
Fluorescence Imaging of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Resistance in Non-Small Cell Lung Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. A Brief Outline of Non-Small Cell Lung Cancer
| EGFR | EGFR is one of the four members of the human epidermal growth factor (HER) family transmembrane receptors (HER1/EGFR, HER2, HER3, and HER4). The prevalence of EGFR oncogene mutations is 50% among Asian patients with lung adenocarcinoma and 15% among Western patients [7]. Exon 19 deletions or L858R point mutations in exon 21 account for 90% of the activating mutations in the tyrosine kinase domain of EGFR, resulting in constitutive activation of EGFR without growth factor-induced stimulation, thus promoting cell proliferation [5]. |
| KRAS | KRAS is the predominantly mutated RAS isoform (85%) and also the most frequent oncogene in NSCLC [8]. KRAS fosters tumour growth via several mechanisms, including by upregulating rate-limiting enzymes involved in amino acid, fatty acid, or nucleotide biosynthesis, and by stimulating scavenging pathways, such as macropinocytosis and autophagy [9,10], which, in turn provide building blocks for the anabolic routes, also maintaining the energy levels and the cell’s redox potential [11]. |
| BRAF | BRAF is a proto-oncogene encoding a serine-threonine protein kinase acting downstream of the RAS/RAF/ERK signalling pathway. BRAF carries signals from membrane receptors (such as EGFR) to the nucleus of the cell to regulate DNA transcription [12]. BRAF is an oncogene located on chromosome 7 involved in several cell functions, including growth, proliferation, survival, and differentiation. Immunotherapy is beginning to show promise as an active therapy in BRAF-mutated NSCLC [13]. |
| ALK | The ALK gene encodes the ALK tyrosine kinase receptor and is associated with many types of cancers, including NSCLC [14]. There are three types of ALK mutations: rearrangement (ALK-R), amplification (ALK-A), and point mutation. ALK gene rearrangement is a driving mutation underlying the development of NSCLC [15], which appears to be more common in younger patients and never or light smokers diagnosed with adenocarcinoma. ALK can phosphorylate STAT3 and PI3K independently of ERK to antagonise apoptosis and promote cell survival [16]. |
| TP53 | The TP53 gene encodes a DNA damage check point p53 protein, which is at the heart of the cellular decision to proliferate or activate programmed cell death. It regulates the transcription of ~500 genes [17], including cell cycle regulatory genes and transcription factors, and DNA repair genes [18]. Over 50% of human cancers carry loss of function mutations in TP53, with the mutant form acting as a dominant-negative inhibitor towards the wild-type moiety. When chromosomal abnormalities or environment stresses become overwhelming, p53 can arrest cell-cycle progression and induce apoptosis. TP53 alterations carry a worse prognosis in NSCLC [19]. |
| MYC | A family of three human proto-oncogenes (c-MYC, l-MYC, and n-MYC) code for transcription factors [20]. In normal cells, depending on nucleotide pools’ levels, growth signals, glucose, or oxygenation, elevated MYC expression can cause apoptosis. Transformed cells can, however, adapt to constitutively elevated levels of MYC expression, resist its apoptotic effects, and only respond to MYC pro-proliferative signals either via loss of growth suppression surveillance mechanisms (e.g., TP53 mutation) and/or by gain of pro-survival signals. MYC is a metastasis gene for NSCLC [21]. |
2. EGFR’s Role in the Development of NSCLC Tumours
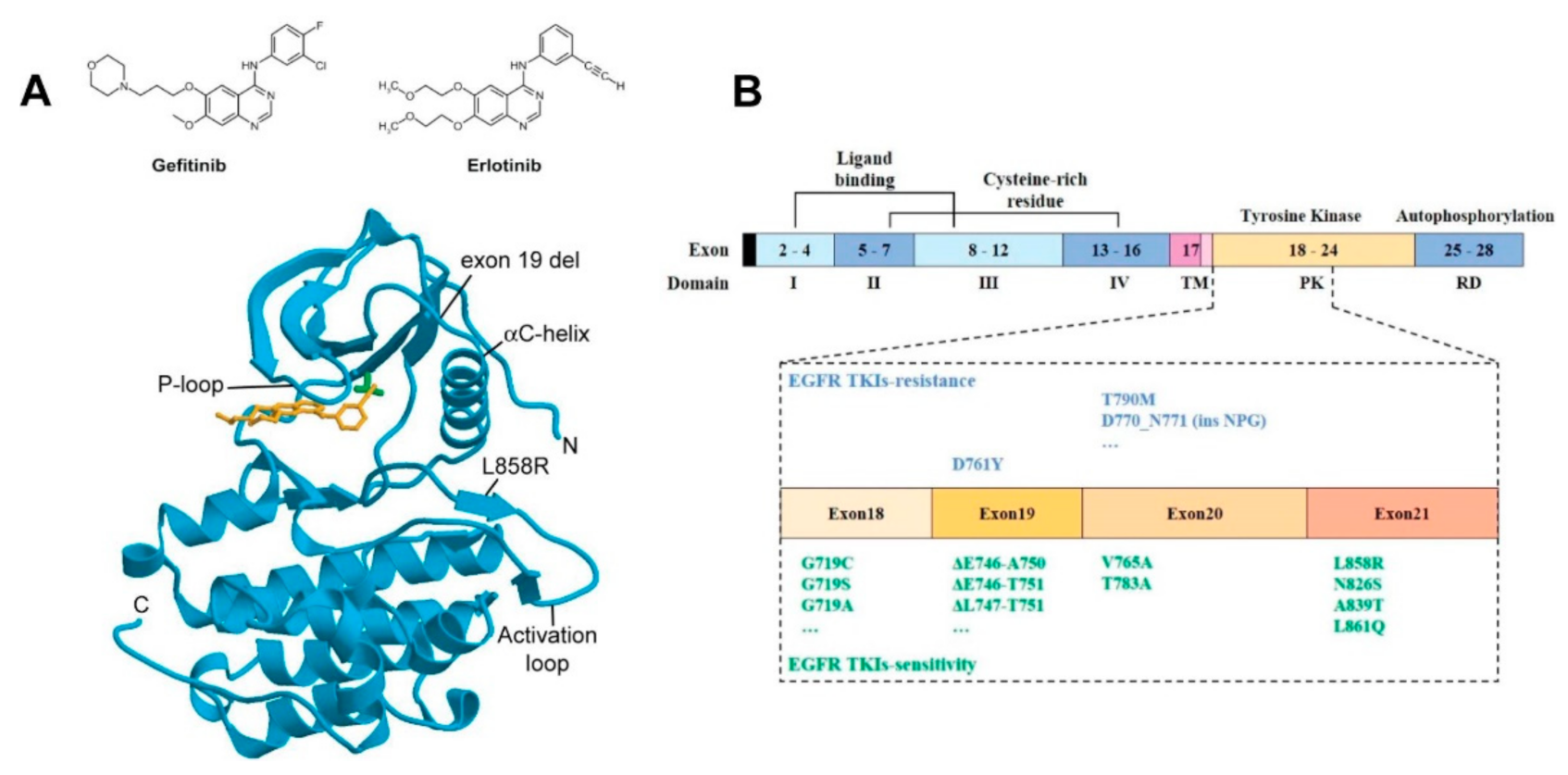
2.1. EGFR Structure and Signalling Pathways
2.2. TKI Treatments Induce Apoptosis via the Mitochondrial Intrinsic Pathway
2.3. The Development of Resistance to TKI Treatment
3. At the Interface between Autophagy and Resistance to TKIs
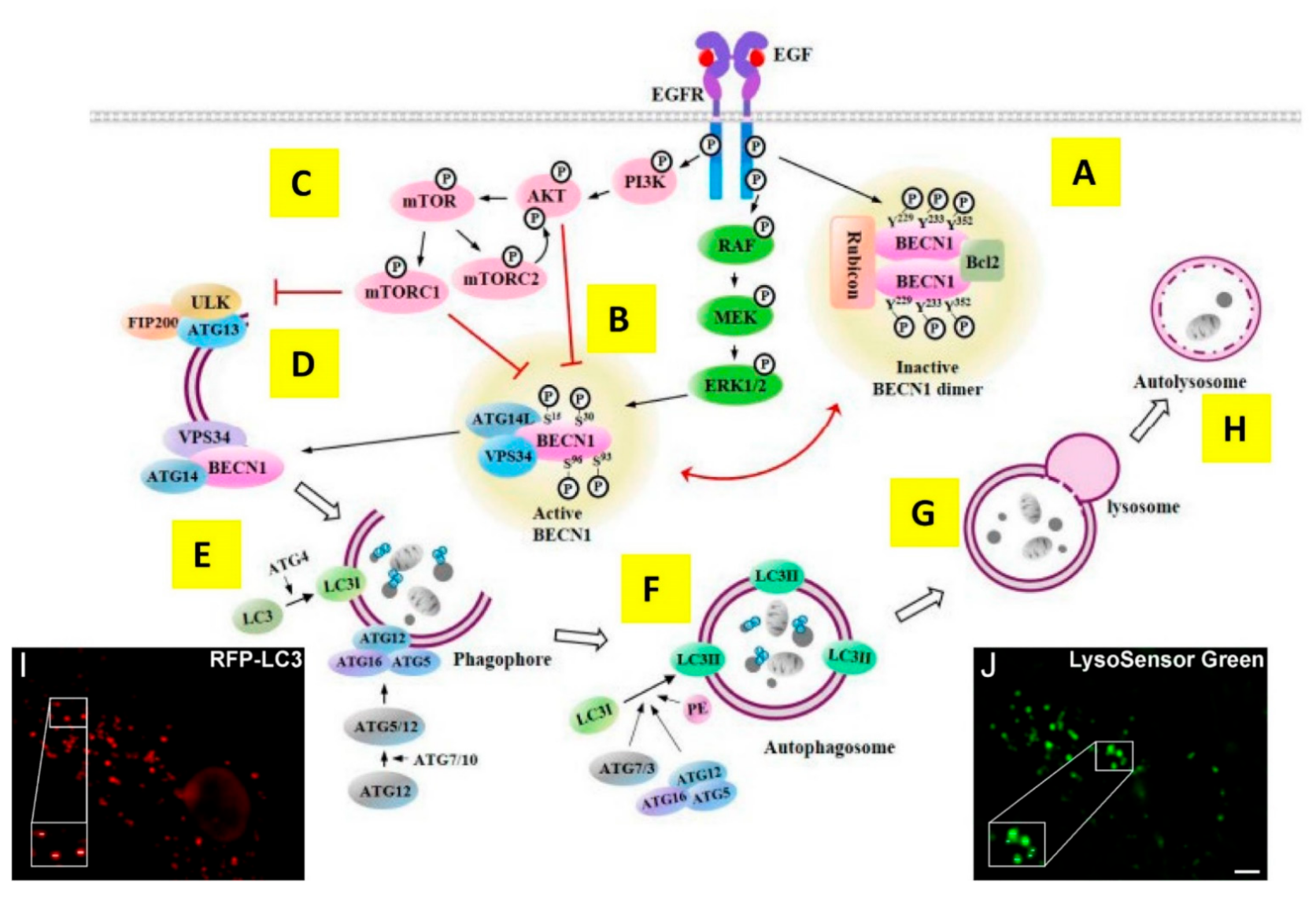
3.1. A Brief Overview of (Macro) Autophagy
3.2. Autophagy and Its Relationship with Apoptosis and Cancer Progression
3.3. The Dual-Edged Sword of Targeting Autophagy
4. Endocytosis and Autophagy: Friends or Foes?
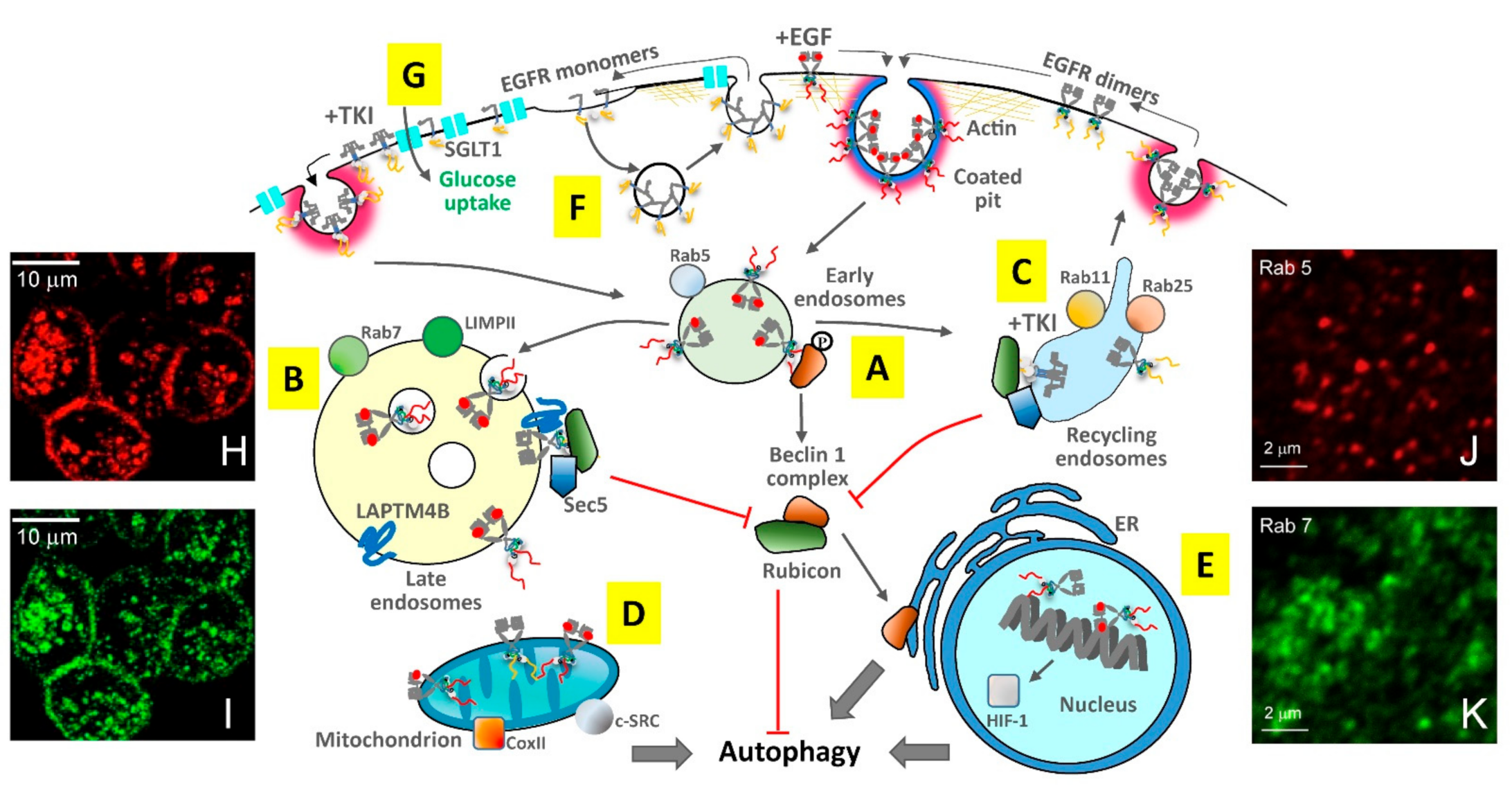
4.1. Endocytosis Walks ‘Hand-In-Hand’ with Autophagy
4.2. Clathrin-Mediated Endocytosis Distributes Egfr throughout Vesicles and Organelles
4.3. Endocytic Trafficking Underpins the TKI Response
4.4. Compartmentalised EGFR Interactions Balance the Regulation of Autophagy
4.5. Endocytosis Underpins Different Responses to TKIs Depending on the EGF Stimulus
5. A Perspective from a Structural Viewpoint
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Inamura, K. Lung Cancer: Understanding Its Molecular Pathology and the 2015 WHO Classification. Front. Oncol. 2017, 7, 193. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Qi, Y.; Wampfler, J.A.; Jatoi, A.; Garces, Y.I.; Busta, A.J.; Mandrekar, S.J.; Yang, P. Effect of cigarette smoking on quality of life in small cell lung cancer patients. Eur. J. Cancer 2012, 48, 1593–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Dai, W. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, 5, 165. [Google Scholar] [CrossRef]
- Soria, J.C.; Mok, T.S.; Cappuzzo, F.; Janne, P.A. EGFR-mutated oncogene-addicted non-small cell lung cancer: Current trends and future prospects. Cancer Treat. Rev. 2012, 38, 416–430. [Google Scholar] [CrossRef]
