
Targeting Cell Cycle Checkpoint Kinases to Overcome Intrinsic Radioresistance in Brain Tumor Cells


Abstract
:Simple Summary
Abstract
1. Introduction
2. GBM Heterogeneity and GSC-Related Therapy Resistance
3. The DNA Damage Response (DDR)
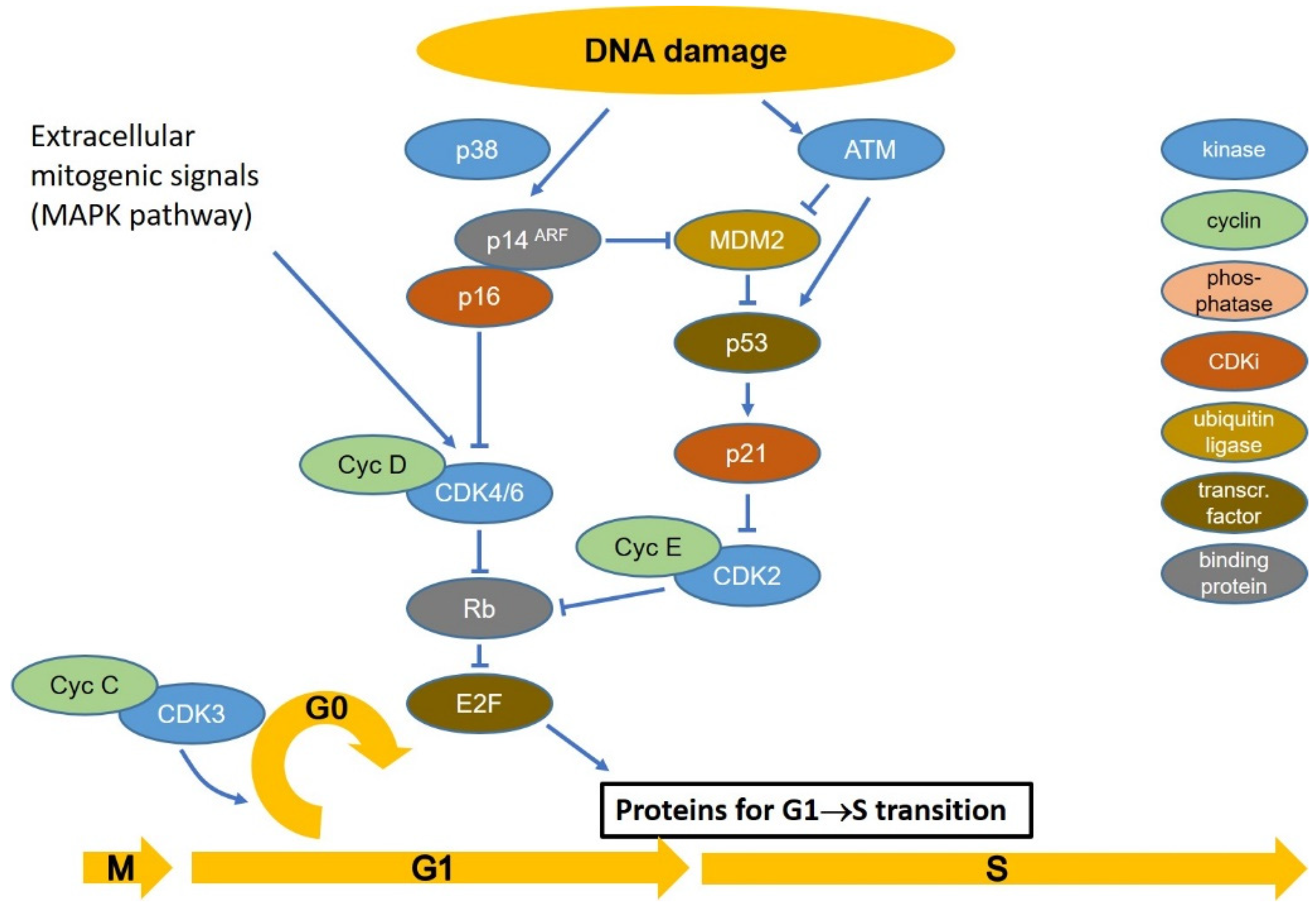
4. Cell Cycle Checkpoints
5. Inhibitors of Checkpoint Control
6. Preclinical Studies
6.1. ATM and ATR
6.2. CHK1 and CHK2
6.3. WEE1
7. Clinical Studies
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McFaline-Figueroa, J.R.; Lee, E.Q. Brain Tumors. Am. J. Med. 2018, 131, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Udaka, Y.T.; Packer, R.J. Pediatric Brain Tumors. Neurol. Clin. 2018, 36, 533–556. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, F.; Turriziani, M.; Tortorelli, G.; Avvisati, G.; Torino, F.; De Vecchis, L. Triazene compounds: Mechanism of action and related DNA repair systems. Pharmacol. Res. 2007, 56, 275–287. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef] [Green Version]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. (R. Coll. Radiol.) 2013, 25, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [Green Version]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair 2019, 78, 128–141. [Google Scholar] [CrossRef]
- Elmore, K.B.; Schaff, L.R. DNA Repair Mechanisms and Therapeutic Targets in Glioma. Curr. Oncol. Rep. 2021, 23, 87. [Google Scholar] [CrossRef] [PubMed]
- Ou, A.; Yung, W.K.A.; Majd, N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 351. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Yong, R.L.; Paddison, P.; Zhu, J. Comparison of glioblastoma (GBM) molecular classification methods. Semin. Cancer Biol. 2018, 53, 201–211. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56 e46. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.R.; Mangesius, J.; Skvortsova, I.-I.; Ganswindt, U. The Role of Cancer Stem Cells in Radiation Resistance. Front. Oncol. 2020, 10, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, H.; Liu, T.; Huang, M.; Butter, P.P.; Li, C.; Zhang, L.; Kao, G.D.; Gong, Y.; Maity, A.; et al. Temporal DNA-PK activation drives genomic instability and therapy resistance in glioma stem cells. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Harding, A.; Kozlov, S.; Kijas, A.W.; Ensbey, K.S.; Walker, D.G.; Lavin, M.F. A role for homologous recombination and abnormal cell-cycle progression in radioresistance of glioma-initiating cells. Mol. Cancer 2012, 11, 1863–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, C.; Weterings, E.; Mahadevan, D.; Sanan, A.; Weinand, M.; Stea, B. Expression Levels of RAD51 Inversely Correlate with Survival of Glioblastoma Patients. Cancers 2021, 13, 5358. [Google Scholar] [CrossRef]
- Narayan, R.S.; Fedrigo, C.A.; Stalpers, L.J.; Baumert, B.G.; Sminia, P. Targeting the Akt-pathway to improve radiosensitivity in glioblastoma. Curr. Pharm. Des. 2013, 19, 951–957. [Google Scholar] [CrossRef]
- Kumar, V.; Vashishta, M.; Kong, L.; Wu, X.; Lu, J.J.; Guha, C.; Dwarakanath, B.S. The Role of Notch, Hedgehog, and Wnt Signaling Pathways in the Resistance of Tumors to Anticancer Therapies. Front. Cell Dev. Biol. 2021, 9, 650772. [Google Scholar] [CrossRef]
