.; Shi, J.; Chung, J.; Haque, R. Aromatase inhibitors, tamoxifen, and endometrial cancer in breast cancer survivors. Cancer 2015, 121, 2147–2155. [Google Scholar] [CrossRef] [Green Version]
- Altman, A.D.; Nelson, G.S.; Chu, P.; Nation, J.; Ghatage, P. Uterine sarcoma and aromatase inhibitors: Tom Baker cancer centre experience and review of the literature. Int. J. Gynecol. Cancer 2012, 22, 1006–1012. [Google Scholar] [CrossRef]
- Thanopoulou, E.; Judson, I. Hormonal therapy in gynecological sarcomas. Expert Rev. Anticancer Ther. 2012, 12, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Bellone, S.; Shah, H.R.; McKenney, J.K.; Stone, P.J.B.; Santin, A.D. Recurrent endometrial carcinoma regression with the use of the aromatase inhibitor anastrozole. Am. J. Obstet. Gynecol. 2008, 199, e7–e10. [Google Scholar] [CrossRef] [PubMed]
- Pink, D.; Lindner, T.; Mrozek, A.; Kretzschmar, A.; Thuss-Patience, P.C.; Dörken, B.; Reichardt, P. Harm or benefit of hormonal treatment in metastatic low-grade endometrial stromal sarcoma: Single center experience with 10 cases and review of the literature. Gynecol. Oncol. 2006, 101, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Spano, J.P.; Soria, J.C.; Kambouchner, M.; Piperno-Neuman, S.; Morin, F.; Morere, J.F.; Martin, A.; Breau, J.L. Long-term survival of patients given hormonal therapy for metastatic endometrial stromal sarcoma. Med. Oncol. 2003, 20, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Thigpen, J.T. The efficacy of hormonal treatment for residual or recurrent low-grade endometrial stromal sarcoma. A retrospective study. Yearb. Oncol. 2009, 2009, 57–58. [Google Scholar] [CrossRef]
- Ramirez, P.T.; Frumovitz, M.; Bodurka, D.C.; Sun, C.C.; Levenback, C. Hormonal therapy for the management of grade 1 endometrial adenocarcinoma: A literature review. Gynecol. Oncol. 2004, 95, 133–138. [Google Scholar] [CrossRef]
- Chu, M.C.; Mor, G.; Lim, C.; Zheng, W.; Parkash, V.; Schwartz, P.E. Low-grade endometrial stromal sarcoma: Hormonal aspects. Gynecol. Oncol. 2003, 90, 170–176. [Google Scholar] [CrossRef]
- Mizuno, M.; Yatabe, Y.; Nawa, A.; Nakanishi, T. Long-term medroxyprogesterone acetate therapy for low-grade endometrial stromal sarcoma. Int. J. Clin. Oncol. 2012, 17, 348–354. [Google Scholar] [CrossRef]
- O’Cearbhaill, R.; Zhou, Q.; Iasonos, A.; Soslow, R.A.; Leitao, M.M.; Aghajanian, C.; Hensley, M.L. Treatment of advanced uterine leiomyosarcoma with aromatase inhibitors. Gynecol. Oncol. 2010, 116, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiser, A.L.; Hamid, A.M.; Blanchard, R. Recurrence of prolactin-producing endometrial stromal sarcoma with sex-cord stromal component treated with progestin and aromatase inhibitor. Gynecol. Oncol. 2004, 94, 567–571. [Google Scholar] [CrossRef]
- Murphy, N.J.; Wallace, D.L. Gonadotropin releasing hormone (GnRH) agonist therapy for reduction of leiomyoma volume. Gynecol. Oncol. 1993, 49, 266–267. [Google Scholar] [CrossRef]
- Wang, C.B.; Wang, C.J.; Huang, H.J.; Hsueh, S.; Chou, H.H.; Soong, Y.K.; Lai, C.H. Fertility-preserving treatment in young patients with endometrial adenocarcinoma. Cancer 2002, 94, 2192–2198. [Google Scholar] [CrossRef]