- Chang, Y.S.; Tu, S.J.; Chen, Y.C.; Liu, T.Y.; Lee, Y.T.; Yen, J.C.; Fang, H.Y.; Chang, J.G. Mutation profile of non-small cell lung cancer revealed by next generation sequencing. Respir. Res. 2021, 22, 3. [Google Scholar] [CrossRef]
- Chevallier, M.; Borgeaud, M.; Addeo, A.; Friedlaender, A. Oncogenic driver mutations in non-small cell lung cancer: Past, present and future. World J. Clin. Oncol. 2021, 12, 217–237. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Jayashankar, V.; Edinger, A.L. Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nat. Commun. 2020, 11, 1121. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [Green Version]
- Kerr, E.M.; Martins, C.P. Metabolic rewiring in mutant Kras lung cancer. FEBS J. 2018, 285, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.G.; Zhang, S.M.; Ding, X.X.; He, B.; Zhang, H.Q. Driver genes in non-small cell lung cancer: Characteristics, detection methods, and targeted therapies. Oncotarget 2017, 8, 57680–57692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, C.G.; Andelkovic, V.; Ladwa, R.; Pavlakis, N.; Zhou, C.; Hirsch, F.; Richard, D.; O’Byrne, K. Targeting BRAF mutations in non-small cell lung cancer. Transl. Lung Cancer Res. 2019, 8, 1119–1124. [Google Scholar] [CrossRef]
- Du, X.; Shao, Y.; Qin, H.F.; Tai, Y.H.; Gao, H.J. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac. Cancer 2018, 9, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Devarakonda, S.; Morgensztern, D.; Govindan, R. Genomic alterations in lung adenocarcinoma. Lancet Oncol. 2015, 16, e342–e351. [Google Scholar] [CrossRef]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Harris, C.C. p53: Traffic cop at the crossroads of DNA repair and recombination. Nat. Rev. Mol. Cell Biol. 2005, 6, 44–55. [Google Scholar] [CrossRef]
- Mogi, A.; Kuwano, H. TP53 mutations in nonsmall cell lung cancer. J. Biomed. Biotechnol. 2011, 2011, 583929. [Google Scholar] [CrossRef] [Green Version]
- McMahon, S.B. MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [Green Version]
- Rapp, U.R.; Korn, C.; Ceteci, F.; Karreman, C.; Luetkenhaus, K.; Serafin, V.; Zanucco, E.; Castro, I.; Potapenko, T. MYC is a metastasis gene for non-small-cell lung cancer. PLoS ONE 2009, 4, e6029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Kohler, J.; Schuler, M. Afatinib, erlotinib and gefitinib in the first-line therapy of EGFR mutation-positive lung adenocarcinoma: A review. Onkologie 2013, 36, 510–518. [Google Scholar] [CrossRef]
- Dimou, A.T.; Syrigos, K.N.; Agarwal, S.; Lozovatsky, L.; Zolota, V.; Anagnostou, V.K.; Rimm, D. Frequency of L858R and D746-750 EGFR mutations in 411 Caucasian patients with non-small cell lung cancer measured by mutation-specific antibodies. J. Clin. Oncol. 2010, 28, 1587. [Google Scholar] [CrossRef]
- Li, A.R.; Chitale, D.; Riely, G.J.; Pao, W.; Miller, V.A.; Zakowski, M.F.; Rusch, V.; Kris, M.G.; Ladanyi, M. EGFR mutations in lung adenocarcinomas: Clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. J. Mol. Diagn. 2008, 10, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Somwar, R.; Politi, K.; Balak, M.; Chmielecki, J.; Jiang, X.; Pao, W. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007, 4, e294. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Maemondo, M.; Minegishi, Y.; Inoue, A.; Kobayashi, K.; Harada, M.; Okinaga, S.; Morikawa, N.; Oizumi, S.; Tanaka, T.; Isobe, H.; et al. First-line gefitinib in patients aged 75 or older with advanced non-small cell lung cancer harboring epidermal growth factor receptor mutations: NEJ 003 study. J. Thorac. Oncol. 2012, 7, 1417–1422. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Khan, I.; Upadhyay, S.; Lewanski, C.; Falk, S.; Skailes, G.; Marshall, E.; Woll, P.J.; Hatton, M.; Lal, R.; et al. First-line erlotinib in patients with advanced non-small-cell lung cancer unsuitable for chemotherapy (TOPICAL): A double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2012, 13, 1161–1170. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wei, Y.; Yu, D.; Xu, J.; Peng, J. Gefitinib provides similar effectiveness and improved safety than erlotinib for east Asian populations with advanced non-small cell lung cancer: A meta-analysis. BMC Cancer 2018, 18, 780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Yu, C.J.; Kim, S.W.; Lin, M.C.; Sriuranpong, V.; Tsai, C.M.; Lee, J.S.; Kang, J.H.; Chan, K.C.; Perez-Moreno, P.; et al. First-Line Erlotinib Therapy Until and Beyond Response Evaluation Criteria in Solid Tumors Progression in Asian Patients With Epidermal Growth Factor Receptor Mutation-Positive Non-Small-Cell Lung Cancer: The ASPIRATION Study. JAMA Oncol. 2016, 2, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Manasanch, E.E.; Wilkerson, J.; Fojo, T. Gefitinib and erlotinib in metastatic non-small cell lung cancer: A meta-analysis of toxicity and efficacy of randomized clinical trials. Oncologist 2015, 20, 400–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.; Cools, J.; Gilliland, D.G. EGFR inhibition in non-small cell lung cancer: Resistance, once again, rears its ugly head. PLoS Med. 2005, 2, e75. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.; Kim, M.; Jung, H.S.; Kim, Y.; Jeoung, D. Targeting Autophagy for Overcoming Resistance to Anti-EGFR Treatments. Cancers 2019, 11, 1374. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, C.N. Epidermal growth factor receptor in non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 110–118. [Google Scholar] [CrossRef]