- Aldaz, P.; Arozarena, I. Tyrosine Kinase Inhibitors in Adult Glioblastoma: An (Un)Closed Chapter? Cancers 2021, 13, 5799. [Google Scholar] [CrossRef]
- Schafer, N.; Gielen, G.H.; Rauschenbach, L.; Kebir, S.; Till, A.; Reinartz, R.; Simon, M.; Niehusmann, P.; Kleinschnitz, C.; Herrlinger, U.; et al. Longitudinal heterogeneity in glioblastoma: Moving targets in recurrent versus primary tumors. J. Transl. Med. 2019, 17, 96. [Google Scholar] [CrossRef] [Green Version]
- Van den Bent, M.J.; Gao, Y.; Kerkhof, M.; Kros, J.M.; Gorlia, T.; van Zwieten, K.; Prince, J.; van Duinen, S.; Sillevis Smitt, P.A.; Taphoorn, M.; et al. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro Oncol. 2015, 17, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, A.; Loeffler, J.S.; Dyson, N.J. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002, 62, 200–207. [Google Scholar] [PubMed]
- Wang, M.; Maier, P.; Wenz, F.; Giordano, F.A.; Herskind, C. Mitogenic signalling in the absence of epidermal growth factor receptor activation in a human glioblastoma cell line. J. NeuroOncol. 2013, 115, 323–331. [Google Scholar] [CrossRef]
- Lauko, A.; Lo, A.; Ahluwalia, M.S.; Lathia, J.D. Cancer cell heterogeneity & plasticity in glioblastoma and brain tumors. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- Nakanishi, M.; Shimada, M.; Niida, H. Genetic instability in cancer cells by impaired cell cycle checkpoints. Cancer Sci. 2006, 97, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Lawrence, T.S. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- De Bacco, F.; D’Ambrosio, A.; Casanova, E.; Orzan, F.; Neggia, R.; Albano, R.; Verginelli, F.; Cominelli, M.; Poliani, P.L.; Luraghi, P.; et al. MET inhibition overcomes radiation resistance of glioblastoma stem-like cells. EMBO Mol. Med. 2016, 8, 550–568. [Google Scholar] [CrossRef] [Green Version]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Muller, G.A. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 638–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.; Clemente-Blanco, A. Cell Cycle and DNA Repair Regulation in the Damage Response: Protein Phosphatases Take Over the Reins. Int. J. Mol. Sci. 2020, 21, 446. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [Green Version]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Latif, C.; Harvey, S.H.; O’Connell, M.J. Ensuring the stability of the genome: DNA damage checkpoints. Sci. World J. 2001, 1, 684–702. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Rollins, B.J. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell 2004, 117, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Deckbar, D.; Jeggo, P.A.; Lobrich, M. Understanding the limitations of radiation-induced cell cycle checkpoints. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 271–283. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Sowd, G.A.; Li, N.Y.; Fanning, E. ATM and ATR activities maintain replication fork integrity during SV40 chromatin replication. PLoS Pathog. 2013, 9, e1003283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharm. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Landsverk, K.S.; Patzke, S.; Rein, I.D.; Stokke, C.; Lyng, H.; De Angelis, P.M.; Stokke, T. Three independent mechanisms for arrest in G2 after ionizing radiation. Cell Cycle 2011, 10, 819–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palii, S.S.; Cui, Y.; Innes, C.L.; Paules, R.S. Dissecting cellular responses to irradiation via targeted disruptions of the ATM-CHK1-PP2A circuit. Cell Cycle 2013, 12, 1105–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiff, T.; Walker, S.A.; Cerosaletti, K.; Goodarzi, A.A.; Petermann, E.; Concannon, P.; O’Driscoll, M.; Jeggo, P.A. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006, 25, 5775–5782. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.L.; Agarwal, A.; Taylor, W.R.; Stark, G.R. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc. Natl. Acad. Sci. USA 1995, 92, 8493–8497. [Google Scholar] [CrossRef] [Green Version]
- Eliezer, Y.; Argaman, L.; Kornowski, M.; Roniger, M.; Goldberg, M. Interplay between the DNA damage proteins MDC1 and ATM in the regulation of the spindle assembly checkpoint. J. Biol. Chem. 2014, 289, 8182–8193. [Google Scholar] [CrossRef] [Green Version]
- Mackay, D.R.; Ullman, K.S. ATR and a Chk1-Aurora B pathway coordinate postmitotic genome surveillance with cytokinetic abscission. Mol. Biol. Cell 2015, 26, 2217–2226. [Google Scholar] [CrossRef]
- Smith, E.; Dejsuphong, D.; Balestrini, A.; Hampel, M.; Lenz, C.; Takeda, S.; Vindigni, A.; Costanzo, V. An ATM- and ATR-dependent checkpoInt. inactivates spindle assembly by targeting CEP63. Nat. Cell Biol. 2009, 11, 278–285. [Google Scholar] [CrossRef] [Green Version]
- Mustofa, M.K.; Tanoue, Y.; Tateishi, C.; Vaziri, C.; Tateishi, S. Roles of Chk2/CHEK2 in guarding against environmentally induced DNA damage and replication-stress. Environ. Mol. Mutagenesis 2020, 61, 730–735. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Pham, M.M.; Tan, D.S.P.; Yap, T.A. Targeting the replication stress response through synthetic lethal strategies in cancer medicine. Trends Cancer 2021, 7, 930–957. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA Replication Stress for Cancer Therapy. Genes 2016, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Kelly, R.; Martin, Y.; Koundrioukoff, S.; Tanenbaum, M.E.; Smits, V.A.; Medema, R.H.; Debatisse, M.; Freire, R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 2011, 194, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, K.; Mahajan, N.P. WEE1 tyrosine kinase, a novel epigenetic modifier. Trends Genet. 2013, 29, 394–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, R.; Yu, H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004, 23, 2016–2027. [Google Scholar] [CrossRef] [Green Version]
- Sasai, K.; Treekitkarnmongkol, W.; Kai, K.; Katayama, H.; Sen, S. Functional Significance of Aurora Kinases-p53 Protein Family Interactions in Cancer. Front. Oncol. 2016, 6, 247. [Google Scholar] [CrossRef] [Green Version]
- Kabeche, L.; Nguyen, H.D.; Buisson, R.; Zou, L. A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science 2018, 359, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Spoerri, L.; Oo, Z.Y.; Larsen, J.E.; Haass, N.K.; Gabrielli, B.; Pavey, S. Cell Cycle CheckpoInt. and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities. In Stress Response Pathways in Cancer: From Molecular Targets to Novel Therapeutics; Wondrak, G.T., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 29–49. [Google Scholar]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal. Transduct Target. 2017, 2, 17040. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Dube, C.; Gibert, M., Jr.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [Green Version]