- Kung, F.T.; Chen, W.J.; Chou, H.H.; Ko, S.F.; Chang, S.Y. Conservative management of early endometrial adenocarcinoma with repeat curettage and hormone therapy under assistance of hysteroscopy and laparoscopy. Hum. Reprod. 1997, 12, 1649–1653. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.Y.; Jung, S.M.; Ng, K.K.; Chang, Y.C.; Lai, C.H. Ovarian metastasis in a nulliparous woman with endometrial adenocarcinoma failing conservative hormonal treatment. Gynecol. Oncol. 2005, 97, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.H.; Chen, C.H.; Yu, M.H. Conservative therapy of stage I endometrial adenocarcinoma and atypical endometrial hyperplasia for the preservation of fertility. Int. J. Gynecol. Obstet. 2006, 92, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Rakib, A.; Islam, M.A.; Khanam, B.H.; Faiz, F.B.; Paul, A.; Chy, M.N.U.; Bhuiya, N.M.M.A.; Uddin, M.M.N.; Ullah, S.M.A.; et al. In vivo and in vitro pharmacological activities of Tacca integrifolia rhizome and investigation of possible lead compounds against breast cancer through in silico approaches. Clin. Phytoscience 2019, 5, 36. [Google Scholar] [CrossRef]
- Milewich, L.; Porter, J.C. In situ steroid sulfatase activity in human epithelial carcinoma cells of vaginal, ovarian, and endometrial origin. J. Clin. Endocrinol. Metab. 1987, 65, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.; Nachtigall, L.; Ulrich, L.G.; Eugster-Hausmann, M.; Gut, R. Endometrial safety of ultra-low-dose estradiol vaginal tablets. Obstet. Gynecol. 2010, 116, 876–883. [Google Scholar] [CrossRef]
- Minkin, M.J.; Maamari, R.; Reiter, S. Improved compliance and patient satisfaction with estradiol vaginal tablets in postmenopausal women previously treated with another local estrogen therapy. Int. J. Womens. Health 2013, 5, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Alvisi, S.; Baldassarre, M.; Martelli, V.; Gava, G.; Seracchioli, R.; Meriggiola, M.C. Effects of ospemifene on vaginal epithelium of post-menopausal women. Gynecol. Endocrinol. 2017, 33, 946–950. [Google Scholar] [CrossRef]
- Bachmann, G.A.; Komi, J.O. Ospemifene effectively treats vulvovaginal atrophy in postmenopausal women: Results from a pivotal phase 3 study. Menopause 2010, 17, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Jacob, L.; Kostev, K.; Kalder, M. Prescription of hormone replacement therapy prior to and after the diagnosis of gynecological cancers in German patients. J. Cancer Res. Clin. Oncol. 2020, 146, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Portman, D.J.; Bachmann, G.A.; Simon, J.A. Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy. Menopause 2013, 20, 623–630. [Google Scholar] [CrossRef]
- Kagan, R.; Williams, R.S.; Pan, K.; Mirkin, S.; Pickar, J.H. A randomized, placebo-and active-controlled trial of bazedoxifene/ conjugated estrogens for treatment of moderate to severe vulvar/vaginal atrophy in postmenopausal women. Menopause 2010, 17, 281–289. [Google Scholar] [CrossRef]
- Bachmann, G.; Bobula, J.; Mirkin, S. Effects of bazedoxifene/conjugated estrogens on quality of life in postmenopausal women with symptoms of vulvar/vaginal atrophy. Climacteric 2010, 13, 132–140. [Google Scholar] [CrossRef]
- Mirkin, S.; Simon, J.A.; Liu, J.H.; Archer, D.F.; Castro, P.D.; Graham, S.; Bernick, B.; Komm, B. Evaluation of endometrial progesterone receptor expression after 12 weeks of exposure to a low-dose vaginal estradiol insert. Menopause 2021, 28, 998–1003. [Google Scholar] [CrossRef]
- Capriglione, S.; Plotti, F.; Montera, R.; Luvero, D.; Lopez, S.; Scaletta, G.; Aloisi, A.; Serra, G.B.; Angioli, R. Role of paroxetine in the management of hot flashes in gynecological cancer survivors: Results of the first randomized single-center controlled trial. Gynecol. Oncol. 2016, 143, 584–588. [Google Scholar] [CrossRef]