- Xu, N.; Fang, W.; Mu, L.; Tang, Y.; Gao, L.; Ren, S.; Cao, D.; Zhou, L.; Zhang, A.; Liu, D.; et al. Overexpression of wildtype EGFR is tumorigenic and denotes a therapeutic target in non-small cell lung cancer. Oncotarget 2016, 7, 3884–3896. [Google Scholar] [CrossRef]
- Thomas, R.; Weihua, Z. Rethink of EGFR in Cancer with Its Kinase Independent Function on Board. Front. Oncol. 2019, 9, 800. [Google Scholar] [CrossRef]
- Jo, U.; Park, K.H.; Whang, Y.M.; Sung, J.S.; Won, N.H.; Park, J.K.; Kim, Y.H. EGFR endocytosis is a novel therapeutic target in lung cancer with wild-type EGFR. Oncotarget 2014, 5, 1265–1278. [Google Scholar] [CrossRef] [Green Version]
- Crowley, L.C.; Marfell, B.J.; Scott, A.P.; Waterhouse, N.J. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, prot087288. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Wang, X.; Xu, P.; Liu, G.; Eden, H.S.; Chen, X. Molecular imaging of apoptosis: From micro to macro. Theranostics 2015, 5, 559–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shearer, L.J.; Petersen, N.O. Distribution and Co-localization of endosome markers in cells. Heliyon 2019, 5, e02375. [Google Scholar] [CrossRef] [Green Version]
- Jovic, M.; Sharma, M.; Rahajeng, J.; Caplan, S. The early endosome: A busy sorting station for proteins at the crossroads. Histol. Histopathol. 2010, 25, 99–112. [Google Scholar] [CrossRef]
- Mu, F.T.; Callaghan, J.M.; Steele-Mortimer, O.; Stenmark, H.; Parton, R.G.; Campbell, P.L.; McCluskey, J.; Yeo, J.P.; Tock, E.P.; Toh, B.H. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine “fingers” and contains a calmodulin-binding IQ motif. J. Biol. Chem. 1995, 270, 13503–13511. [Google Scholar] [CrossRef] [Green Version]
- Willoughby, P.M.; Allen, M.; Yu, J.; Korytnikov, R.; Chen, T.; Liu, Y.; So, I.; Macpherson, N.; Mitchell, J.A.; Fernandez-Gonzalez, R.; et al. The recycling endosome protein Rab25 coordinates collective cell movements in the zebrafish surface epithelium. eLife 2021, 10, e66060. [Google Scholar] [CrossRef]
- Vanlandingham, P.A.; Ceresa, B.P. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Biol. Chem. 2009, 284, 12110–12124. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.T.; Xie, Y.X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.H. Revisiting LAMP1 as a marker for degradative autophagy-lysosomal organelles in the nervous system. Autophagy 2018, 14, 1472–1474. [Google Scholar] [CrossRef]
- Nishimura, Y.; Bereczky, B.; Ono, M. The EGFR inhibitor gefitinib suppresses ligand-stimulated endocytosis of EGFR via the early/late endocytic pathway in non-small cell lung cancer cell lines. Histochem. Cell Biol. 2007, 127, 541–553. [Google Scholar] [CrossRef]
- Tynan, C.J.; Clarke, D.T.; Coles, B.C.; Rolfe, D.J.; Martin-Fernandez, M.L.; Webb, S.E. Multicolour single molecule imaging in cells with near infra-red dyes. PLoS ONE 2012, 7, e36265. [Google Scholar] [CrossRef] [PubMed]
- England, C.G.; Luo, H.; Cai, W. HaloTag technology: A versatile platform for biomedical applications. Bioconjug. Chem. 2015, 26, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Bosch, P.J.; Correa, I.R., Jr.; Sonntag, M.H.; Ibach, J.; Brunsveld, L.; Kanger, J.S.; Subramaniam, V. Evaluation of fluorophores to label SNAP-tag fused proteins for multicolor single-molecule tracking microscopy in live cells. Biophys. J. 2014, 107, 803–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecker, M.; Redpath, G.M.I.; Nicovich, P.R.; Rossy, J. Quantitative visualization of endocytic trafficking through photoactivation of fluorescent proteins. Mol. Biol. Cell 2021, 32, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Varella-Garcia, M.; Diebold, J.; Eberhard, D.A.; Geenen, K.; Hirschmann, A.; Kockx, M.; Nagelmeier, I.; Ruschoff, J.; Schmitt, M.; Arbogast, S.; et al. EGFR fluorescence in situ hybridisation assay: Guidelines for application to non-small-cell lung cancer. J. Clin. Pathol. 2009, 62, 970–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Backues, S.K.; Chen, D.; Ruan, J.; Xie, Z.; Klionsky, D.J. Estimating the size and number of autophagic bodies by electron microscopy. Autophagy 2014, 10, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Orhon, I.; Reggiori, F. Assays to Monitor Autophagy Progression in Cell Cultures. Cells 2017, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Ling, Y.H.; Sironi, J.; Schwartz, E.L.; Perez-Soler, R.; Piperdi, B. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J. Thorac. Oncol. 2013, 8, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Bains, M.; Heidenreich, K.A. Live-cell imaging of autophagy induction and autophagosome-lysosome fusion in primary cultured neurons. Methods Enzymol. 2009, 453, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Sankaranarayanan, S.; De Angelis, D.; Rothman, J.E.; Ryan, T.A. The use of pHluorins for optical measurements of presynaptic activity. Biophys. J. 2000, 79, 2199–2208. [Google Scholar] [CrossRef] [Green Version]
- Martin-Fernandez, M.L.; Clarke, D.T. Single molecule fluorescence detection and tracking in mammalian cells: The state-of-the-art and future perspectives. Int. J. Mol. Sci. 2012, 13, 14742–14765. [Google Scholar] [CrossRef] [PubMed]
