
Multifaceted Interplay between Hormones, Growth Factors and Hypoxia in the Tumor Microenvironment

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Importance of Hormones and Growth Factors (GFs) in Tumor Onset and Progression
1.1. Hormone and GF-Mediated Regulation of Intracellular Signaling Cascades
1.2. Hormones and GFs in Tumor Growth and Progression
2. Hypoxia-Inducible Factors
2.1. HIF-Dependent Regulatory Mechanisms
2.2. Oxygen-Dependent Regulation of HIF Signaling
3. Modulation of HIF Signaling by Hormones and GFs
3.1. Hormone-Dependent Regulation of HIF Expression and Signaling
3.2. GF-Mediated Regulation of HIF Expression and Signaling
4. The Contribution of Hypoxia to the Immune-Excluded Phenotype
4.1. Physical Barriers
4.2. Functional Barriers
5. Perspectives on the Interplay between Hypoxia and Immunotherapies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACT | Adoptive cell therapy |
| bFGF | Basic FGF |
| CAFs | Cancer-associated fibroblasts |
| CAs | Carbonic anhydrases |
| Tcm | Central memory T cells |
| CSF1 | Colony-Stimulating Factor 1 |
| CXCR4 | CXC chemokine receptor 4 |
| cAMP | Cyclic adenosine monophosphate |
| CTLs | Cytotoxic T cells |
| CTLA-4 | Cytotoxic T-lymphocyte associated protein 4 |
| DCIS | Ductal carcinoma in situ |
| Tem | Effector memory T cells |
| EGFR | EGF receptor |
| EPCs | Endothelial progenitor cells |
| EGFs | Epidermal Growth Factors |
| EMT | Epithelial-mesenchymal transition |
| ER | Estrogen Receptor |
| EREs | Estrogen responsive elements |
| ERRα | Estrogen-related receptor-α |
| ECM | Extracellular matrix |
| FIH | Factor inhibiting HIF |
| FIH | Factors Inhibiting HIF |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| GFRs | GF receptors |
| GLUTs | Glucose transport |
| GPER | G-protein estrogen receptor |
| GFs | Growth factors |
| GH | Growth Hormone |
| GHR | Growth Hormone Receptor |
| HGF | Hepatocyte growth factor |
| HAPs | Hypoxia-activated prodrugs |
| HIFs | Hypoxia-inducible factors |
| HiTAsystem | Hypoxia-inducible transcription amplification system |
| HREs | Hypoxia-regulated elements |
| IDO | Indoleamine 2,3 dioxygenase |
| IGF-IR | Insulin-like growth factor 1 receptor |
| IGFs | Insulin-like Growth Factors |
| IL- | Interleukin |
| KS | Kaposi sarcoma |
| LAG3 | Lymphocyte-activation gene 3 |
| MHC-I | Major histocompatibility class-I |
| MMPs | Matrix metalloproteinases |
| mAbs | Monoclonal antibodies |
| TIM3 | Mucin-domain containing-3 |
| Mcl-1 | Myeloid cell leukemia-1 |
| MDSCs | Myeloid-derived suppressor cells |
| HNSCC | Neck squamous cell carcinoma |
| NRGs | Neuregulins |
| NO | Nitric oxide |
| NSCLC | Non-small cell lung cancer |
| Nrf2 | Nuclear erythroid 2 p45–related factor 2 |
| PDGF | Platelet-derived growth factor |
| PRs | Progesterone Receptors |
| PD-1 | Programmed cell death protein 1 |
| PHDs | Prolyl hydrolases |
| PHDs | Prolyl hydroxylases domains |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| STATs | Signal Transducers and Activator of Transcription |
| SCF | Stem cell factor |
| SHs | Steroid ex hormones |
| SO | Superoxide |
| TGF-α | Transforming Growth Factor-α |
| TGF-β | Transforming Growth Factor-β |
| TMs | Transition metals |
| TME | Tumor microenvironment |
| TNF-α | Tumor necrosis factor α |
| TAMs | Tumor-associated macrophages |
| TKIs | Tyrosine kinase inhibitors |
| VEGFs | Vascular Endothelial Growth Factors |
| pVHL | Von Hippel-Lindau tumor suppressor |
References
- Reznikov, A. Hormonal Impact on Tumor Growth and Progression. Exp. Oncol. 2015, 37, 162–172. [Google Scholar] [CrossRef]
- Hollinger, J.O.; Alvarez-Urena, P.; Ducheyne, P.; Srinivasan, A.; Baskin, J.; Waters, H.; Gruber, R. Comprehensive Biomaterials II. 6.2 Bone Tissue Engineering: Growth Factors and Cytokines; Elsevier: Amsterdam, The Netherlands, 2017; pp. 20–53. [Google Scholar]
- Lee, E.Y.; Parry, G.; Bissell, M.J. Modulation of Secreted Proteins of Mouse Mammary Epithelial Cells by the Collagenous Substrata. J. Cell Biol. 1984, 98, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, L.M.; Parris, E.E.; Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Witsch, E.; Sela, M.; Yarden, Y. Roles for Growth Factors in Cancer Progression. Physiology 2010, 25, 85–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sporn, M.B.; Todaro, G.J. Autocrine Secretion and Malignant Transformation of Cells. N. Engl. J. Med. 1980, 303, 878–880. [Google Scholar] [CrossRef]
- Yeung, S.; Gagel, R.; Kufe, D.; Pollock, R.; Weichselbaum, R. Endocrine Paraneoplastic Syndromes (“ectopic” Hormone Production). In Holland-Frei Cancer Medicine, 6th ed.; BC Decker Inc.: Hamilton, ON, USA, 2003. [Google Scholar]
- Hinson, J.; Raven, P.; Chew, S. Receptors and Hormone Action. In The Endocrine System; Elsevier: Amsterdam, The Netherlands, 2010; pp. 15–26. [Google Scholar]
- Stone, W.L.; Leavitt, L.; Varacallo, M. Physiology, Growth Factor; StatPearls Publishing: Treasure Island, FL, USA, 2017. [Google Scholar]
- Chen, Y.; Takeshita, A.; Ozaki, K.; Kitano, S.; Hanazawa, S. Transcriptional Regulation by Transforming Growth Factor β of the Expression of Retinoic Acid and Retinoid X Receptor Genes in Osteoblastic Cells Is Mediated through AP-1. J. Biol. Chem. 1996, 271, 31602–31606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beato, M.; Chávez, S.; Truss, M. Transcriptional Regulation by Steroid Hormones. Steroids 1996, 61, 240–251. [Google Scholar] [CrossRef]
- Ing, N.H. Steroid Hormones Regulate Gene Expression Posttranscriptionally by Altering the Stabilities of Messenger RNAs. Biol. Reprod. 2005, 72, 1290–1296. [Google Scholar] [CrossRef] [Green Version]
- Boguszewski, C.L.; da Silva Boguszewski, M.C. Growth Hormone’s Links to Cancer. Endocr. Rev. 2019, 40, 558–574. [Google Scholar] [CrossRef]
- Sherbet, G.V. Hormonal Influences on Cancer Progression and Prognosis. Vitam. Horm. 2005, 71, 147–200. [Google Scholar]
- Waters, M.J. The Growth Hormone Receptor. Growth Horm. IGF Res. 2016, 28, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, J.K.; Liu, D.-X.; Wu, Z.-S.; Zhu, T.; Lobie, P.E. Growth Hormone and Cancer: An Update on Progress. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Wu, Q.; Sun, A.; Liu, X.; Fan, Y.; Deng, X. Cancer Cell Glycocalyx and Its Significance in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 2484. [Google Scholar] [CrossRef] [Green Version]
- Miranda, O.; Farooqui, M.; Siegfried, J.M. Status of Agents Targeting the HGF/c-Met Axis in Lung Cancer. Cancers 2018, 10, 280. [Google Scholar] [CrossRef] [Green Version]
- Cevenini, A.; Orrù, S.; Mancini, A.; Alfieri, A.; Buono, P.; Imperlini, E. Molecular Signatures of the Insulin-like Growth Factor 1-Mediated Epithelial-Mesenchymal Transition in Breast, Lung and Gastric Cancers. Int. J. Mol. Sci. 2018, 19, 2411. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Huang, H.-J.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.-D.; Huang, C.-M.; Gill, G.N.; Steven Wiley, H.; Cavenee, W.K. The Enhanced Tumorigenic Activity of a Mutant Epidermal Growth Factor Receptor Common in Human Cancers Is Mediated by Threshold Levels of Constitutive Tyrosine Phosphorylation and Unattenuated Signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Muleris, M.; Dutrillaux, B.; Malfoy, B. The Insulin-like Growth Factor I Receptor Gene Is the Target for the 15q26 Amplicon in Breast Cancer. Genes Chromosomes Cancer 1994, 11, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Hurd, S.D.; Winnier, A.R.; Johnson, M.D.; Fendly, B.M.; Forbes, J.T. Anti-Transforming Growth Factor (TGF)-Beta Antibodies Inhibit Breast Cancer Cell Tumorigenicity and Increase Mouse Spleen Natural Killer Cell Activity. Implications for a Possible Role of Tumor Cell/host TGF-Beta Interactions in Human Breast Cancer Progression. J. Clin. Investig. 1993, 92, 2569–2576. [Google Scholar]