- Santen, R.J. Menopausal hormone therapy and breast cancer. J. Steroid Biochem. Mol. Biol. 2014, 142, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Lower, E.E.; Blau, R.; Gazder, P.; Stahl, D.L. The effect of estrogen usage on the subsequent hormone receptor status of primary breast cancer. Breast Cancer Res. Treat. 1999, 58, 205–210. [Google Scholar] [CrossRef]
- Legha, S.S.; Davis, H.L.; Muggia, F.M. Hormonal therapy of breast cancer: New approaches and concepts. Ann. Intern. Med. 1978, 88, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Vidula, N.; Ma, C. Improving Response to Hormone Therapy in Breast Cancer: New Targets, New Therapeutic Options. Am. Soc. Clin. Oncol. Educ. B. 2016, 36, e40–e54. [Google Scholar] [CrossRef] [PubMed]
- Schenck-Gustafsson, K.; Brincat, M.; Erel, C.T.; Gambacciani, M.; Lambrinoudaki, I.; Moen, M.H.; Tremollieres, F.; Rozenberg, S.V.S.; Rees, M. EMAS position statement: Managing the menopause in the context of coronary heart disease. Maturitas 2011, 68, 94–97. [Google Scholar] [CrossRef]
- Lakoski, S.G.; Herrington, D.M. Effects of hormone therapy on C-reactive protein and IL-6 in postmenopausal women: A review article. Climacteric 2005, 8, 317–326. [Google Scholar] [CrossRef]
- Rauf, A.; Abu-Izneid, T.; Khalil, A.A.; Imran, M.; Shah, Z.A.; Emran, T.B.; Mitra, S.; Khan, Z.; Alhumaydhi, F.A.; Aljohani, A.S.; et al. Berberine as a potential anticancer agent: A comprehensive review. Molecules 2021, 26, 7368. [Google Scholar] [CrossRef]
- Reeves, G.K.; Beral, V.; Green, J.; Gathani, T.; Bull, D. Hormonal therapy for menopause and breast-cancer risk by histological type: A cohort study and meta-analysis. Lancet Oncol. 2006, 7, 910–918. [Google Scholar] [CrossRef]
- Ye, F.; Wen, J.; Yang, A.; Wang, Y.; Li, N.; Yu, P.; Wei, W.; Tang, J. The Influence of Hormone Therapy on secondary diabetes mellitus in Breast Cancer: A Meta-analysis. Clin. Breast Cancer 2021, 22, e48–e58. [Google Scholar] [CrossRef]
- Heit, J.A.; Silverstein, M.D.; Mohr, D.N.; Petterson, T.M.; O’Fallon, W.M.; Melton, L.J. Risk factors for deep vein thrombosis and pulmonary embolism: A population-based case-control study. Arch. Intern. Med. 2002, 160, 809–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carugno, J. Clinical management of vaginal bleeding in postmenopausal women. Climacteric 2020, 23, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Beral, V.; Reeves, G.; Bull, D.; Green, J. Breast cancer risk in relation to the interval between menopause and starting hormone therapy. J. Natl. Cancer Inst. 2011, 103, 296–305. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Name of Hormone | Formulation Name and Dose | Observation Time | Study Model | Results | References |
|---|---|---|---|---|---|
| ER antagonist | 0.15 mL Faslodex and 1.5 mg of raloxifene | Faslodex twice a week for a month and raloxifene for a month, 5 days a week | K14E7 and K14E6 transgenic mice | Raloxifene, ER antagonist and selective ER modulator, efficiently clears cancer and its precursor lesions in both the cervix and the vagina | [79] |
| Non-steroid synthetic estrogen with synthetic progestagen | Dienestrol—1 tablet (5 mg) and Chlormadinon—1 tablet (2 mg) | 5 years | 120 patients after surgery and/or radiotherapy | Only 20% and 32% incidence of cancer recurrences and survival without cancer symptoms was found in 80% and 65% of cases, respectively in the hormonally treated group and in the control group | [90] |