- Gil del Alcazar, C.R.; Hardebeck, M.C.; Mukherjee, B.; Tomimatsu, N.; Gao, X.; Yan, J.; Xie, X.J.; Bachoo, R.; Li, L.; Habib, A.A.; et al. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clin. Cancer Res. 2014, 20, 1235–1248. [Google Scholar] [CrossRef] [Green Version]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2015, 9, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Raso, A.; Vecchio, D.; Cappelli, E.; Ropolo, M.; Poggi, A.; Nozza, P.; Biassoni, R.; Mascelli, S.; Capra, V.; Kalfas, F.; et al. Characterization of glioma stem cells through multiple stem cell markers and their specific sensitization to double-strand break-inducing agents by pharmacological inhibition of ataxia telangiectasia mutated protein. Brain Pathol 2012, 22, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.M.; Stricker, S.H.; Halavach, H.; Poetsch, A.R.; Cresswell, G.; Kelly, G.; Kanu, N.; Marino, S.; Luscombe, N.M.; Pollard, S.M.; et al. Inactivation of the ATMIN/ATM pathway protects against glioblastoma formation. eLife 2016, 5, e08711. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Stringer, B.W.; Kozlov, S.; Fazry, S.; Bruce, Z.C.; Ensbey, K.S.; Walker, D.G.; Boyd, A.W.; et al. Increased sensitivity to ionizing radiation by targeting the homologous recombination pathway in glioma initiating cells. Mol. Oncol. 2014, 8, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.U.; Carruthers, R.; Gilmour, L.; Yildirim, S.; Watts, C.; Chalmers, A.J. Selective Inhibition of Parallel DNA Damage Response Pathways Optimizes Radiosensitization of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 4416–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [Green Version]
- Vecchio, D.; Daga, A.; Carra, E.; Marubbi, D.; Raso, A.; Mascelli, S.; Nozza, P.; Garre, M.L.; Pitto, F.; Ravetti, J.L.; et al. Pharmacokinetics, pharmacodynamics and efficacy on pediatric tumors of the glioma radiosensitizer KU60019. Int. J. Cancer 2015, 136, 1445–1457. [Google Scholar] [CrossRef]
- Vecchio, D.; Daga, A.; Carra, E.; Marubbi, D.; Baio, G.; Neumaier, C.E.; Vagge, S.; Corvo, R.; Pia Brisigotti, M.; Louis Ravetti, J.; et al. Predictability, efficacy and safety of radiosensitization of glioblastoma-initiating cells by the ATM inhibitor KU-60019. Int. J. Cancer 2014, 135, 479–491. [Google Scholar] [CrossRef]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Pass, M.; et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 2018. [Google Scholar] [CrossRef] [Green Version]
- Karlin, J.; Allen, J.; Ahmad, S.F.; Hughes, G.; Sheridan, V.; Odedra, R.; Farrington, P.; Cadogan, E.B.; Riches, L.C.; Garcia-Trinidad, A.; et al. Orally Bioavailable and Blood-Brain Barrier-Penetrating ATM Inhibitor (AZ32) Radiosensitizes Intracranial Gliomas in Mice. Mol. Cancer 2018, 17, 1637–1647. [Google Scholar] [CrossRef] [Green Version]