- Low-Nam, S.T.; Lidke, K.A.; Cutler, P.J.; Roovers, R.C.; van Bergen en Henegouwen, P.M.; Wilson, B.S.; Lidke, D.S. ErbB1 dimerization is promoted by domain co-confinement and stabilized by ligand binding. Nat. Struct. Mol. Biol. 2011, 18, 1244–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolfe, D.J.; McLachlan, C.I.; Hirsch, M.; Needham, S.R.; Tynan, C.J.; Webb, S.E.; Martin-Fernandez, M.L.; Hobson, M.P. Automated multidimensional single molecule fluorescence microscopy feature detection and tracking. Eur. Biophys. J. 2011, 40, 1167–1186. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.L.; Tynan, C.J.; Webb, S.E. A ‘pocket guide’ to total internal reflection fluorescence. J. Microsc. 2013, 252, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanetti-Domingues, L.C.; Tynan, C.J.; Rolfe, D.J.; Clarke, D.T.; Martin-Fernandez, M. Hydrophobic fluorescent probes introduce artifacts into single molecule tracking experiments due to non-specific binding. PLoS ONE 2013, 8, e74200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanetti-Domingues, L.C.; Martin-Fernandez, M.L.; Needham, S.R.; Rolfe, D.J.; Clarke, D.T. A systematic investigation of differential effects of cell culture substrates on the extent of artifacts in single-molecule tracking. PLoS ONE 2012, 7, e45655. [Google Scholar] [CrossRef]
- Lv, C.; Yang, C.; Ding, D.; Sun, Y.; Wang, R.; Han, D.; Tan, W. Endocytic Pathways and Intracellular Transport of Aptamer-Drug Conjugates in Live Cells Monitored by Single-Particle Tracking. Anal. Chem. 2019, 91, 13818–13823. [Google Scholar] [CrossRef]
- Izeddin, I.; Recamier, V.; Bosanac, L.; Cisse, I.I.; Boudarene, L.; Dugast-Darzacq, C.; Proux, F.; Benichou, O.; Voituriez, R.; Bensaude, O.; et al. Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. eLife 2014, 3, e02230. [Google Scholar] [CrossRef] [Green Version]
- Harootunian, A.; Betzig, E.; Isaacson, M.; Lewis, A. Superresolution Fluorescence Near-Field Scanning Optical Microscopy. Appl. Phys. Lett. 1986, 49, 674–676. [Google Scholar] [CrossRef]
- Manzo, C.; van Zanten, T.S.; Garcia-Parajo, M.F. Nanoscale fluorescence correlation spectroscopy on intact living cell membranes with NSOM probes. Biophys. J. 2011, 100, L8–L10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Bateman, B.; Zanetti-Domingues, L.C.; Moores, A.N.; Astbury, S.; Spindloe, C.; Darrow, M.C.; Romano, M.; Needham, S.R.; Beis, K.; et al. Solid immersion microscopy images cells under cryogenic conditions with 12 nm resolution. Commun. Biol. 2019, 2, 74. [Google Scholar] [CrossRef] [PubMed]
- Sekar, R.B.; Periasamy, A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 2003, 160, 629–633. [Google Scholar] [CrossRef] [Green Version]
- Förster, T. Delocalized excitation and excitation transfer. In Modern Quantum Chemistry; Sinanoglu, O., Ed.; Academic Press Inc.: New York, NY, USA, 1965; Volume 2, pp. 93–137. [Google Scholar]
- Martin-Fernandez, M.; Clarke, D.T.; Tobin, M.J.; Jones, S.V.; Jones, G.R. Preformed oligomeric epidermal growth factor receptors undergo an ectodomain structure change during signaling. Biophys. J. 2002, 82, 2415–2427. [Google Scholar] [CrossRef]
- Kiuchi, T.; Ortiz-Zapater, E.; Monypenny, J.; Matthews, D.R.; Nguyen, L.K.; Barbeau, J.; Coban, O.; Lawler, K.; Burford, B.; Rolfe, D.J.; et al. The ErbB4 CYT2 variant protects EGFR from ligand-induced degradation to enhance cancer cell motility. Sci. Signal. 2014, 7, ra78. [Google Scholar] [CrossRef]
- Webb, S.E.; Roberts, S.K.; Needham, S.R.; Tynan, C.J.; Rolfe, D.J.; Winn, M.D.; Clarke, D.T.; Barraclough, R.; Martin-Fernandez, M.L. Single-molecule imaging and fluorescence lifetime imaging microscopy show different structures for high- and low-affinity epidermal growth factor receptors in A431 cells. Biophys. J. 2008, 94, 803–819. [Google Scholar] [CrossRef] [Green Version]
- Webb, S.E.; Needham, S.R.; Roberts, S.K.; Martin-Fernandez, M.L. Multidimensional single-molecule imaging in live cells using total-internal-reflection fluorescence microscopy. Opt. Lett. 2006, 31, 2157–2159. [Google Scholar] [CrossRef]
- Gadella, T.W., Jr.; Jovin, T.M. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 1995, 129, 1543–1558. [Google Scholar] [CrossRef] [Green Version]
- Webb, S.E.; Rolfe, D.J.; Needham, S.R.; Roberts, S.K.; Clarke, D.T.; McLachlan, C.I.; Hobson, M.P.; Martin-Fernandez, M.L. Simultaneous widefield single molecule orientation and FRET microscopy in cells. Opt. Express 2008, 16, 20258–20265. [Google Scholar] [CrossRef]
- Tynan, C.J.; Roberts, S.K.; Rolfe, D.J.; Clarke, D.T.; Loeffler, H.H.; Kastner, J.; Winn, M.D.; Parker, P.J.; Martin-Fernandez, M.L. Human epidermal growth factor receptor (EGFR) aligned on the plasma membrane adopts key features of Drosophila EGFR asymmetry. Mol. Cell Biol. 2011, 31, 2241–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needham, S.R.; Hirsch, M.; Rolfe, D.J.; Clarke, D.T.; Zanetti-Domingues, L.C.; Wareham, R.; Martin-Fernandez, M.L. Measuring EGFR separations on cells with ~10 nm resolution via fluorophore localization imaging with photobleaching. PLoS ONE 2013, 8, e62331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanetti-Domingues, L.C.; Hirsch, M.; Tynan, C.J.; Rolfe, D.J.; Boyadzhiev, T.V.; Scherer, K.M.; Clarke, D.T.; Martin-Fernandez, M.L.; Needham, S.R. Determining the geometry of oligomers of the human epidermal growth factor family on cells with 7 nm resolution. Prog. Biophys. Mol. Biol. 2015, 118, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Needham, S.R.; Roberts, S.K.; Arkhipov, A.; Mysore, V.P.; Tynan, C.J.; Zanetti-Domingues, L.C.; Kim, E.T.; Losasso, V.; Korovesis, D.; Hirsch, M.; et al. EGFR oligomerization organizes kinase-active dimers into competent signalling platforms. Nat. Commun. 2016, 7, 13307. [Google Scholar] [CrossRef] [PubMed]