| Estrogen and progesterone | HT formulation (estrogen alone, progesterone alone, combination of estrogen/progesterone) | 3 groups (≤1, 2–4, ≥5 years) | 261 ICC and 804 CIN3/CIS cases of post- and perimenopausal women | Significantly decreased the risk of ICC in peri- and postmenopausal women, but menopausal estrogens alone were associated with an increased risk of CIN3/CIS and combined HT was inversely associated with ICC | [81] |
| ERT | — | 1–10 years | 645 women | Exogenous estrogens decreased the risk of cervical cancer | [78] |
| Estrogens progestin | Oral conjugated equine estrogens, 0.625 mg/day, plus medroxyprogesterone acetate, 2.5 mg/day | 2 years | 2561 women | Did not significantly affect the incidence of cytologic abnormalities | [76] |
| Anti-estrogen | Triphenylethylene antiestrogen tamoxifen | 10 days (20 or 40 mg/day) | 19 patients | Certain cervical carcinomas had changes in their proliferation and differentiation levels following tamoxifen administration | [82] |
| Anti-estrogen | Tamoxifen | 10 mg per orally twice a day | 34 patients | The objective response rate was 11.1%, so tamoxifen appears to have minimal activity in non-squamous cell carcinoma of the cervix | [83] |
| Progestagen | Combined oral contraceptives | <5 years and >5 years | 16,573 women | The risk of invasive cervical cancer increased with increasing duration of use, not for short time use | [84] |
| Estrone, estradiol and progesterone | SHBG (20 nmol·L−1) estradiol (5 pg·mL−1) estrone (5 pg·mL−1) estrone sulphate (100 pg·mL−1) DHEAS 10 μg·dl−1 and progesterone 100·pg·mL−1 | 5–10 years of study | 11,742 women | Elevated plasma levels of endogenous estrogens or progesterone decrease the risk of cervical neoplasia | [85] |
| HRT | — | 1 January, 2005 to 31 December 31 2015 | 222 women | 48% patients received counseling for HRT and then improved efforts to reduce disparities in the distribution of survivorship care | [86] |
| ER modulator, Tamoxifen | 5% dextran-charcoal treated fetal bovine serum (D5) and 0, 1, 2.5, 5, 7.5, or 10 μM tamoxifen | 6 days | In vitro growth of three cell lines derived from carcinoma of the uterine cervix (HeLa, CaSki, ME-180) | Inhibited cell growth of the cervical carcinoma cell lines; 2.5 μM tamoxifen induced more than 60% growth inhibition where 5 μM tamoxifen was cytotoxic | [87] |
| Estrogens and progestogens | Contraceptives (G03A), estrogens (G03C), progestogens (G03D), and progestogens and estrogens in combination (G03F). | 0.5 to 1 year after diagnosis | 171 (67%) of 257 women had at least one dispensing of HT (Hormonal Therapy) | Fewer than half of cervical cancer survivors with therapy-induced early menopause used HT | [88] |
| Estradiol | 30 to 35 μg ethinylestradiol | From 1995 to 2014 | women aged 15 to 49 | The risk pattern among any hormonal and combined contraceptive users generally increased with longer duration of use and declined after stopping | [89] |
| Name of Hormone | Formulation Name and Dose | Observation time | Study Model | Results | References |
|---|---|---|---|---|---|
| GnRHa | Leuprolide acetate (1 mg) | 8 weeks | 23 patients | Showed evidence of antitumor activity against refractory grade 1 epithelial adenocarcinoma of the ovary | [96] |
| Aromatase inhibitor | Letrozole (2.5 mg daily) at the time of CA125 relapse | 12 weeks | 60 patients | Produced disease stabilization and CA125 responses that in turn are linked to higher levels of ER expression | [97] |
| Aromatase inhibitor | Letrozole at a dose of 2.5 mg once a day | 27 patients | The aromatase inhibitor letrozole is an agent with some activity and limited toxicity for relapsed ovarian cancer. | [98] | |