- Meijnen, P.; Peterse, J.L.; Antonini, N.; Rutgers, E.J.T.; van de Vijver, M.J. Immunohistochemical Categorisation of Ductal Carcinoma in Situ of the Breast. Br. J. Cancer 2008, 98, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of Putative Progenitor Endothelial Cells for Angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the Cytoskeleton, and Cancer Cell Invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, T.; Chen, P.; Goodly, L.J.; Wells, A. EGF Receptor Signaling Enhances In Vivo Invasiveness of DU-145 Human Prostate Carcinoma Cells. Clin. Exp. Metastasis 1996, 14, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhu, F.; Zhang, H.; Chen, D.; Zhang, X.; Gao, Q.; Li, Y. Conditional Ablation of TGF-β Signaling Inhibits Tumor Progression and Invasion in an Induced Mouse Bladder Cancer Model. Sci. Rep. 2016, 6, 29479. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G. Matrix Metalloproteinases in Angiogenesis: A Moving Target for Therapeutic Intervention. J. Clin. Investig. 1999, 103, 1237–1241. [Google Scholar] [CrossRef] [Green Version]
- Wells, A.; Kassis, J.; Solava, J.; Turner, T.; Lauffenburger, D.A. Growth Factor-Induced Cell Motility in Tumor Invasion. Acta Oncol. 2002, 41, 124–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Mendonsa, A.M.; Na, T.-Y.; Gumbiner, B.M. E-Cadherin in Contact Inhibition and Cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef]
- Fan, Y.; Shen, B.; Tan, M.; Mu, X.; Qin, Y.; Zhang, F.; Liu, Y. TGF-β–Induced Upregulation of malat1 Promotes Bladder Cancer Metastasis by Associating with suz12. Clin. Cancer Res. 2014, 20, 1531–1541. [Google Scholar] [CrossRef] [Green Version]
- Atlas, E.; Cardillo, M.; Mehmi, I.; Zahedkargaran, H. Heregulin Is Sufficient for the Promotion of Tumorigenicity and Metastasis of Breast Cancer Cells In Vivo. Mol. Cancer Res. 2003, 1, 165–175. [Google Scholar]
- Katt, M.E.; Wong, A.D.; Searson, P.C. Dissemination from a Solid Tumor: Examining the Multiple Parallel Pathways. Trends Cancer Res. 2018, 4, 20–37. [Google Scholar] [CrossRef]
- Padua, D.; Zhang, X.H.-F.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massagué, J. TGFβ Primes Breast Tumors for Lung Metastasis Seeding through Angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Tarin, D. Cell and Tissue Interactions in Carcinogenesis and Metastasis and Their Clinical Significance. Semin. Cancer Biol. 2011, 21, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Huysentruyt, L.C. On the Origin of Cancer Metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [Green Version]
- Aharinejad, S.; Paulus, P.; Sioud, M.; Hofmann, M.; Zins, K.; Schafer, R.; Stanley, E.R.; Abraham, D. Experimental Therapeutics, Molecular Targets, and Chemical Biology-Colony-Stimulating Factor-1 Blockade by Antisense Oligonucleotides and Small Interfering RNAs Suppresses Growth of Human Mammary. Cancer Res. 2004, 64, 5378–5384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyden, D.; Hattori, K.; Dias, S.; Costa, C.; Blaikie, P.; Butros, L.; Chadburn, A.; Heissig, B.; Marks, W.; Witte, L.; et al. Impaired Recruitment of Bone-Marrow-Derived Endothelial and Hematopoietic Precursor Cells Blocks Tumor Angiogenesis and Growth. Nat. Med. 2001, 7, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Baeriswyl, V.; Christofori, G. The Angiogenic Switch in Carcinogenesis. Semin. Cancer Biol. 2009, 19, 329–337. [Google Scholar] [CrossRef]
- Ortiz-Prado, E.; Dunn, J.F.; Vasconez, J.; Castillo, D.; Viscor, G. Partial Pressure of Oxygen in the Human Body: A General Review. Am. J. Blood Res. 2019, 9, 1–14. [Google Scholar] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Hlatky, M.A.; Quertermous, T.; Boothroyd, D.B.; Priest, J.R.; Glassford, A.J.; Myers, R.M.; Fortmann, S.P.; Iribarren, C.; Tabor, H.K.; Assimes, T.L.; et al. Polymorphisms in Hypoxia Inducible Factor 1 and the Initial Clinical Presentation of Coronary Disease. Am. Heart J. 2007, 154, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Jha, N.K.; Jha, S.K.; Sharma, R.; Kumar, D.; Ambasta, R.K.; Kumar, P. Hypoxia-Induced Signaling Activation in Neurodegenerative Diseases: Targets for New Therapeutic Strategies. J. Alzheimers Dis. 2018, 62, 15–38. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Zhao, F.; Zhou, Y.; He, B.; Huang, W.; Zhang, F.; Long, Y.-G.; Xia, X.; Liu, M.-L.; Jiang, Y.-H. Systematic Review and Meta-Analysis of the Impact of Hypoxia on Infarcted Myocardium: Better or Worse? Cell. Physiol. Biochem. 2018, 51, 949–960. [Google Scholar] [CrossRef]
- Levenson, N.I.; Adolph, R.J.; Romhilt, D.W.; Gabel, M.; Sodd, V.J.; August, L.S. Effects of Myocardial Hypoxia and Ischemia on Myocardial Scintigraphy. Am. J. Cardiol. 1975, 35, 251–257. [Google Scholar] [CrossRef]
- Vaupel, P.; Schlenger, K.; Knoop, C.; Höckel, M. Oxygenation of Human Tumors: Evaluation of Tissue Oxygen Distribution in Breast Cancers by Computerized O2 Tension Measurements. Cancer Res. 1991, 51, 3316–3322. [Google Scholar] [PubMed]
- Braun, R.D.; Lanzen, J.L.; Snyder, S.A.; Dewhirst, M.W. Comparison of Tumor and Normal Tissue Oxygen Tension Measurements Using OxyLite or Microelectrodes in Rodents. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2533–H2544. [Google Scholar] [CrossRef] [PubMed]
- McKeown, S.R. Defining Normoxia, Physoxia and Hypoxia in Tumours-Implications for Treatment Response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- Thomlinson, R.H.; Gray, L.H. The Histological Structure of Some Human Lung Cancers and the Possible Implications for Radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.D. The Detection and Measurement of Hypoxic Cells in Solid Tumors. Cancer 1984, 54, 2441–2449. [Google Scholar] [CrossRef]
- Nordsmark, M.; Bentzen, S.M.; Overgaard, J. Measurement of Human Tumour Oxygenation Status by a Polarographic Needle Electrode. An Analysis of Inter- and Intratumour Heterogeneity. Acta Oncol. 1994, 33, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Forster, J.C.; Harriss-Phillips, W.M.; Douglass, M.J.; Bezak, E. A Review of the Development of Tumor Vasculature and Its Effects on the Tumor Microenvironment. Hypoxia 2017, 5, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Bayer, C.; Vaupel, P. Acute versus Chronic Hypoxia in Tumors: Controversial Data Concerning Time Frames and Biological Consequences. Strahlenther. Onkol. 2012, 188, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, D.J.; Durand, R.E.; Olive, P.L. Acute Hypoxia in Tumors: Implications for Modifiers of Radiation Effects. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1279–1282. [Google Scholar] [CrossRef]