- Frosina, G.; Profumo, A.; Marubbi, D.; Marcello, D.; Ravetti, J.L.; Daga, A. ATR kinase inhibitors NVP-BEZ235 and AZD6738 effectively penetrate the brain after systemic administration. Radiat. Oncol. 2018, 13, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.; Gilmour, L.; et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, J.F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Wakimoto, H.; et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10, 2910. [Google Scholar] [CrossRef] [PubMed]
- Bredel, M.; Pollack, I.F.; Freund, J.M.; Rusnak, J.; Lazo, J.S. Protein kinase C inhibition by UCN-01 induces apoptosis in human glioma cells in a time-dependent fashion. J. NeuroOncol. 1999, 41, 9–20. [Google Scholar] [CrossRef]
- Tang, Y.Y.; Grant, S.; Dent, P. Enhancing CHK1 inhibitor lethality in glioblastoma. Cancer Biol. 2012, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Burdak-Rothkamm, S.; Rothkamm, K.; McClelland, K.; Al Rashid, S.T.; Prise, K.M. BRCA1, FANCD2 and Chk1 are potential molecular targets for the modulation of a radiation-induced DNA damage response in bystander cells. Cancer Lett. 2015, 356, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Mir, S.E.; De Witt Hamer, P.C.; Krawczyk, P.M.; Balaj, L.; Claes, A.; Niers, J.M.; Van Tilborg, A.A.; Zwinderman, A.H.; Geerts, D.; Kaspers, G.J.; et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell 2010, 18, 244–257. [Google Scholar] [CrossRef] [Green Version]
- Patties, I.; Kallendrusch, S.; Bohme, L.; Kendzia, E.; Oppermann, H.; Gaunitz, F.; Kortmann, R.D.; Glasow, A. The Chk1 inhibitor SAR-020106 sensitizes human glioblastoma cells to irradiation, to temozolomide, and to decitabine treatment. J. Exp. Clin. Cancer Res. 2019, 38, 420. [Google Scholar] [CrossRef] [Green Version]
- Gogineni, V.R.; Nalla, A.K.; Gupta, R.; Dinh, D.H.; Klopfenstein, J.D.; Rao, J.S. Chk2-mediated G2/M cell cycle arrest maintains radiation resistance in malignant meningioma cells. Cancer Lett. 2011, 313, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Sarcar, B.; Kahali, S.; Prabhu, A.H.; Shumway, S.D.; Xu, Y.; Demuth, T.; Chinnaiyan, P. Targeting radiation-induced G(2) checkpoInt. activation with the Wee-1 inhibitor MK-1775 in glioblastoma cell lines. Mol. Cancer 2011, 10, 2405–2414. [Google Scholar] [CrossRef] [Green Version]
- Pokorny, J.L.; Calligaris, D.; Gupta, S.K.; Iyekegbe, D.O., Jr.; Mueller, D.; Bakken, K.K.; Carlson, B.L.; Schroeder, M.A.; Evans, D.L.; Lou, Z.; et al. The Efficacy of the Wee1 Inhibitor MK-1775 Combined with Temozolomide Is Limited by Heterogeneous Distribution across the Blood-Brain Barrier in Glioblastoma. Clin. Cancer Res. 2015, 21, 1916–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.; Hashizume, R.; Yang, X.; Kolkowitz, I.; Olow, A.K.; Phillips, J.; Smirnov, I.; Tom, M.W.; Prados, M.D.; James, C.D.; et al. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro Oncol. 2014, 16, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Caretti, V.; Hiddingh, L.; Lagerweij, T.; Schellen, P.; Koken, P.W.; Hulleman, E.; van Vuurden, D.G.; Vandertop, W.P.; Kaspers, G.J.; Noske, D.P.; et al. WEE1 kinase inhibition enhances the radiation response of diffuse intrinsic pontine gliomas. Mol. Cancer 2013, 12, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The_UniProt_Consortium. UniProtKB—Q13315 (ATM_HUMAN). Available online: www.uniprot.org/uniprot/Q13315 (accessed on 14 November 2021).