| Progestin | Megestrol acetate: 800 mg/day for 1 month followed by 400 mg/day as maintenance treatment | 1 month | 72 patients | This study does not suggest that the overall 10% benefit from hormonal therapy for chemotherapy refractory ovarian cancer will improve by increasing the dose | [99] |
| Estrogen, progesterone | - | 2933 women | PR expression and ER expression were associated with improved disease-specific survival | [100] | |
| Aromatase inhibitor | Oral everolimus 10 mg daily and letrozole 2.5 mg daily | 12 weeks of therapy with the combination of everolimus and letrozole | 20 patients | Associated with a promising (47%) progression-free survival rate in patients with ER-positive relapsed high-grade ovarian cancer | [101] |
| Gonadotropin | Injection of single dose gonadotropin daily | 2003–2008 | 100 infertile clomiphene-citrate resistance women with POCS | Pregnancy and abortion rate in infertile women of PCOS receiving gonadotropin as a treatment for induction of ovulation seems to be more effective | [103] |
| E1S | 20 pmol of [3H] E1S in 100 mL of Tris-HCl | (15 and 60 min) incubation time | 12 postmenopausal women | Conversion of circulating E1S to E2 by the tumor tissue could be one important reason for elevated S-E2 levels in postmenopausal women with non-estrogen-producing ovarian tumors | [104] |
| Name of Hormone | Formulation Name and Dose | Observation Time | Study Model | Results | References |
|---|---|---|---|---|---|
| Aromatase Inhibitor | Anastrozole (1 mg/day), exemestane (25 mg/day)-1 patient, letrozole (2.5 mg/day) | 29.2 months | Patients with uterine sarcoma (4 patients with ESS, endometrial stromal sarcoma, and 3 patients with LMS) | Effective in the treatment of endometrial stromal sarcomas | [117] |
| Progesterone | Medroxy progesterone acetate (36 patients; 44%) or megestrol acetate(28 patients; 35%), progestins | 24weeks | 81 patients | When disease recurs, carcinoma extending beyond the uterus is rare in patients reported with well-differentiated endometrial adenocarcinoma who undergo treatment with a progestational agent | [123] |
| Estrogen, progestin, aromatase inhibitors | ERT, tamoxifen, progestins, aromatase inhibitors | 4 to 164 months | 800 patients | MPA and letrozole, in particular, are highly effective and lead to sustained disease control in most cases | [120] |
| Progestin, Aromatase inhibitors | Megestrol acetate (MA), Aromatase inhibitor (letrozole) | 4+ to 252+ months (median 48+ months). | 11 patients | Hormonal treatment for measurable residual or recurrent low-grade ESS has a high response rate and should be considered as the treatment of choice for patients in which recurrent disease cannot easily be eliminated | [122] |
| Exogenous or endogenous estrogen and progestins | Megestrol acetate 160 mg, progestins | 100 months (range, 2–258) | 22 patients | ERT was detrimental in patients with low-grade endometrial stromal sarcoma, but progestin therapy should be routinely considered for adjuvant therapy and for the treatment of recurrent endometrial stromal sarcomas | [124] |
| Progesterone | Medroxy progesterone acetate (MPA) | Dosing period 64 months (range 28–92 months) but follow-up period was 117 months | 13 patients | MPA therapy might be considered as a therapeutic option for residual or recurrent low-grade ESS and perhaps chosen as a first-line therapy | [125] |