- Chaplin, D.J.; Olive, P.L.; Durand, R.E. Intermittent Blood Flow in a Murine Tumor: Radiobiological Effects. Cancer Res. 1987, 47, 597–601. [Google Scholar]
- Rofstad, E.K.; Gaustad, J.-V.; Egeland, T.A.M.; Mathiesen, B.; Galappathi, K. Tumors Exposed to Acute Cyclic Hypoxic Stress Show Enhanced Angiogenesis, Perfusion and Metastatic Dissemination. Int. J. Cancer 2010, 127, 1535–1546. [Google Scholar] [CrossRef]
- Kato, Y.; Yashiro, M.; Fuyuhiro, Y.; Kashiwagi, S.; Matsuoka, J.; Hirakawa, T.; Noda, S.; Aomatsu, N.; Hasegawa, T.; Matsuzaki, T.; et al. Effects of Acute and Chronic Hypoxia on the Radiosensitivity of Gastric and Esophageal Cancer Cells. Anticancer Res. 2011, 31, 3369–3375. [Google Scholar] [PubMed]
- Martin, J.D.; Fukumura, D.; Duda, D.G.; Boucher, Y.; Jain, R.K. Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb. Perspect. Med. 2016, 6, a027094. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Giaccia, A.J. The Unique Physiology of Solid Tumors: Opportunities (and Problems) for Cancer Therapy. Cancer Res. 1998, 58, 1408–1416. [Google Scholar]
- Semenza, G.L. Hypoxia, Clonal Selection, and the Role of HIF-1 in Tumor Progression. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 71–103. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 and Tumor Progression: Pathophysiology and Therapeutics. Trends Mol. Med. 2002, 8, S62–S67. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of Hypoxia-Inducible Factor 1alpha in Common Human Cancers and Their Metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The Expression and Distribution of the Hypoxia-Inducible Factors HIF-1alpha and HIF-2alpha in Normal Human Tissues, Cancers, and Tumor-Associated Macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-Inducible Factor 1 Is a Basic-Helix-Loop-Helix-PAS Heterodimer Regulated by Cellular O2 Tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Semenza, G.L. Purification and Characterization of Hypoxia-Inducible Factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.-J.; Sataur, A.; Wang, L.; Chen, H.; Simon, M.C. The N-Terminal Transactivation Domain Confers Target Gene Specificity of Hypoxia-Inducible Factors HIF-1alpha and HIF-2alpha. Mol. Biol. Cell 2007, 18, 4528–4542. [Google Scholar] [CrossRef] [Green Version]
- Salnikow, K.; Aprelikova, O.; Ivanov, S.; Tackett, S.; Kaczmarek, M.; Karaczyn, A.; Yee, H.; Kasprzak, K.S.; Niederhuber, J. Regulation of Hypoxia-Inducible Genes by ETS1 Transcription Factor. Carcinogenesis 2008, 29, 1493–1499. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and Inhibitory Domains of Hypoxia-Inducible Factor 1alpha. Modulation of Transcriptional Activity by Oxygen Tension. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef] [Green Version]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional Regulation by Hypoxia Inducible Factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential Roles of Hypoxia-Inducible Factor 1alpha (HIF-1alpha) and HIF-2alpha in Hypoxic Gene Regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, N.; Maemura, K.; Imai, Y.; Harada, T.; Kawanami, D.; Nojiri, T.; Manabe, I.; Nagai, R. Endothelial PAS Domain Protein 1 Gene Promotes Angiogenesis through the Transactivation of Both Vascular Endothelial Growth Factor and Its Receptor, Flt-1. Circ. Res. 2004, 95, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, M.Y.; Lemos, R., Jr.; Liu, X.; Powis, G. The Hypoxia-Associated Factor Switches Cells from HIF-1α- to HIF-2α-Dependent Signaling Promoting Stem Cell Characteristics, Aggressive Tumor Growth and Invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [Green Version]
- Koh, M.Y.; Powis, G. Passing the Baton: The HIF Switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavadas, M.A.S.; Taylor, C.T.; Cheong, A. Acquisition of Temporal HIF Transcriptional Activity Using a Secreted Luciferase Assay. Methods Mol. Biol. 2018, 1742, 37–44. [Google Scholar] [PubMed]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular Characterization and Chromosomal Localization of a Third Alpha-Class Hypoxia Inducible Factor Subunit, HIF3alpha. Gene Expr. 1998, 7, 205–213. [Google Scholar]
- Makino, Y.; Cao, R.; Svensson, K.; Bertilsson, G.; Asman, M.; Tanaka, H.; Cao, Y.; Berkenstam, A.; Poellinger, L. Inhibitory PAS Domain Protein Is a Negative Regulator of Hypoxia-Inducible Gene Expression. Nature 2001, 414, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Kanopka, A.; Wilson, W.J.; Tanaka, H.; Poellinger, L. Inhibitory PAS Domain Protein (IPAS) Is a Hypoxia-Inducible Splicing Variant of the Hypoxia-Inducible Factor-3alpha Locus. J. Biol. Chem. 2002, 277, 32405–32408. [Google Scholar] [CrossRef] [Green Version]
- Maynard, M.A.; Evans, A.J.; Hosomi, T.; Hara, S.; Jewett, M.A.S.; Ohh, M. Human HIF-3alpha4 Is a Dominant-Negative Regulator of HIF-1 and Is down-Regulated in Renal Cell Carcinoma. FASEB J. 2005, 19, 1396–1406. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A Nuclear Factor Induced by Hypoxia via de Novo Protein Synthesis Binds to the Human Erythropoietin Gene Enhancer at a Site Required for Transcriptional Activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS Domain Protein 1 (EPAS1), a Transcription Factor Selectively Expressed in Endothelial Cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Cavadas, M.A.S.; Mesnieres, M.; Crifo, B.; Manresa, M.C.; Selfridge, A.C.; Keogh, C.E.; Fabian, Z.; Scholz, C.C.; Nolan, K.A.; Rocha, L.M.A.; et al. REST Is a Hypoxia-Responsive Transcriptional Repressor. Sci. Rep. 2016, 6, 31355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, Y.; Malek, S.N.; Zheng, P.; Liu, Y. Targeting HIF1α Eliminates Cancer Stem Cells in Hematological Malignancies. Cell Stem Cell 2011, 8, 399–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, P.; Leav, I.; Pursell, B.; Bae, D.; Yang, X.; Taglienti, C.A.; Gouvin, L.M.; Sharma, V.M.; Mercurio, A.M. ERbeta Impedes Prostate Cancer EMT by Destabilizing HIF-1alpha and Inhibiting VEGF-Mediated Snail Nuclear Localization: Implications for Gleason Grading. Cancer Cell 2010, 17, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.E.; Bindra, R.S.; Glazer, P.M.; Harris, A.L. Hypoxia-Induced Genetic Instability—A Calculated Mechanism Underlying Tumor Progression. J. Mol. Med. 2007, 85, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Corle, C.; Seagroves, T.N.; Johnson, R.S. Hypoxia-Inducible Factor-1alpha Is a Key Regulator of Metastasis in a Transgenic Model of Cancer Initiation and Progression. Cancer Res. 2007, 67, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L. Regulation of Tumor pH and the Role of Carbonic Anhydrase 9. Cancer Metastasis Rev. 2007, 26, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, D.; Ohta, A.; Sitkovsky, M. Hypoxia-Dependent Anti-Inflammatory Pathways in Protection of Cancerous Tissues. Cancer Metastasis Rev. 2007, 26, 273–279. [Google Scholar] [CrossRef]
- Chan, D.A.; Giaccia, A.J. Hypoxia, Gene Expression, and Metastasis. Cancer Metastasis Rev. 2007, 26, 333–339. [Google Scholar] [CrossRef]