- Armata, H.L.; Golebiowski, D.; Jung, D.Y.; Ko, H.J.; Kim, J.K.; Sluss, H.K. Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis. Mol. Cell Biol. 2010, 30, 5787–5794. [Google Scholar] [CrossRef] [Green Version]
- Lavin, M.F. Radiosensitivity and oxidative signalling in ataxia telangiectasia: An update. Radiother. Oncol. 1998, 47, 113–123. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Wu, Z.; Wang, L.; Wu, Y.; Li, D.; Ma, U.; Shao, J.; Yu, H.; Wang, D. Silencing of ATM expression by siRNA technique contributes to glioma stem cell radiosensitivity in vitro and in vivo. Oncol. Rep. 2017, 38, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.C.; Quek, H.; Offenhauser, C.; Fazry, S.; Boyd, A.; Lavin, M.; Roberts, T.; Day, B. ATM inhibition prevents interleukin-6 from contributing to the proliferation of glioblastoma cells after ionizing radiation. J. NeuroOncol. 2018, 138, 509–518. [Google Scholar] [CrossRef]
- Sinha, S.; Ghildiyal, R.; Mehta, V.S.; Sen, E. ATM-NFkappaB axis-driven TIGAR regulates sensitivity of glioma cells to radiomimetics in the presence of TNFalpha. Cell Death Dis 2013, 4, e615. [Google Scholar] [CrossRef] [Green Version]
- Nadkarni, A.; Shrivastav, M.; Mladek, A.C.; Schwingler, P.M.; Grogan, P.T.; Chen, J.; Sarkaria, J.N. ATM inhibitor KU-55933 increases the TMZ responsiveness of only inherently TMZ sensitive GBM cells. J. NeuroOncol. 2012, 110, 349–357. [Google Scholar] [CrossRef] [Green Version]
- The_UniProt_Consortium. UniProtKB—D6RIG7 (D6RIG7_HUMAN). Available online: https://www.uniprot.org/uniprot/D6RIG7 (accessed on 14 November 2021).
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar] [CrossRef]
- Middleton, F.K.; Patterson, M.J.; Elstob, C.J.; Fordham, S.; Herriott, A.; Wade, M.A.; Mccormick, A.; Edmodson, R.; May, F.E.B.; Allan, J.M.; et al. Common cancer-associated imbalances in the DNA damage response confer sensitivity to single agent ATR inhibition. Oncotarget 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Xie, G.; Zhou, G.; Cheng, Y.; Zhang, G.; Yao, G.; Chen, Y.; Li, Y.; Zhao, G. NVP-BEZ235, a novel dual PI3K-mTOR inhibitor displays anti-glioma activity and reduces chemoresistance to temozolomide in human glioma cells. Cancer Lett. 2015, 367, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suva, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Noorbakhsh, S.I.; Sundaram, R.K.; Kalathil, A.N.; Ganesa, S.; Jia, L.; Breslin, H.; Burgenske, D.M.; Gilad, O.; Sarkaria, J.N.; et al. Temozolomide Sensitizes MGMT-Deficient Tumor Cells to ATR Inhibitors. Cancer Res. 2019, 79, 4331–4338. [Google Scholar] [CrossRef]
- The_UniProt_Consortium. UniProtKB-O14757 (CHK1_HUMAN). Available online: www.uniprot.org/uniprot/O14757 (accessed on 14 November 2021).
- The_UniProt_Consortium. UniProtKB-Q683Z8 (Q683Z8_HUMAN). Available online: www.uniprot.org/uniprot/Q683Z8 (accessed on 14 November 2021).
- Wu, J.; Lai, G.; Wan, F.; Xiao, Z.; Zeng, L.; Wang, X.; Ye, F.; Lei, T. Knockdown of CheckpoInt. Kinase 1 Is Associated with the Increased Radiosensitivity of Glioblastoma Stem-Like Cells. Tohoku J. Exp. Med. 2012, 226, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Carrassa, L.; Broggini, M.; Erba, E.; Damia, G. Chk1, but not Chk2, is involved in the cellular response to DNA damaging agents: Differential activity in cells expressing or not p53. Cell Cycle 2004, 3, 1177–1181. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoInt. and inhibition of homologous recombinational DNA repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef] [Green Version]
- Pollack, I.F.K.S.; Lazo, J.S. Blocking of glioma proliferation in vitro and in vivo and potentiating the effects of BCNU and cisplatin: UCN-01, a selective protein kinase C inhibitor. J. Neurosurg. 1996, 1024–1032. [Google Scholar] [CrossRef]
- Signore, M.; Pelacchi, F.; di Martino, S.; Runci, D.; Biffoni, M.; Giannetti, S.; Morgante, L.; De Majo, M.; Petricoin, E.F.; Stancato, L.; et al. Combined PDK1 and CHK1 inhibition is required to kill glioblastoma stem-like cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1223. [Google Scholar] [CrossRef] [Green Version]