| Aromatase | Aromatase inhibitors used were letrozole (in 74% of patients), anastrozole (21%), and exemestane (6%) | Between 1998 and 2008 | 40 patients | Aromatase inhibitors achieved objective response in only 9%. Progression free survival was longer among patients with ER and/or PR positive tumors than among patients with ER and PR negative tumors | [126] |
| GnRH agonist | GnRH agonist, leuprolide acetate, adriamycin, cisplatin, ifosfamide etc. | 15 months | A patient with menorrhagia, dysmenorrhea, and an enlarged uterus | GnRH therapy mask the symptoms of leiomyosarcomas, e.g., rapidly enlarging uterine mass, pelvic pain, uterovaginal bleeding | [133] |
| Progestin and aromatase | Three cycles of BEP (bleomycin, etoposide, cisplatin), anastrozole and megestrol acetate | 2 years | 48-year-old woman was diagnosed with stage I endometrial stroma sarcoma | Endometrial stromal sarcoma with sex-cord stromal component may be hormonally functional and can be cured by treating with progestin and aromatase inhibitor | [127] |
| Estrogen, progesterone | Megestrol acetate and tamoxifen | 6 months | A 22-year-old nullipara | 1 year after the last curettage, there is no evidence of disease | [130] |
| Estrogen, progesterone | Combinations of megestrol acetate (160 mg/day), tamoxifen (30 mg/day), and GnRHa | 6 months | 9 patients with clinically diagnosed endometrial adenocarcinoma stage IA, grade 1 | Of the 9 patients, 8 (88.9%) achieved complete remission after hormone therapy. All nine patients have been alive without evidence of disease | [129] |
| Estrogen, progesterone, (GnRHa) | Megestrol (1-month), tamoxifen (20 mg/day) and depot leuprolide acetate subcutaneous injection (3.75 mg/month) | 6-months | A 36-year-old nulliparous woman | This case report signals a warning that negative clinical investigations are not reassuring for a relapsing endometrial adenocarcinoma failing conservative hormonal treatment | [131] |
| Estrogen, progestin | 500 mg of oral medroxyprogesterone for 6 months, twice weekly | 9 months (range, 3–18 months) | 2 women | The quarterly interval for D&Cs was satisfactory with medroxyprogesterone treatment, and the patients’ desire not to undergo hysterectomy was met | [132] |
| Name of Hormone | Formulation Name and Dose | Observation Time | Study Model | Results | References |
|---|---|---|---|---|---|
| Estrogen (estrone sulfate) | Vaginal promestriene 10 mg soft vaginal suppository daily for one month | 1–6 months | 17 patients (after menopause) | The level of circulating E1S was not significantly affected by vaginal promestriene treatment, but a wide range of levels was noted pre- and post-treatment in individual patients | [66] |
| E1S | ElS and DS (2.25 μCi, 15 μM) | Incubation time for at least 2 h and with cell number up to 3.2 × 106 cells/mL | AC-258 cell line, squamous vaginal carcinoma cells (line EC-82) | Played a role in the development of tumors of the ovary and vagina | [134] |
| Estradiol | Ultra-low-dose 10-microgram 17β-estradiol vaginal tablets | 52 weeks | (n = 205) in a randomized, double-blind, placebo-controlled trial | There was no increased risk of endometrial hyperplasia and carcinoma in postmenopausal women | [135] |
| Estrogen | Local estrogen therapy (LET) in form of estradiol vaginal tablets, 10 μg, estradiol cream, 0.1 mg estradiol/g USP | 5-week period, from 6 March 2012 through 9 April 2012 | 423 women | There was greater compliance with vaginal tablets than with vaginal cream; respondents preferred their current treatment with the vaginal tablet | [136] |