- Moeller, B.J.; Richardson, R.A.; Dewhirst, M.W. Hypoxia and Radiotherapy: Opportunities for Improved Outcomes in Cancer Treatment. Cancer Metastasis Rev. 2007, 26, 241–248. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Kshitiz; Gilkes, D.M.; Rey, S.; Wong, C.C.; Luo, W.; Kim, D.-H.; Dang, C.V.; Levchenko, A.; Semenza, G.L. A Nontranscriptional Role for HIF-1α as a Direct Inhibitor of DNA Replication. Sci. Signal. 2013, 6, ra10. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of Hypoxia-Inducible Factor 1alpha Is Mediated by an O2-Dependent Degradation Domain via the Ubiquitin-Proteasome Pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, J.F.; Tian, Y.M.; Ratcliffe, P.J.; Pugh, C.W. Oxygen-Regulated and Transactivating Domains in Endothelial PAS Protein 1: Comparison with Hypoxia-Inducible Factor-1alpha. J. Biol. Chem. 1999, 274, 2060–2071. [Google Scholar] [CrossRef] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha Targeted for VHL-Mediated Destruction by Proline Hydroxylation: Implications for O2 Sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-Alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A Novel Protein That Interacts with HIF-1alpha and VHL to Mediate Repression of HIF-1 Transcriptional Activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Semenza, G.L. General Involvement of Hypoxia-Inducible Factor 1 in Transcriptional Response to Hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Semenza, G.L. Characterization of Hypoxia-Inducible Factor 1 and Regulation of DNA Binding Activity by Hypoxia. J. Biol. Chem. 1993, 268, 21513–21518. [Google Scholar] [CrossRef]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A Novel bHLH-PAS Factor with Close Sequence Similarity to Hypoxia-Inducible Factor 1alpha Regulates the VEGF Expression and Is Potentially Involved in Lung and Vascular Development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [Green Version]
- Flamme, I.; Fröhlich, T.; von Reutern, M.; Kappel, A.; Damert, A.; Risau, W. HRF, a Putative Basic Helix-Loop-Helix-PAS-Domain Transcription Factor Is Closely Related to Hypoxia-Inducible Factor-1 Alpha and Developmentally Expressed in Blood Vessels. Mech. Dev. 1997, 63, 51–60. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer Res. 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.A.; Dunning, S.P.; Bunn, H.F. Regulation of the Erythropoietin Gene: Evidence That the Oxygen Sensor Is a Heme Protein. Science 1988, 242, 1412–1415. [Google Scholar] [CrossRef]
- Salnikow, K.; An, W.G.; Melillo, G.; Blagosklonny, M.V.; Costa, M. Nickel-Induced Transformation Shifts the Balance between HIF-1 and p53 Transcription Factors. Carcinogenesis 1999, 20, 1819–1823. [Google Scholar] [CrossRef] [Green Version]
- Befani, C.; Mylonis, I.; Gkotinakou, I.-M.; Georgoulias, P.; Hu, C.-J.; Simos, G.; Liakos, P. Cobalt Stimulates HIF-1-Dependent but Inhibits HIF-2-Dependent Gene Expression in Liver Cancer Cells. Int. J. Biochem. Cell Biol. 2013, 45, 2359–2368. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarek, M.; Timofeeva, O.A.; Karaczyn, A.; Malyguine, A.; Kasprzak, K.S.; Salnikow, K. The Role of Ascorbate in the Modulation of HIF-1alpha Protein and HIF-Dependent Transcription by chromium(VI) and nickel(II). Free Radic. Biol. Med. 2007, 42, 1246–1257. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Chen, H.; Huang, X.; Costa, M. Effects of 12 Metal Ions on Iron Regulatory Protein 1 (IRP-1) and Hypoxia-Inducible Factor-1 Alpha (HIF-1alpha) and HIF-Regulated Genes. Toxicol. Appl. Pharmacol. 2006, 213, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salnikow, K.; Donald, S.P.; Bruick, R.K.; Zhitkovich, A.; Phang, J.M.; Kasprzak, K.S. Depletion of Intracellular Ascorbate by the Carcinogenic Metals Nickel and Cobalt Results in the Induction of Hypoxic Stress. J. Biol. Chem. 2004, 279, 40337–40344. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarek, M.; Cachau, R.E.; Topol, I.A.; Kasprzak, K.S.; Ghio, A.; Salnikow, K. Metal Ions-Stimulated Iron Oxidation in Hydroxylases Facilitates Stabilization of HIF-1 Alpha Protein. Toxicol. Sci. 2009, 107, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive Oxygen Species Generated at Mitochondrial Complex III Stabilize Hypoxia-Inducible Factor-1alpha during Hypoxia: A Mechanism of O2 Sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial Complex III Is Required for Hypoxia-Induced ROS Production and Cellular Oxygen Sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Kaewpila, S.; Venkataraman, S.; Buettner, G.R.; Oberley, L.W. Manganese Superoxide Dismutase Modulates Hypoxia-Inducible Factor-1 Alpha Induction via Superoxide. Cancer Res. 2008, 68, 2781–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, Y.L.; Dufour, E.; Dassa, E.P.; Rustin, P.; Jacobs, H.T.; Taylor, C.T.; Hagen, T. Stabilization of Hypoxia-Inducible Factor-1alpha Protein in Hypoxia Occurs Independently of Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2010, 285, 31277–31284. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.S.; Chandel, N.S. The Qo Site of the Mitochondrial Complex III Is Required for the Transduction of Hypoxic Signaling via Reactive Oxygen Species Production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasabe, E.; Yang, Z.; Ohno, S.; Yamamoto, T. Reactive Oxygen Species Produced by the Knockdown of Manganese-Superoxide Dismutase up-Regulate Hypoxia-Inducible Factor-1alpha Expression in Oral Squamous Cell Carcinoma Cells. Free Radic. Biol. Med. 2010, 48, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Masson, N.; Singleton, R.S.; Sekirnik, R.; Trudgian, D.C.; Ambrose, L.J.; Miranda, M.X.; Tian, Y.-M.; Kessler, B.M.; Schofield, C.J.; Ratcliffe, P.J. The FIH Hydroxylase Is a Cellular Peroxide Sensor That Modulates HIF Transcriptional Activity. EMBO Rep. 2012, 13, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Sandau, K.B.; Fandrey, J.; Brüne, B. Accumulation of HIF-1alpha under the Influence of Nitric Oxide. Blood 2001, 97, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.A.; Gaston, B.; Johns, R.A. Normoxic Stabilization of Hypoxia-Inducible Factor-1 Expression and Activity: Redox-Dependent Effect of Nitrogen Oxides. Mol. Pharmacol. 2000, 58, 1197–1203. [Google Scholar] [CrossRef]
- Sumbayev, V.V.; Budde, A.; Zhou, J.; Brüne, B. HIF-1 Alpha Protein as a Target for S-Nitrosation. FEBS Lett. 2003, 535, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Metzen, E.; Zhou, J.; Jelkmann, W.; Fandrey, J.; Brüne, B. Nitric Oxide Impairs Normoxic Degradation of HIF-1alpha by Inhibition of Prolyl Hydroxylases. Mol. Biol. Cell 2003, 14, 3470–3481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, N.; van der Vliet, A. Interactions between Nitric Oxide and Hypoxia-Inducible Factor Signaling Pathways in Inflammatory Disease. Nitric Oxide 2011, 25, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-Inducible Factor-1a by Reactive Oxygen Species: New Developments in an Old Debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Li, J.; Brown, L.F.; Hibberd, M.G.; Grossman, J.D.; Morgan, J.P.; Simons, M. VEGF, Flk-1, and Flt-1 Expression in a Rat Myocardial Infarction Model of Angiogenesis. Am. J. Physiol. 1996, 270, H1803–H1811. [Google Scholar] [CrossRef]