- Zanetti-Domingues, L.C.; Korovesis, D.; Needham, S.R.; Tynan, C.J.; Sagawa, S.; Roberts, S.K.; Kuzmanic, A.; Ortiz-Zapater, E.; Jain, P.; Roovers, R.C.; et al. The architecture of EGFR’s basal complexes reveals autoinhibition mechanisms in dimers and oligomers. Nat. Commun. 2018, 9, 4325. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Downward, J.; Yarden, Y.; Mayes, E.; Scrace, G.; Totty, N.; Stockwell, P.; Ullrich, A.; Schlessinger, J.; Waterfield, M.D. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 1984, 307, 521–527. [Google Scholar] [CrossRef]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J.; et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef]
- Chen, J.; Zeng, F.; Forrester, S.J.; Eguchi, S.; Zhang, M.Z.; Harris, R.C. Expression and Function of the Epidermal Growth Factor Receptor in Physiology and Disease. Physiol. Rev. 2016, 96, 1025–1069. [Google Scholar] [CrossRef]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000Res 2016, 5, F1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.S.; Leahy, D.J.; Lemmon, M.A. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Zhu, H.J.; Walker, F.; Frenkel, M.J.; Hoyne, P.A.; et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 2002, 110, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Cantor, A.J.; Shah, N.H.; Kuriyan, J. Deep mutational analysis reveals functional trade-offs in the sequences of EGFR autophosphorylation sites. Proc. Natl. Acad. Sci. USA 2018, 115, E7303–E7312. [Google Scholar] [CrossRef] [Green Version]
- Endres, N.F.; Das, R.; Smith, A.W.; Arkhipov, A.; Kovacs, E.; Huang, Y.; Pelton, J.G.; Shan, Y.; Shaw, D.E.; Wemmer, D.E.; et al. Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 2013, 152, 543–556. [Google Scholar] [CrossRef] [Green Version]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Batzer, A.G.; Rotin, D.; Urena, J.M.; Skolnik, E.Y.; Schlessinger, J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell Biol. 1994, 14, 5192–5201. [Google Scholar] [CrossRef]
- Molina, J.R.; Adjei, A.A. The Ras/Raf/MAPK pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Whitman, M.; Downes, C.P.; Keeler, M.; Keller, T.; Cantley, L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 1988, 332, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Bari, M.A.A.; Xu, P. Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann. N. Y. Acad. Sci. 2020, 1467, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef] [Green Version]
- Chung, B.M.; Dimri, M.; George, M.; Reddi, A.L.; Chen, G.; Band, V.; Band, H. The role of cooperativity with Src in oncogenic transformation mediated by non-small cell lung cancer-associated EGF receptor mutants. Oncogene 2009, 28, 1821–1832. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kalyankrishna, S.; Wislez, M.; Thilaganathan, N.; Saigal, B.; Wei, W.; Ma, L.; Wistuba, I.I.; Johnson, F.M.; Kurie, J.M. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am. J. Pathol. 2007, 170, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Harada, D.; Takigawa, N.; Kiura, K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers 2014, 6, 708–722. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Budkevich, I.N.; Zhivotovsky, B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 2018, 9, 117. [Google Scholar] [CrossRef]
- Chang, G.C.; Yu, C.T.; Tsai, C.H.; Tsai, J.R.; Chen, J.C.; Wu, C.C.; Wu, W.J.; Hsu, S.L. An epidermal growth factor inhibitor, Gefitinib, induces apoptosis through a p53-dependent upregulation of pro-apoptotic molecules and downregulation of anti-apoptotic molecules in human lung adenocarcinoma A549 cells. Eur. J. Pharmacol. 2008, 600, 37–44. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.B.; Halmos, B.; Kumar, A.; Schumer, S.T.; Huberman, M.S.; Boggon, T.J.; Tenen, D.G.; Kobayashi, S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007, 4, 1669–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.; Creighton, C.J.; Stivers, D.; Fujimoto, N.; Kurie, J.M. Transcriptional profiling of non-small cell lung cancer cells with activating EGFR somatic mutations. PLoS ONE 2007, 2, e1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetti, S.; Molina, M.A.; Queralt, C.; de Aguirre, I.; Mayo, C.; Bertran-Alamillo, J.; Sanchez, J.J.; Gonzalez-Larriba, J.L.; Jimenez, U.; Isla, D.; et al. Detection of EGFR mutations with mutation-specific antibodies in stage IV non-small-cell lung cancer. J. Transl. Med. 2010, 8, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, K.; Okamoto, I.; Okamoto, W.; Tanaka, K.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; Nishio, K.; Nakagawa, K. Role of survivin in EGFR inhibitor-induced apoptosis in non-small cell lung cancers positive for EGFR mutations. Cancer Res. 2010, 70, 10402–10410. [Google Scholar] [CrossRef] [Green Version]
- Westphal, D.; Kluck, R.M.; Dewson, G. Building blocks of the apoptotic pore: How Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014, 21, 196–205. [Google Scholar] [CrossRef] [Green Version]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Maes, M.E.; Schlamp, C.L.; Nickells, R.W. Live-cell imaging to measure BAX recruitment kinetics to mitochondria during apoptosis. PLoS ONE 2017, 12, e0184434. [Google Scholar] [CrossRef] [Green Version]