| Estrogen | ER modulator (ospemifene) | 1 month | 32 postmenopausal women | Increased maturation, and ERα expression of the vaginal mucosa. These changes partially explained the improvement of symptoms of vaginal atrophy and cancer | [137] |
| ER modulator | Ospemifene 30 or 60 mg/day | 12 weeks | 826 postmenopausal women were randomized | Effective and well tolerated for the treatment of the symptoms of vaginal dryness and dyspareunia associated with vulvovaginal atrophy over | [138] |
| Name of Hormone | Formulation Name and Dose | Observation Time | Study Model | Results | References |
|---|---|---|---|---|---|
| Estrogen, progesterone | Hormone replacement therapy | 12 months | 7189 women | Significantly decreased the proportion of women receiving at least one HRT prescription. The majority of vulvar cancers are not estrogen-dependent, and the prescription of HRT is not contraindicated after the diagnosis of this type of cancer | [139] |
| ER modulator | Oral ospemifene 60 mg/day | 12 weeks | 605 women | Effective for the treatment of vulvar and vaginal atrophy and cancer in postmenopausal women with dyspareunia | [140] |
| ER modulator | BZA 20 mg/CE 0.625 mg and 0.45 mg, BZA 20 mg | 12 weeks | Healthy postmenopausal women (n = 664; aged 40–65 y) | Effective in treating moderate to severe VVA and vaginal symptoms. | [141] |
| Estrogen and ER modulator | Conjugated estrogens 0.625 mg and 0.45 mg/BZA 20 mg | 2 years | 1583 and 1583 postmenopausal women respectively | Conjugated estrogens/BZA provides endometrial protection without increasing breast pain/density, vaginal bleeding, or ovarian cysts | [75] |
| Estrogen and ER modulator | BZA 20 mg/CE 0.45 or 0.625 mg, BZA 20 mg. | 12 weeks | Postmenopausal, non-hysterectomized women (n = 652) with symptoms of moderate to severe vulvar/vaginal atrophy | Shown to significantly improve sexual function and quality-of-life measures in symptomatic postmenopausal women | [142] |
| Estradiol | Vaginal 4 μg and 10 μg estradiol (E2) | 12 weeks | 25 eligible women | Because of endometrial progesterone receptor expression, vaginal E2 would not be expected to stimulate endometrial hyperplasia leading to moderate to severe dyspareunia which is a symptom of vulvar and vaginal atrophy and cancer | [143] |
| Selective serotonin-reuptake inhibitors (SSRIs) | 7.5 mg oral paroxetine or placebo daily | 16 weeks | 80 women | Paroxetine significantly reduced hot flashes in weekly frequency and severity in gynecological cancer survivors | [144] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitra, S.; Lami, M.S.; Ghosh, A.; Das, R.; Tallei, T.E.; Fatimawali; Islam, F.; Dhama, K.; Begum, M.Y.; Aldahish, A.; et al. Hormonal Therapy for Gynecological Cancers: How Far Has Science Progressed toward Clinical Applications? Cancers 2022, 14, 759. https://doi.org/10.3390/cancers14030759
Mitra S, Lami MS, Ghosh A, Das R, Tallei TE, Fatimawali, Islam F, Dhama K, Begum MY, Aldahish A, et al. Hormonal Therapy for Gynecological Cancers: How Far Has Science Progressed toward Clinical Applications? Cancers. 2022; 14(3):759. https://doi.org/10.3390/cancers14030759
Chicago/Turabian StyleMitra, Saikat, Mashia Subha Lami, Avoy Ghosh, Rajib Das, Trina Ekawati Tallei, Fatimawali, Fahadul Islam, Kuldeep Dhama, M. Yasmin Begum, Afaf Aldahish, and et al. 2022. "Hormonal Therapy for Gynecological Cancers: How Far Has Science Progressed toward Clinical Applications?" Cancers 14, no. 3: 759. https://doi.org/10.3390/cancers14030759