- Kim, C.-H.; Cho, Y.-S.; Chun, Y.-S.; Park, J.-W.; Kim, M.-S. Early Expression of Myocardial HIF-1alpha in Response to Mechanical Stresses: Regulation by Stretch-Activated Channels and the Phosphatidylinositol 3-Kinase Signaling Pathway. Circ. Res. 2002, 90, E25–E33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.; Shyu, K.-G.; Wang, B.-W.; Kuan, P. Regulation of Hypoxia-Inducible Factor-1alpha by Cyclical Mechanical Stretch in Rat Vascular Smooth Muscle Cells. Clin. Sci. 2003, 105, 447–456. [Google Scholar] [CrossRef] [Green Version]
- Milkiewicz, M.; Doyle, J.L.; Fudalewski, T.; Ispanovic, E.; Aghasi, M.; Haas, T.L. HIF-1alpha and HIF-2alpha Play a Central Role in Stretch-Induced but Not Shear-Stress-Induced Angiogenesis in Rat Skeletal Muscle. J. Physiol. 2007, 583, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Deroo, B.J.; Korach, K.S. Estrogen Receptors and Human Disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Seton-Rogers, S. Breast Cancer: Untangling the Role of Progesterone Receptors. Nat. Rev. Cancer 2015, 15, 456. [Google Scholar] [CrossRef]
- D’Uva, G.; Lauriola, M. Towards the Emerging Crosstalk: ERBB Family and Steroid Hormones. Semin. Cell Dev. Biol. 2016, 50, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; De Marco, P.; De Francesco, E.M.; Chimento, A.; Pezzi, V.; Maggiolini, M. Cross-Talk between GPER and Growth Factor Signaling. J. Steroid Biochem. Mol. Biol. 2013, 137, 50–56. [Google Scholar] [CrossRef]
- Hawsawi, Y.; El-Gendy, R.; Twelves, C.; Speirs, V.; Beattie, J. Insulin-like Growth Factor—Oestradiol Crosstalk and Mammary Gland Tumourigenesis. Biochim. Biophys. Acta 2013, 1836, 345–353. [Google Scholar] [CrossRef]
- Bartella, V.; De Marco, P.; Malaguarnera, R.; Belfiore, A.; Maggiolini, M. New Advances on the Functional Cross-Talk between Insulin-like Growth Factor-I and Estrogen Signaling in Cancer. Cell. Signal. 2012, 24, 1515–1521. [Google Scholar] [CrossRef]
- Liang, J.; Shang, Y. Estrogen and Cancer. Annu. Rev. Physiol. 2013, 75, 225–240. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.G.; Zeng, Q.; Tse, G.M. Estrogen and Its Receptors in Cancer. Med. Res. Rev. 2008, 28, 954–974. [Google Scholar] [CrossRef]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björnström, L.; Sjöberg, M. Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Hammes, S.R.; Levin, E.R. Minireview: Recent Advances in Extranuclear Steroid Receptor Actions. Endocrinology 2011, 152, 4489–4495. [Google Scholar] [CrossRef] [Green Version]
- Prossnitz, E.R.; Barton, M. The G-Protein-Coupled Estrogen Receptor GPER in Health and Disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty Years of the G Protein-Coupled Estrogen Receptor GPER: Historical and Personal Perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a Therapeutic Target in Cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Caldon, C.E.; Sutherland, R.L.; Musgrove, E. Cell Cycle Proteins in Epithelial Cell Differentiation: Implications for Breast Cancer. Cell Cycle 2010, 9, 1918–1928. [Google Scholar] [CrossRef]
- Gompel, A.; Somaï, S.; Chaouat, M.; Kazem, A.; Kloosterboer, H.J.; Beusman, I.; Forgez, P.; Mimoun, M.; Rostène, W. Hormonal Regulation of Apoptosis in Breast Cells and Tissues. Steroids 2000, 65, 593–598. [Google Scholar] [CrossRef]
- Miller, V.M.; Duckles, S.P. Vascular Actions of Estrogens: Functional Implications. Pharmacol. Rev. 2008, 60, 210–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendrik, C.; Dabrosin, C. Estradiol Increases IL-8 Secretion of Normal Human Breast Tissue and Breast Cancer In Vivo. J. Immunol. 2009, 182, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, G.; Saarinen, N.; Abrahamsson, A.; Dabrosin, C. Tamoxifen, Flaxseed, and the Lignan Enterolactone Increase Stroma- and Cancer Cell-Derived IL-1Ra and Decrease Tumor Angiogenesis in Estrogen-Dependent Breast Cancer. Cancer Res. 2011, 71, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Belguise, K.; Kersual, N.; Kirsch, K.H.; Mineva, N.D.; Galtier, F.; Chalbos, D.; Sonenshein, G.E. Oestrogen Signalling Inhibits Invasive Phenotype by Repressing RelB and Its Target BCL2. Nat. Cell Biol. 2007, 9, 470–478. [Google Scholar] [CrossRef] [Green Version]
- McCune, K.; Mehta, R.; Thorat, M.A.; Badve, S.; Nakshatri, H. Loss of ERα and FOXA1 Expression in a Progression Model of Luminal Type Breast Cancer: Insights from PyMT Transgenic Mouse Model. Oncol. Rep. 2010, 24, 1233–1239. [Google Scholar]
- Guttilla, I.K.; Adams, B.D.; White, B.A. ERα, microRNAs, and the Epithelial-Mesenchymal Transition in Breast Cancer. Trends Endocrinol. Metab. 2012, 23, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.P.; Lappano, R.; Albanito, L.; Madeo, A.; Maggiolini, M.; Picard, D. Estrogenic GPR30 Signalling Induces Proliferation and Migration of Breast Cancer Cells through CTGF. EMBO J. 2009, 28, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Filardo, E.J. A Role for G-Protein Coupled Estrogen Receptor (GPER) in Estrogen-Induced Carcinogenesis: Dysregulated Glandular Homeostasis, Survival and Metastasis. J. Steroid Biochem. Mol. Biol. 2018, 176, 38–48. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. GPER Is Involved in the Functional Liaison between Breast Tumor Cells and Cancer-Associated Fibroblasts (CAFs). J. Steroid Biochem. Mol. Biol. 2018, 176, 49–56. [Google Scholar] [CrossRef]
- Lappano, R.; Talia, M.; Cirillo, F.; Rigiracciolo, D.C.; Scordamaglia, D.; Guzzi, R.; Miglietta, A.M.; De Francesco, E.M.; Belfiore, A.; Sims, A.H.; et al. The IL1β-IL1R Signaling Is Involved in the Stimulatory Effects Triggered by Hypoxia in Breast Cancer Cells and Cancer-Associated Fibroblasts (CAFs). J. Exp. Clin. Cancer Res. 2020, 39, 153. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER Mediates Activation of HIF1α/VEGF Signaling by Estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Lappano, R.; Santolla, M.F.; Marsico, S.; Caruso, A.; Maggiolini, M. HIF-1α/GPER Signaling Mediates the Expression of VEGF Induced by Hypoxia in Breast Cancer Associated Fibroblasts (CAFs). Breast Cancer Res. 2013, 15, R64. [Google Scholar] [CrossRef] [Green Version]
- De Marco, P.; Lappano, R.; De Francesco, E.M.; Cirillo, F.; Pupo, M.; Avino, S.; Vivacqua, A.; Abonante, S.; Picard, D.; Maggiolini, M. GPER Signalling in Both Cancer-Associated Fibroblasts and Breast Cancer Cells Mediates a Feedforward IL1β/IL1R1 Response. Sci. Rep. 2016, 6, 24354. [Google Scholar] [CrossRef] [Green Version]
- Egloff, A.M.; Rothstein, M.E.; Seethala, R.; Siegfried, J.M.; Grandis, J.R.; Stabile, L.P. Cross-Talk between Estrogen Receptor and Epidermal Growth Factor Receptor in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2009, 15, 6529–6540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzino, M.; Morelli, C.; Garofalo, C.; Panno, M.L.; Mauro, L.; Andò, S.; Sisci, D. Interaction between Estrogen Receptor Alpha and insulin/IGF Signaling in Breast Cancer. Curr. Cancer Drug Targets 2008, 8, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, J.M.; Farooqui, M.; Rothenberger, N.J.; Dacic, S.; Stabile, L.P. Interaction between the Estrogen Receptor and Fibroblast Growth Factor Receptor Pathways in Non-Small Cell Lung Cancer. Oncotarget 2017, 8, 24063–24076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santolla, M.F.; Vivacqua, A.; Lappano, R.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Brunetti, G.; Miglietta, A.M.; Belfiore, A.; et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression. Cells 2019, 8, 223. [Google Scholar] [CrossRef] [Green Version]