- Lomonosova, E.; Chinnadurai, G. BH3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27 (Suppl. 1), S2–S19. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Wang, X. Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J. Biol. Chem. 2000, 275, 31199–31203. [Google Scholar] [CrossRef] [Green Version]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Wang, X.; Jin, H. EGFR-TKI resistance in NSCLC patients: Mechanisms and strategies. Am. J. Cancer Res. 2014, 4, 411–435. [Google Scholar] [PubMed]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.; Lester, J.F. Tyrosine Kinase Inhibitors for the Treatment of EGFR Mutation-Positive Non-Small-Cell Lung Cancer: A Clash of the Generations. Clin. Lung Cancer 2020, 21, e216–e228. [Google Scholar] [CrossRef]
- Robichaux, J.P.; Le, X.; Vijayan, R.S.K.; Hicks, J.K.; Heeke, S.; Elamin, Y.Y.; Lin, H.Y.; Udagawa, H.; Skoulidis, F.; Tran, H.; et al. Structure-based classification predicts drug response in EGFR-mutant NSCLC. Nature 2021, 597, 732–737. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Gao, W.; Su, M.; Nice, E.C.; Zhang, W.; Lin, J.; Xie, N. Adaptive Mechanisms of Tumor Therapy Resistance Driven by Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 641469. [Google Scholar] [CrossRef]
- Coker, K.J.; Staros, J.V.; Guyer, C.A. A kinase-negative epidermal growth factor receptor that retains the capacity to stimulate DNA synthesis. Proc. Natl. Acad. Sci. USA 1994, 91, 6967–6971. [Google Scholar] [CrossRef] [Green Version]
- Eldredge, E.R.; Korf, G.M.; Christensen, T.A.; Connolly, D.C.; Getz, M.J.; Maihle, N.J. Activation of c-fos gene expression by a kinase-deficient epidermal growth factor receptor. Mol. Cell Biol. 1994, 14, 7527–7534. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.A.; Sooro, M.A.; Zhang, P. Autophagic Regulation of p62 is Critical for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Delorme-Axford, E.; Backues, S.K.; Klionsky, D.J. Atg41/Icy2 regulates autophagosome formation. Autophagy 2015, 11, 2288–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, C.M.; Garcia-Haro, L.; Sparks, C.A.; Guertin, D.A. mTOR-dependent cell survival mechanisms. Cold Spring Harb. Perspect. Biol. 2012, 4, 8771. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation—At the Center of Autophagy Regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, C.; Chen, X.; Grandis, J.R.; Duvvuri, U. EGFR tyrosine kinase inhibition induces autophagy in cancer cells. Cancer Biol. Ther. 2012, 13, 1417–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadlakonda, L.; Dash, A.; Pasupuleti, M.; Anil Kumar, K.; Reddanna, P. The Paradox of Akt-mTOR Interactions. Front. Oncol. 2013, 3, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Tooze, S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020, 6, 32. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Eskelinen, E.L.; Deretic, V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes... wait, I’m confused. Autophagy 2014, 10, 549–551. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Rello-Varona, S.; Lissa, D.; Shen, S.; Niso-Santano, M.; Senovilla, L.; Marino, G.; Vitale, I.; Jemaa, M.; Harper, F.; Pierron, G.; et al. Autophagic removal of micronuclei. Cell Cycle 2012, 11, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Autophagy and Mitophagy in Cellular Damage Control. Redox Biol. 2013, 1, 19–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.N.; Leung, M.C.; Rooney, J.P.; Sendoel, A.; Hengartner, M.O.; Kisby, G.E.; Bess, A.S. Mitochondria as a target of environmental toxicants. Toxicol. Sci. 2013, 134, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A. Apoptosis and autophagy: Regulatory connections between two supposedly different processes. Apoptosis 2008, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Niso-Santano, M.; Adjemian, S.; Takehara, T.; Malik, S.A.; Minoux, H.; Souquere, S.; Marino, G.; Lachkar, S.; Senovilla, L.; et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol. Cell 2012, 48, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Pan, H.; Chen, Y.; Sun, J.; Wang, Y.; Li, J.; Ge, W.; Feng, L.; Lin, X.; Wang, X.; et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS ONE 2011, 6, e18691. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Whitmarsh-Everiss, T.; Laraia, L. Small molecule probes for targeting autophagy. Nat. Chem. Biol. 2021, 17, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellman, I.; Yarden, Y. Endocytosis and cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a016949. [Google Scholar] [CrossRef] [Green Version]
- Elkin, S.R.; Lakoduk, A.M.; Schmid, S.L. Endocytic pathways and endosomal trafficking: A primer. Wien Med. Wochenschr. 2016, 166, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- Bakker, J.; Spits, M.; Neefjes, J.; Berlin, I. The EGFR odyssey—From activation to destruction in space and time. J. Cell Sci. 2017, 130, 4087–4096. [Google Scholar] [CrossRef] [Green Version]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Tan, X.; Thapa, N.; Sun, Y.; Anderson, R.A. A kinase-independent role for EGF receptor in autophagy initiation. Cell 2015, 160, 145–160. [Google Scholar] [CrossRef] [Green Version]