- Kruger, K.; Geist, K.; Stuhldreier, F.; Schumacher, L.; Blumel, L.; Remke, M.; Wesselborg, S.; Stork, B.; Klocker, N.; Bormann, S.; et al. Multiple DNA damage-dependent and DNA damage-independent stress responses define the outcome of ATR/Chk1 targeting in medulloblastoma cells. Cancer Lett. 2018, 430, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.M.; Pieper, R.O. Abrogation of the Chk1-mediated G(2) checkpoInt. pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res. 2005, 5843–5849. [Google Scholar]
- Hirose, Y.M.; Mirzoeva, O.K.; Berger, M.S.; Pieper, R.O. Akt activation suppresses Chk2-mediated, methylating agent-induced G2 arrest and protects from temozolomide-induced mitotic catastrophe and cellular senescence. Cancer Res. 2005, 4861–4869. [Google Scholar] [CrossRef] [Green Version]
- Hauge, S.; Macurek, L.; Syljuåsen, R.G. p21 limits S phase DNA damage caused by the Wee1 inhibitor MK1775. Cell Cycle 2019, 18, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.S.; Gasol, A.; Slangen, P.L.G.; Cornelissen, F.M.G.; Lagerweij, T.; Veldman, H.; Dik, R.; van den Berg, J.; Slotman, B.J.; Wurdinger, T.; et al. Identification of MEK162 as a Radiosensitizer for the Treatment of Glioblastoma. Mol. Cancer 2018, 17, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, P.; Sun, L.; Robbins, Y.; Clavijo, P.E.; Friedman, J.; Silvin, C.; Van Waes, C.; Cook, J.; Mitchell, J.; Allen, C. Enhancing direct cytotoxicity and response to immune checkpoInt. blockade following ionizing radiation with Wee1 kinase inhibition. Oncoimmunology 2019, 8, e1638207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golden, E.B.; Pellicciotta, I.; Demaria, S.; Barcellos-Hoff, M.H.; Formenti, S.C. The convergence of radiation and immunogenic cell death signaling pathways. Front. Oncol. 2012, 2, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herskind, C.; Wenz, F.; Giordano, F.A. Immunotherapy Combined with Large Fractions of Radiotherapy: Stereotactic Radiosurgery for Brain Metastases-Implications for Intraoperative Radiotherapy after Resection. Front. Oncol. 2017, 7, 147. [Google Scholar] [CrossRef] [Green Version]
- Van Bijsterveldt, L.; Durley, S.C.; Maughan, T.S.; Humphrey, T.C. The Challenge of Combining Chemo- and Radiotherapy with CheckpoInt. Kinase Inhibitors. Clin. Cancer Res. 2021, 27, 937–962. [Google Scholar] [CrossRef]
- Metselaar, D.S.; du Chatinier, A.; Stuiver, I.; Kaspers, G.J.L.; Hulleman, E. Radiosensitization in Pediatric High-Grade Glioma: Targets, Resistance and Developments. Front. Oncol. 2021, 11, 662209. [Google Scholar] [CrossRef]
- NIH. U.S. National Institutes of Health: ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 10 January 2022).
- Sanai, N.; Li, J.; Boerner, J.; Stark, K.; Wu, J.; Kim, S.; Derogatis, A.; Mehta, S.; Dhruv, H.D.; Heilbrun, L.K.; et al. Phase 0 Trial of AZD1775 in First-Recurrence Glioblastoma Patients. Clin. Cancer Res. 2018, 24, 3820–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vulpen, M.; Kal, H.B.; Taphoorn, M.J.; El-Sharouni, S.Y. Changes in blood-brain barrier permeability induced by radiotherapy: Implications for timing of chemotherapy? (Review). Oncol. Rep. 2002, 9, 683–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Study to Assess the Safety and Tolerability of AZD1390 Given with Radiation Therapy in Patients with Brain Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03423628 (accessed on 10 January 2022).
- Evaluation of LY2606368 Therapy in Combination with Cyclophosphamide or Gemcitabine for Children and Adolescents With Refractory or Recurrent Group 3/Group 4 or SHH Medulloblastoma Brain Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT04023669 (accessed on 10 January 2022).
- A Phase 0 Study of AZD1775 in Recurrent GBM Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT02207010 (accessed on 10 January 2022).
- Adavosertib, Radiation Therapy, and Temozolomide in Treating Patients with Newly Diagnosed or Recurrent Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT01849146 (accessed on 10 January 2022).
- Adavosertib and Local Radiation Therapy in Treating Children with Newly Diagnosed Diffuse Intrinsic Pontine Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT01922076 (accessed on 10 January 2022).
- Adavosertib and Irinotecan Hydrochloride in Treating Younger Patients with Relapsed or Refractory Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT02095132 (accessed on 10 January 2022).