- De Marco, P.; Bartella, V.; Vivacqua, A.; Lappano, R.; Santolla, M.F.; Morcavallo, A.; Pezzi, V.; Belfiore, A.; Maggiolini, M. Insulin-like Growth Factor-I Regulates GPER Expression and Function in Cancer Cells. Oncogene 2013, 32, 678–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal Growth Factor-Related Peptides and Their Receptors in Human Malignancies. Crit. Rev. Oncol. Hematol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Gullick, W.J. The Epidermal Growth Factor System of Ligands and Receptors in Cancer. Eur. J. Cancer 2009, 45 (Suppl. S1), 205–210. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin Receptor Isoforms and Insulin Receptor/insulin-like Growth Factor Receptor Hybrids in Physiology and Disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, M. Insulin and Insulin-like Growth Factor Signalling in Neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast Growth Factor Signalling: From Development to Cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.; Langhans, S.A. Epidermal Growth Factor Signaling in Transformed Cells. Int. Rev. Cell Mol. Biol. 2015, 314, 1–41. [Google Scholar] [PubMed] [Green Version]
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.-Z.; Roychowdhury, S. Fibroblast Growth Factor Receptors in Cancer: Genetic Alterations, Diagnostics, Therapeutic Targets and Mechanisms of Resistance. Br. J. Cancer 2021, 124, 880–892. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Lennartsson, J.; Westermark, B. Involvement of Platelet-Derived Growth Factor Ligands and Receptors in Tumorigenesis. J. Intern. Med. 2018, 283, 16–44. [Google Scholar] [CrossRef] [Green Version]
- Franovic, A.; Gunaratnam, L.; Smith, K.; Robert, I.; Patten, D.; Lee, S. Translational up-Regulation of the EGFR by Tumor Hypoxia Provides a Nonmutational Explanation for Its Overexpression in Human Cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 13092–13097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.; Gunaratnam, L.; Morley, M.; Franovic, A.; Mekhail, K.; Lee, S. Silencing of Epidermal Growth Factor Receptor Suppresses Hypoxia-Inducible Factor-2-Driven VHL-/- Renal Cancer. Cancer Res. 2005, 65, 5221–5230. [Google Scholar] [CrossRef] [Green Version]
- Franovic, A.; Holterman, C.E.; Payette, J.; Lee, S. Human Cancers Converge at the HIF-2alpha Oncogenic Axis. Proc. Natl. Acad. Sci. USA 2009, 106, 21306–21311. [Google Scholar] [CrossRef] [Green Version]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) Signaling Increases the Rate of Hypoxia-Inducible Factor 1alpha (HIF-1alpha) Synthesis: Novel Mechanism for HIF-1-Mediated Vascular Endothelial Growth Factor Expression. Mol. Cell. Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, R.; Hirota, K.; Fan, F.; Jung, Y.D.; Ellis, L.M.; Semenza, G.L. Insulin-like Growth Factor 1 Induces Hypoxia-Inducible Factor 1-Mediated Vascular Endothelial Growth Factor Expression, Which Is Dependent on MAP Kinase and Phosphatidylinositol 3-Kinase Signaling in Colon Cancer Cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.-H.; Bingle, L.; Gong, L.-H.; Wang, Y.-X.; Corke, K.P.; Fang, W.-G. Basic FGF Augments Hypoxia Induced HIF-1-Alpha Expression and VEGF Release in T47D Breast Cancer Cells. Pathology 2007, 39, 396–400. [Google Scholar] [CrossRef]
- Bos, R.; van Diest, P.J.; de Jong, J.S.; van der Groep, P.; van der Valk, P.; van der Wall, E. Hypoxia-Inducible Factor-1alpha Is Associated with Angiogenesis, and Expression of bFGF, PDGF-BB, and EGFR in Invasive Breast Cancer. Histopathology 2005, 46, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Hirami, Y.; Aoe, M.; Tsukuda, K.; Hara, F.; Otani, Y.; Koshimune, R.; Hanabata, T.; Nagahiro, I.; Sano, Y.; Date, H.; et al. Relation of Epidermal Growth Factor Receptor, Phosphorylated-Akt, and Hypoxia-Inducible Factor-1alpha in Non-Small Cell Lung Cancers. Cancer Lett. 2004, 214, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabjeesh, N.J.; Willard, M.T.; Frederickson, C.E.; Zhong, H.; Simons, J.W. Androgens Stimulate Hypoxia-Inducible Factor 1 Activation via Autocrine Loop of Tyrosine Kinase Receptor/phosphatidylinositol 3′-Kinase/protein Kinase B in Prostate Cancer Cells. Clin. Cancer Res. 2003, 9, 2416–2425. [Google Scholar] [PubMed]
- Hua, K.; Din, J.; Cao, Q.; Feng, W.; Zhang, Y.; Yao, L.; Huang, Y.; Zhao, Y.; Feng, Y. Estrogen and Progestin Regulate HIF-1alpha Expression in Ovarian Cancer Cell Lines via the Activation of Akt Signaling Transduction Pathway. Oncol. Rep. 2009, 21, 893–898. [Google Scholar] [PubMed] [Green Version]
- Von Wahlde, M.-K.; Hülsewig, C.; Ruckert, C.; Götte, M.; Kiesel, L.; Bernemann, C. The Anti-Androgen Drug Dutasteride Renders Triple Negative Breast Cancer Cells More Sensitive to Chemotherapy via Inhibition of HIF-1α-/VEGF-Signaling. Gynecol. Endocrinol. 2015, 31, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Ragnum, H.B.; Røe, K.; Holm, R.; Vlatkovic, L.; Nesland, J.M.; Aarnes, E.-K.; Ree, A.H.; Flatmark, K.; Seierstad, T.; Lilleby, W.; et al. Hypoxia-Independent Downregulation of Hypoxia-Inducible Factor 1 Targets by Androgen Deprivation Therapy in Prostate Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 753–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajoria, S.; Hanly, E.; Nicolini, A.; George, A.L.; Geliebter, J.; Shin, E.J.; Suriano, R.; Carpi, A.; Tiwari, R.K. Interlinking of Hypoxia and Estrogen in Thyroid Cancer Progression. Curr. Med. Chem. 2014, 21, 1351–1360. [Google Scholar] [CrossRef]
- George, A.L.; Rajoria, S.; Suriano, R.; Mittleman, A.; Tiwari, R.K. Hypoxia and Estrogen Are Functionally Equivalent in Breast Cancer-Endothelial Cell Interdependence. Mol. Cancer 2012, 11, 80. [Google Scholar] [CrossRef] [Green Version]
- Dewangan, J.; Kaushik, S.; Rath, S.K.; Balapure, A.K. Centchroman Regulates Breast Cancer Angiogenesis via Inhibition of HIF-1α/VEGFR2 Signalling Axis. Life Sci. 2018, 193, 9–19. [Google Scholar] [CrossRef]
- Fuady, J.H.; Gutsche, K.; Santambrogio, S.; Varga, Z.; Hoogewijs, D.; Wenger, R.H. Estrogen-Dependent Downregulation of Hypoxia-Inducible Factor (HIF)-2α in Invasive Breast Cancer Cells. Oncotarget 2016, 7, 31153–31165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiehl, D.P.; Bordoli, M.R.; Abreu-Rodríguez, I.; Wollenick, K.; Schraml, P.; Gradin, K.; Poellinger, L.; Kristiansen, G.; Wenger, R.H. Non-Canonical HIF-2α Function Drives Autonomous Breast Cancer Cell Growth via an AREG-EGFR/ErbB4 Autocrine Loop. Oncogene 2012, 31, 2283–2297. [Google Scholar] [CrossRef] [Green Version]
- Seifeddine, R.; Dreiem, A.; Tomkiewicz, C.; Fulchignoni-Lataud, M.-C.; Brito, I.; Danan, J.-L.; Favaudon, V.; Barouki, R.; Massaad-Massade, L. Hypoxia and Estrogen Co-Operate to Regulate Gene Expression in T-47D Human Breast Cancer Cells. J. Steroid Biochem. Mol. Biol. 2007, 104, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jubb, A.M.; Pike, L.; Buffa, F.M.; Turley, H.; Baban, D.; Leek, R.; Gatter, K.C.; Ragoussis, J.; Harris, A.L. The Histone Demethylase JMJD2B Is Regulated by Estrogen Receptor Alpha and Hypoxia, and Is a Key Mediator of Estrogen Induced Growth. Cancer Res. 2010, 70, 6456–6466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Harris, A.L.; Davidoff, A.M. Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer. Int. J. Mol. Sci. 2018, 19, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawazu, M.; Saso, K.; Tong, K.I.; McQuire, T.; Goto, K.; Son, D.-O.; Wakeham, A.; Miyagishi, M.; Mak, T.W.; Okada, H. Histone Demethylase JMJD2B Functions as a Co-Factor of Estrogen Receptor in Breast Cancer Proliferation and Mammary Gland Development. PLoS ONE 2011, 6, e17830. [Google Scholar] [CrossRef]