- Demory, M.L.; Boerner, J.L.; Davidson, R.; Faust, W.; Miyake, T.; Lee, I.; Huttemann, M.; Douglas, R.; Haddad, G.; Parsons, S.J. Epidermal growth factor receptor translocation to the mitochondria: Regulation and effect. J. Biol. Chem. 2009, 284, 36592–36604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weihua, Z.; Tsan, R.; Huang, W.C.; Wu, Q.; Chiu, C.H.; Fidler, I.J.; Hung, M.C. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008, 13, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Kirkpatrick, D.; Jiang, X.; Gygi, S.; Sorkin, A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol. Cell 2006, 21, 737–748. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Kessler, D.; Gruen, G.C.; Heider, D.; Morgner, J.; Reis, H.; Schmid, K.W.; Jendrossek, V. The action of small GTPases Rab11 and Rab25 in vesicle trafficking during cell migration. Cell Physiol. Biochem. 2012, 29, 647–656. [Google Scholar] [CrossRef]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Che, T.F.; Lin, C.W.; Wu, Y.Y.; Chen, Y.J.; Han, C.L.; Chang, Y.L.; Wu, C.T.; Hsiao, T.H.; Hong, T.M.; Yang, P.C. Mitochondrial translocation of EGFR regulates mitochondria dynamics and promotes metastasis in NSCLC. Oncotarget 2015, 6, 37349–37366. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.N.; Yamaguchi, H.; Hsu, J.M.; Hung, M.C. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene 2010, 29, 3997–4006. [Google Scholar] [CrossRef] [Green Version]
- Brand, T.M.; Iida, M.; Luthar, N.; Starr, M.M.; Huppert, E.J.; Wheeler, D.L. Nuclear EGFR as a molecular target in cancer. Radiother. Oncol. 2013, 108, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Rush, J.S.; Ceresa, B.P. RAB7 and TSG101 are required for the constitutive recycling of unliganded EGFRs via distinct mechanisms. Mol. Cell. Endocrinol. 2013, 381, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shtiegman, K.; Kochupurakkal, B.S.; Zwang, Y.; Pines, G.; Starr, A.; Vexler, A.; Citri, A.; Katz, M.; Lavi, S.; Ben-Basat, Y.; et al. Defective ubiquitinylation of EGFR mutants of lung cancer confers prolonged signaling. Oncogene 2007, 26, 6968–6978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, B.M.; Raja, S.M.; Clubb, R.J.; Tu, C.; George, M.; Band, V.; Band, H. Aberrant trafficking of NSCLC-associated EGFR mutants through the endocytic recycling pathway promotes interaction with Src. BMC Cell Biol. 2009, 10, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Park, Y.S.; Sung, J.S.; Lee, J.W.; Lee, S.B.; Kim, Y.H. Clathrin-mediated EGFR endocytosis as a potential therapeutic strategy for overcoming primary resistance of EGFR TKI in wild-type EGFR non-small cell lung cancer. Cancer Med. 2021, 10, 372–385. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshioka, K.; Takiguchi, S.; Bereczky, B.; Nakabeppu, Y.; Itoh, K. A Role for SNX1 in the Regulation of EGF-Dependent Phosphorylated EGFR Endocytosis Via the Early/Late Endocytic Pathway in a Gefitinib-Sensitive Human Lung Cancer Cells. Curr. Signal Transduct. Ther. 2011, 6, 383–395. [Google Scholar] [CrossRef]
- Chung, B.M.; Tom, E.; Zutshi, N.; Bielecki, T.A.; Band, V.; Band, H. Nexus of signaling and endocytosis in oncogenesis driven by non-small cell lung cancer-associated epidermal growth factor receptor mutants. World J. Clin. Oncol. 2014, 5, 806–823. [Google Scholar] [CrossRef] [Green Version]
- Tooze, S.A.; Abada, A.; Elazar, Z. Endocytosis and autophagy: Exploitation or cooperation? Cold Spring Harb. Perspect. Biol. 2014, 6, a018358. [Google Scholar] [CrossRef]
- Traxler, P.; Allegrini, P.R.; Brandt, R.; Brueggen, J.; Cozens, R.; Fabbro, D.; Grosios, K.; Lane, H.A.; McSheehy, P.; Mestan, J.; et al. AEE788: A dual family epidermal growth factor receptor/ErbB2 and vascular endothelial growth factor receptor tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2004, 64, 4931–4941. [Google Scholar] [CrossRef] [Green Version]
- Leprivier, G.; Rotblat, B. How does mTOR sense glucose starvation? AMPK is the usual suspect. Cell Death Discov. 2020, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Wood, I.S.; Trayhurn, P. Glucose transporters (GLUT and SGLT): Expanded families of sugar transport proteins. Br. J. Nutr. 2003, 89, 3–9. [Google Scholar] [CrossRef]
- Meng, Y.; Wang, L.; Chen, D.; Chang, Y.; Zhang, M.; Xu, J.J.; Zhou, R.; Zhang, Q.Y. LAPTM4B: An oncogene in various solid tumors and its functions. Oncogene 2016, 35, 6359–6365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Urdiroz, M.; Deeks, M.J.; Horton, C.G.; Dawe, H.R.; Jourdain, I. The Exocyst Complex in Health and Disease. Front. Cell Dev. Biol. 2016, 4, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timon-Gomez, A.; Nyvltova, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Chang, Y.; Lu, W.; Sheng, X.; Wang, S.; Xu, H.; Ma, J. Regulation of Autophagy by Glycolysis in Cancer. Cancer Manag. Res. 2020, 12, 13259–13271. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, X.W.; Li, L.; Hu, M.N.; Hu, W.Q.; Zhang, J.Y.; Miao, X.K.; Yang, W.L.; Mou, L.Y. Inhibition of autophagy by YC-1 promotes gefitinib induced apoptosis by targeting FOXO1 in gefitinib-resistant NSCLC cells. Eur. J. Pharmacol. 2021, 908, 174346. [Google Scholar] [CrossRef]
- Menard, L.; Floc’h, N.; Martin, M.J.; Cross, D.A.E. Reactivation of Mutant-EGFR Degradation through Clathrin Inhibition Overcomes Resistance to EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2018, 78, 3267–3279. [Google Scholar] [CrossRef] [Green Version]
- de Wit, M.; Gao, Y.; Mercieca, D.; de Heer, I.; Valkenburg, B.; van Royen, M.E.; Aerts, J.; Sillevis Smitt, P.; French, P. Mutation and drug-specific intracellular accumulation of EGFR predict clinical responses to tyrosine kinase inhibitors. EBioMedicine 2020, 56, 102796. [Google Scholar] [CrossRef]
- Bjorkelund, H.; Gedda, L.; Barta, P.; Malmqvist, M.; Andersson, K. Gefitinib induces epidermal growth factor receptor dimers which alters the interaction characteristics with (1)(2)(5)I-EGF. PLoS ONE 2011, 6, e24739. [Google Scholar] [CrossRef] [Green Version]