- Targeted Therapy Directed by Genetic Testing in Treating Patients with Advanced Refractory Solid Tumors, Lymphomas, or Multiple Myeloma (The MATCH Screening Trial). Available online: https://clinicaltrials.gov/ct2/show/NCT02465060 (accessed on 10 January 2022).



{kind=link}
{kind=link}
| Protein Name (Gene Symbol) | Molecular Weight | Molecular Function | Inhibitor Used for Addressed Target in Combination with RT on Brain Tumor Models | References |
|---|---|---|---|---|
| ATM (ATM) | 350,687 Da | DNA- and ATP-binding Serine/threonine protein kinase | NVP-BEZ235 | [71] |
| KU-55933 | [72,73,74,75,76] | |||
| KU-60019 | [76,77,78,79] | |||
| AZD1390 | [80] | |||
| AZ31 and AZ32 | [81] | |||
| ATR (ATR) | 301,367 Da | DNA- and ATP-binding Serine/threonine protein kinase | NVP-BEZ235 | [82] |
| AZD6738 | [82] | |||
| VE821 | [83,84,85] | |||
| VE822 | [84] | |||
| AZ20 | [84] | |||
| CHK1 (CHEK1) | 54,434 Da | ATP-binding Serine/threonine protein kinase | Gö-6976 | [73] |
| UCN-01 | [86,87,88] | |||
| debromohymenialdisine | [38] | |||
| SAR-020106 | [89] | |||
| SCH900776 | [76] | |||
| CHIR-124 | [76] | |||
| CHK2 (CHEK2) | 60,915 Da | ATP-binding Serine/threonine protein kinase | BML-277 | [73] |
| debromohymenialdisine | [38] | |||
| NSC 109555 | [90] | |||
| WEE1 (WEE1) | 71,597 Da | ATP-binding Serine/threonine protein kinase | MK-1775 | [80,91,92,93,94] |
| PD0166285 | [88] |
| Target | Drug | Phase | Estimated End Date/End Date | Tumor | ClinicalTrials.Gov Identifier | References | Status | Other Treatments |
|---|---|---|---|---|---|---|---|---|
| ATM | AZD1390 | I | 14 December 2023 | Brain tumors, Leptomeningeal disease | NCT03423628 | [129] | Active | RT |
| CHK1 | LY2606368 | I | June 2026 | Medulloblastoma in pediatric patients | NCT04023669 | [130] | Active | Cyclophosphamide Gemcitabine Filgrastim peg-filgrastim |
| WEE1 | AZD1775 (MK-1775) | I | 25 March 2019 | Recurrent glioblastoma | NCT02207010 | [127,131] | Completed | RT |
| WEE1 | AZD1775 (MK-1775) | I | 29 July 2021, primary | Glioblastoma | NCT01849146 | [132] | Active | RT, TMZ |
| WEE1 | AZD1775 (MK-1775) | I | 31 October 2021, primary | Diffuse intrinsic pontine gliomas | NCT01922076 | [133] | Active | RT |
| WEE1 | AZD1775 (MK-1775) | I | 31 December 2021 | Relapsed or refractory solid tumors (including medulloblastoma) | NCT02095132 | [134] | Active | Irinotecan hydrochloride |
| WEE1 | AZD1775 (MK-1775) | II | 30 June 2022 | Advanced refractory solid tumors (including gliomas), lymphomas, or multiple myeloma | NCT02465060 | [135] | Active | Multiple drugs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlatkovic, T.; Veldwijk, M.R.; Giordano, F.A.; Herskind, C. Targeting Cell Cycle Checkpoint Kinases to Overcome Intrinsic Radioresistance in Brain Tumor Cells. Cancers 2022, 14, 701. https://doi.org/10.3390/cancers14030701
Vlatkovic T, Veldwijk MR, Giordano FA, Herskind C. Targeting Cell Cycle Checkpoint Kinases to Overcome Intrinsic Radioresistance in Brain Tumor Cells. Cancers. 2022; 14(3):701. https://doi.org/10.3390/cancers14030701
Chicago/Turabian StyleVlatkovic, Tijana, Marlon R. Veldwijk, Frank A. Giordano, and Carsten Herskind. 2022. "Targeting Cell Cycle Checkpoint Kinases to Overcome Intrinsic Radioresistance in Brain Tumor Cells" Cancers 14, no. 3: 701. https://doi.org/10.3390/cancers14030701