- Yang, J.; AlTahan, A.; Jones, D.T.; Buffa, F.M.; Bridges, E.; Interiano, R.B.; Qu, C.; Vogt, N.; Li, J.-L.; Baban, D.; et al. Estrogen Receptor-α Directly Regulates the Hypoxia-Inducible Factor 1 Pathway Associated with Antiestrogen Response in Breast Cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15172–15177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Generali, D.; Buffa, F.M.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Phosphorylated ERalpha, HIF-1alpha, and MAPK Signaling as Predictors of Primary Endocrine Treatment Response and Resistance in Patients with Breast Cancer. J. Clin. Oncol. 2009, 27, 227–234. [Google Scholar] [CrossRef]
- Generali, D.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Wigfield, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Hypoxia-Inducible Factor-1alpha Expression Predicts a Poor Response to Primary Chemoendocrine Therapy and Disease-Free Survival in Primary Human Breast Cancer. Clin. Cancer Res. 2006, 12, 4562–4568. [Google Scholar] [CrossRef] [Green Version]
- Bos, R.; Zhong, H.; Hanrahan, C.F.; Mommers, E.C.; Semenza, G.L.; Pinedo, H.M.; Abeloff, M.D.; Simons, J.W.; van Diest, P.J.; van der Wall, E. Levels of Hypoxia-Inducible Factor-1 Alpha during Breast Carcinogenesis. J. Natl. Cancer Inst. 2001, 93, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Bialesova, L.; Xu, L.; Gustafsson, J.-Å.; Haldosen, L.-A.; Zhao, C.; Dahlman-Wright, K. Estrogen Receptor β2 Induces Proliferation and Invasiveness of Triple Negative Breast Cancer Cells: Association with Regulation of PHD3 and HIF-1α. Oncotarget 2017, 8, 76622–76633. [Google Scholar] [CrossRef] [Green Version]
- Dey, P.; Velazquez-Villegas, L.A.; Faria, M.; Turner, A.; Jonsson, P.; Webb, P.; Williams, C.; Gustafsson, J.-Å.; Ström, A.M. Estrogen Receptor β2 Induces Hypoxia Signature of Gene Expression by Stabilizing HIF-1α in Prostate Cancer. PLoS ONE 2015, 10, e0128239. [Google Scholar]
- Zou, C.; Yu, S.; Xu, Z.; Wu, D.; Ng, C.-F.; Yao, X.; Yew, D.T.; Vanacker, J.-M.; Chan, F.L. ERRα Augments HIF-1 Signalling by Directly Interacting with HIF-1α in Normoxic and Hypoxic Prostate Cancer Cells. J. Pathol. 2014, 233, 61–73. [Google Scholar] [CrossRef]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of Hypoxia-Inducible Factor 1alpha Expression by the Epidermal Growth Factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP Pathway in Human Prostate Cancer Cells: Implications for Tumor Angiogenesis and Therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Phillips, R.J.; Mestas, J.; Gharaee-Kermani, M.; Burdick, M.D.; Sica, A.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. Epidermal Growth Factor and Hypoxia-Induced Expression of CXC Chemokine Receptor 4 on Non-Small Cell Lung Cancer Cells Is Regulated by the Phosphatidylinositol 3-kinase/PTEN/AKT/mammalian Target of Rapamycin Signaling Pathway and Activation of Hypoxia Inducible Factor-1alpha. J. Biol. Chem. 2005, 280, 22473–22481. [Google Scholar]
- Han, Z.-B.; Ren, H.; Zhao, H.; Chi, Y.; Chen, K.; Zhou, B.; Liu, Y.-J.; Zhang, L.; Xu, B.; Liu, B.; et al. Hypoxia-Inducible Factor (HIF)-1 Alpha Directly Enhances the Transcriptional Activity of Stem Cell Factor (SCF) in Response to Hypoxia and Epidermal Growth Factor (EGF). Carcinogenesis 2008, 29, 1853–1861. [Google Scholar] [CrossRef]
- Zhao, F.-L.; Qin, C.-F. EGF Promotes HIF-1α Expression in Colorectal Cancer Cells and Tumor Metastasis by Regulating Phosphorylation of STAT3. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1055–1062. [Google Scholar] [PubMed]
- Peng, X.-H.; Karna, P.; Cao, Z.; Jiang, B.-H.; Zhou, M.; Yang, L. Cross-Talk between Epidermal Growth Factor Receptor and Hypoxia-Inducible Factor-1alpha Signal Pathways Increases Resistance to Apoptosis by up-Regulating Survivin Gene Expression. J. Biol. Chem. 2006, 281, 25903–25914. [Google Scholar] [CrossRef] [Green Version]
- Swinson, D.E.B.; O’Byrne, K.J. Interactions between Hypoxia and Epidermal Growth Factor Receptor in Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2006, 7, 250–256. [Google Scholar] [CrossRef]
- Hu, W.; Zheng, S.; Guo, H.; Dai, B.; Ni, J.; Shi, Y.; Bian, H.; Li, L.; Shen, Y.; Wu, M.; et al. PLAGL2-EGFR-HIF-1/2α Signaling Loop Promotes HCC Progression and Erlotinib Insensitivity. Hepatology 2021, 73, 674–691. [Google Scholar] [CrossRef]
- Wang, Y.; Roche, O.; Xu, C.; Moriyama, E.H.; Heir, P.; Chung, J.; Roos, F.C.; Chen, Y.; Finak, G.; Milosevic, M.; et al. Hypoxia Promotes Ligand-Independent EGF Receptor Signaling via Hypoxia-Inducible Factor-Mediated Upregulation of Caveolin-1. Proc. Natl. Acad. Sci. USA 2012, 109, 4892–4897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Schneider, A. HIF-2alpha-Mediated Activation of the Epidermal Growth Factor Receptor Potentiates Head and Neck Cancer Cell Migration in Response to Hypoxia. Carcinogenesis 2010, 31, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.; Smith, K.; Gullick, W.J.; Harris, A.L. Mutant Epidermal Growth Factor Receptor Enhances Induction of Vascular Endothelial Growth Factor by Hypoxia and Insulin-like Growth Factor-1 via a PI3 Kinase Dependent Pathway. Br. J. Cancer 2001, 84, 1322–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, A.; Boeckx, C.; Vermorken, J.B.; Van den Weyngaert, D.; Peeters, M.; Lardon, F. The Intriguing Interplay between Therapies Targeting the Epidermal Growth Factor Receptor, the Hypoxic Microenvironment and Hypoxia-Inducible Factors. Curr. Pharm. Des. 2013, 19, 907–917. [Google Scholar] [CrossRef]
- Petit, A.M.; Rak, J.; Hung, M.C.; Rockwell, P.; Goldstein, N.; Fendly, B.; Kerbel, R.S. Neutralizing Antibodies against Epidermal Growth Factor and ErbB-2/neu Receptor Tyrosine Kinases down-Regulate Vascular Endothelial Growth Factor Production by Tumor Cells In Vitro and In Vivo: Angiogenic Implications for Signal Transduction Therapy of Solid Tumors. Am. J. Pathol. 1997, 151, 1523–1530. [Google Scholar]
- Riesterer, O.; Mason, K.A.; Raju, U.; Yang, Q.; Wang, L.; Hittelman, W.N.; Ang, K.K.; Milas, L. Enhanced Response to C225 of A431 Tumor Xenografts Growing in Irradiated Tumor Bed. Radiother. Oncol. 2009, 92, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.J.; Harbison, M.T.; Davis, D.W.; Portera, C.A.; Tsan, R.; McConkey, D.J.; Evans, D.B.; Abbruzzese, J.L.; Hicklin, D.J.; Radinsky, R. Epidermal Growth Factor Receptor Blockade with C225 plus Gemcitabine Results in Regression of Human Pancreatic Carcinoma Growing Orthotopically in Nude Mice by Antiangiogenic Mechanisms. Clin. Cancer Res. 2000, 6, 1936–1948. [Google Scholar]