- Bublil, E.M.; Pines, G.; Patel, G.; Fruhwirth, G.; Ng, T.; Yarden, Y. Kinase-mediated quasi-dimers of EGFR. FASEB J. 2010, 24, 4744–4755. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Mi, L.Z.; Schurpf, T.; Walz, T.; Springer, T.A. Mechanisms for kinase-mediated dimerization of the epidermal growth factor receptor. J. Biol. Chem. 2012, 287, 38244–38253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, P.O.; Hristova, K.; Leahy, D.J. EGFR forms ligand-independent oligomers that are distinct from the active state. J. Biol. Chem. 2020, 295, 13353–13362. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Bharill, S.; Karandur, D.; Peterson, S.M.; Marita, M.; Shi, X.; Kaliszewski, M.J.; Smith, A.W.; Isacoff, E.Y.; Kuriyan, J. Molecular basis for multimerization in the activation of the epidermal growth factor receptor. eLife 2016, 5, e14107. [Google Scholar] [CrossRef]
- Ibach, J.; Radon, Y.; Gelleri, M.; Sonntag, M.H.; Brunsveld, L.; Bastiaens, P.I.; Verveer, P.J. Single Particle Tracking Reveals that EGFR Signaling Activity Is Amplified in Clathrin-Coated Pits. PLoS ONE 2015, 10, e0143162. [Google Scholar] [CrossRef] [Green Version]
- Stang, E.; Blystad, F.D.; Kazazic, M.; Bertelsen, V.; Brodahl, T.; Raiborg, C.; Stenmark, H.; Madshus, I.H. Cbl-dependent ubiquitination is required for progression of EGF receptors into clathrin-coated pits. Mol. Biol. Cell 2004, 15, 3591–3604. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gao, J.; Guo, X.; Tong, T.; Shi, X.; Li, L.; Qi, M.; Wang, Y.; Cai, M.; Jiang, J.; et al. Regulation of EGFR nanocluster formation by ionic protein-lipid interaction. Cell Res. 2014, 24, 959–976. [Google Scholar] [CrossRef]
- Coban, O.; Zanetti-Dominguez, L.C.; Matthews, D.R.; Rolfe, D.J.; Weitsman, G.; Barber, P.R.; Barbeau, J.; Devauges, V.; Kampmeier, F.; Winn, M.; et al. Effect of phosphorylation on EGFR dimer stability probed by single-molecule dynamics and FRET/FLIM. Biophys. J. 2015, 108, 1013–1026. [Google Scholar] [CrossRef] [Green Version]
- Freed, D.M.; Bessman, N.J.; Kiyatkin, A.; Salazar-Cavazos, E.; Byrne, P.O.; Moore, J.O.; Valley, C.C.; Ferguson, K.M.; Leahy, D.J.; Lidke, D.S.; et al. EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 2017, 171, 683–695.e618. [Google Scholar] [CrossRef] [Green Version]
- Clayton, A.H.; Walker, F.; Orchard, S.G.; Henderson, C.; Fuchs, D.; Rothacker, J.; Nice, E.C.; Burgess, A.W. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor-A multidimensional microscopy analysis. J. Biol. Chem. 2005, 280, 30392–30399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abulrob, A.; Lu, Z.; Baumann, E.; Vobornik, D.; Taylor, R.; Stanimirovic, D.; Johnston, L.J. Nanoscale imaging of epidermal growth factor receptor clustering: Effects of inhibitors. J. Biol. Chem. 2010, 285, 3145–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazit, A.; Yaish, P.; Gilon, C.; Levitzki, A. Tyrphostins I: Synthesis and biological activity of protein tyrosine kinase inhibitors. J. Med. Chem. 1989, 32, 2344–2352. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.E.; Mueller, K.L.; Bohin, N.; Ge, Y.; Boerner, J.L. Lipid raft localization of EGFR alters the response of cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J. Cell Physiol. 2011, 226, 2316–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Wilkinson, J.C.; Guyer, C.A.; Staros, J.V. Ligand- and kinase activity-independent cell survival mediated by the epidermal growth factor receptor expressed in 32D cells. Exp. Cell Res. 2003, 282, 121–131. [Google Scholar] [CrossRef]
- Pascolutti, R.; Algisi, V.; Conte, A.; Raimondi, A.; Pasham, M.; Upadhyayula, S.; Gaudin, R.; Maritzen, T.; Barbieri, E.; Caldieri, G.; et al. Molecularly Distinct Clathrin-Coated Pits Differentially Impact EGFR Fate and Signaling. Cell Rep. 2019, 27, 3049–3061.e6. [Google Scholar] [CrossRef] [Green Version]
- Baldys, A.; Raymond, J.R. Critical role of ESCRT machinery in EGFR recycling. Biochemistry 2009, 48, 9321–9323. [Google Scholar] [CrossRef]
- Zhu, H.; Cao, X.; Ali-Osman, F.; Keir, S.; Lo, H.W. EGFR and EGFRvIII interact with PUMA to inhibit mitochondrial translocalization of PUMA and PUMA-mediated apoptosis independent of EGFR kinase activity. Cancer Lett. 2010, 294, 101–110. [Google Scholar] [CrossRef] [Green Version]
- De Boer, P.; Hoogenboom, J.P.; Giepmans, B.N. Correlated light and electron microscopy: Ultrastructure lights up! Nat. Methods 2015, 12, 503–513. [Google Scholar] [CrossRef]
- Herth, F.J.; Ernst, A.; Becker, H.D. Endobronchial ultrasound-guided transbronchial lung biopsy in solitary pulmonary nodules and peripheral lesions. Eur. Respir. J. 2002, 20, 972–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin-Fernandez, M.L. Fluorescence Imaging of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Resistance in Non-Small Cell Lung Cancer. Cancers 2022, 14, 686. https://doi.org/10.3390/cancers14030686
Martin-Fernandez ML. Fluorescence Imaging of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Resistance in Non-Small Cell Lung Cancer. Cancers. 2022; 14(3):686. https://doi.org/10.3390/cancers14030686
Chicago/Turabian StyleMartin-Fernandez, Marisa L. 2022. "Fluorescence Imaging of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Resistance in Non-Small Cell Lung Cancer" Cancers 14, no. 3: 686. https://doi.org/10.3390/cancers14030686