- Li, X.; Lu, Y.; Liang, K.; Pan, T.; Mendelsohn, J.; Fan, Z. Requirement of Hypoxia-Inducible Factor-1alpha down-Regulation in Mediating the Antitumor Activity of the Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Cetuximab. Mol. Cancer Ther. 2008, 7, 1207–1217. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Fan, Z. The Epidermal Growth Factor Receptor Antibody Cetuximab Induces Autophagy in Cancer Cells by Downregulating HIF-1alpha and Bcl-2 and Activating the Beclin 1/hVps34 Complex. Cancer Res. 2010, 70, 5942–5952. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Liang, K.; Lu, Y.; Fan, Z. The Anti-EGFR Antibody Cetuximab Sensitizes Human Head and Neck Squamous Cell Carcinoma Cells to Radiation in Part through Inhibiting Radiation-Induced Upregulation of HIF-1α. Cancer Lett. 2012, 322, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciardiello, F.; Caputo, R.; Bianco, R.; Damiano, V.; Fontanini, G.; Cuccato, S.; De Placido, S.; Bianco, A.R.; Tortora, G. Inhibition of Growth Factor Production and Angiogenesis in Human Cancer Cells by ZD1839 (Iressa), a Selective Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor. Clin. Cancer Res. 2001, 7, 1459–1465. [Google Scholar]
- Hirata, A.; Ogawa, S.-I.; Kometani, T.; Kuwano, T.; Naito, S.; Kuwano, M.; Ono, M. ZD1839 (Iressa) Induces Antiangiogenic Effects through Inhibition of Epidermal Growth Factor Receptor Tyrosine Kinase. Cancer Res. 2002, 62, 2554–2560. [Google Scholar]
- Pore, N.; Jiang, Z.; Gupta, A.; Cerniglia, G.; Kao, G.D.; Maity, A. EGFR Tyrosine Kinase Inhibitors Decrease VEGF Expression by Both Hypoxia-Inducible Factor (HIF)-1-Independent and HIF-1-Dependent Mechanisms. Cancer Res. 2006, 66, 3197–3204. [Google Scholar] [CrossRef] [Green Version]
- Han, J.-Y.; Oh, S.H.; Morgillo, F.; Myers, J.N.; Kim, E.; Hong, W.K.; Lee, H.-Y. Hypoxia-Inducible Factor 1alpha and Antiangiogenic Activity of Farnesyltransferase Inhibitor SCH66336 in Human Aerodigestive Tract Cancer. J. Natl. Cancer Inst. 2005, 97, 1272–1286. [Google Scholar] [CrossRef]
- Catrina, S.-B.; Botusan, I.R.; Rantanen, A.; Catrina, A.I.; Pyakurel, P.; Savu, O.; Axelson, M.; Biberfeld, P.; Poellinger, L.; Brismar, K. Hypoxia-Inducible Factor-1alpha and Hypoxia-Inducible Factor-2alpha Are Expressed in Kaposi Sarcoma and Modulated by Insulin-like Growth Factor-I. Clin. Cancer Res. 2006, 12, 4506–4514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, V.A.; Ashcroft, M. Role of Hypoxia-Inducible Factor (HIF)-1alpha versus HIF-2alpha in the Regulation of HIF Target Genes in Response to Hypoxia, Insulin-like Growth Factor-I, or Loss of von Hippel-Lindau Function: Implications for Targeting the HIF Pathway. Cancer Res. 2006, 66, 6264–6270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, K.M.; Hayat, S.; Chau, N.-M.; Cook, S.; Pouyssegur, J.; Ahmed, A.; Perusinghe, N.; Le Floch, R.; Yang, J.; Ashcroft, M. Selective Inhibition of MEK1/2 Reveals a Differential Requirement for ERK1/2 Signalling in the Regulation of HIF-1 in Response to Hypoxia and IGF-1. Oncogene 2007, 26, 3920–3929. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Sims, A.H.; Maggiolini, M.; Sotgia, F.; Lisanti, M.P.; Clarke, R.B. GPER Mediates the Angiocrine Actions Induced by IGF1 through the HIF-1α/VEGF Pathway in the Breast Tumor Microenvironment. Breast Cancer Res. 2017, 19, 129. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, Q.; Shi, S.; Yen, Y.; Li, X.; Zhang, Y.; Zhou, K.; Le, A.D. Bisphosphonates Suppress Insulin-like Growth Factor 1-Induced Angiogenesis via the HIF-1alpha/VEGF Signaling Pathways in Human Breast Cancer Cells. Int. J. Cancer 2010, 126, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Feng, Y.; Liu, J.; Feng, X.; Zhou, K.; Tang, X. Epigallocatechin-3-Gallate Inhibits IGF-I-Stimulated Lung Cancer Angiogenesis through Downregulation of HIF-1α and VEGF Expression. J. Nutrigenet. Nutr. 2013, 6, 169–178. [Google Scholar] [CrossRef]
- Tang, X.-D.; Zhou, X.; Zhou, K.-Y. Dauricine Inhibits Insulin-like Growth Factor-I-Induced Hypoxia Inducible Factor 1alpha Protein Accumulation and Vascular Endothelial Growth Factor Expression in Human Breast Cancer Cells. Acta Pharmacol. Sin. 2009, 30, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Feldser, D.; Agani, F.; Iyer, N.V.; Pak, B.; Ferreira, G.; Semenza, G.L. Reciprocal Positive Regulation of Hypoxia-Inducible Factor 1alpha and Insulin-like Growth Factor 2. Cancer Res. 1999, 59, 3915–3918. [Google Scholar] [PubMed]
- Mancini, M.; Gariboldi, M.B.; Taiana, E.; Bonzi, M.C.; Craparotta, I.; Pagin, M.; Monti, E. Co-Targeting the IGF System and HIF-1 Inhibits Migration and Invasion by (triple-Negative) Breast Cancer Cells. Br. J. Cancer 2014, 110, 2865–2873. [Google Scholar] [CrossRef] [Green Version]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.-H.; Wang, Y.-X.; Bingle, L.; Gong, L.-H.; Heng, W.-J.; Li, Y.; Fang, W.-G. In Vitro Study of HIF-1 Activation and VEGF Release by bFGF in the T47D Breast Cancer Cell Line under Normoxic Conditions: Involvement of PI-3K/Akt and MEK1/ERK Pathways. J. Pathol. 2005, 205, 530–536. [Google Scholar] [CrossRef]
- Shi, Y.-H.; Wang, Y.-X.; You, J.-F.; Heng, W.-J.; Zhong, H.-H.; Fang, W.-G. Activation of HIF-1 by bFGF in breast cancer: Role of PI-3K and MEK1/ERK pathways. Zhonghua Yi Xue Za Zhi 2004, 84, 1899–1903. [Google Scholar] [PubMed]
- Iqbal, S.; Zhang, S.; Driss, A.; Liu, Z.-R.; Kim, H.-R.C.; Wang, Y.; Ritenour, C.; Zhau, H.E.; Kucuk, O.; Chung, L.W.K.; et al. PDGF Upregulates Mcl-1 through Activation of β-Catenin and HIF-1α-Dependent Signaling in Human Prostate Cancer Cells. PLoS ONE 2012, 7, e30764. [Google Scholar] [CrossRef] [Green Version]
- Kimura, Y.; Inoue, K.; Abe, M.; Nearman, J.; Baranowska-Kortylewicz, J. PDGFRbeta and HIF-1alpha Inhibition with Imatinib and Radioimmunotherapy of Experimental Prostate Cancer. Cancer Biol. Ther. 2007, 6, 1763–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schito, L.; Rey, S.; Tafani, M.; Zhang, H.; Wong, C.C.-L.; Russo, A.; Russo, M.A.; Semenza, G.L. Hypoxia-Inducible Factor 1-Dependent Expression of Platelet-Derived Growth Factor B Promotes Lymphatic Metastasis of Hypoxic Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2012, 109, E2707–E2716. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.K.; Yang, Z.F.; Ho, D.W.; Ng, M.N.; Yeoh, G.C.T.; Poon, R.T.P.; Fan, S.T. An Akt/hypoxia-Inducible Factor-1alpha/platelet-Derived Growth Factor-BB Autocrine Loop Mediates Hypoxia-Induced Chemoresistance in Liver Cancer Cells and Tumorigenic Hepatic Progenitor Cells. Clin. Cancer Res. 2009, 15, 3462–3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of Non-Hodgkin’s Lymphoma with a Defined Ratio of CD8+ and CD4+ CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar]
- Turan, T.; Kannan, D.; Patel, M.; Matthew Barnes, J.; Tanlimco, S.G.; Lu, R.; Halliwill, K.; Kongpachith, S.; Kline, D.E.; Hendrickx, W.; et al. Immune Oncology, Immune Responsiveness and the Theory of Everything. J. Immunother. Cancer 2018, 6, 50. [Google Scholar] [CrossRef] [Green Version]
- Bedognetti, D.; Ceccarelli, M.; Galluzzi, L.; Lu, R.; Palucka, K.; Samayoa, J.; Spranger, S.; Warren, S.; Wong, K.-K.; Ziv, E.; et al. Correction to: Toward a Comprehensive View of Cancer Immune Responsiveness: A Synopsis from the SITC Workshop. J. Immunother. Cancer 2019, 7, 167. [Google Scholar] [CrossRef] [Green Version]
- Pietrobon, V.; Marincola, F.M. Hypoxia and the Phenomenon of Immune Exclusion. J. Transl. Med. 2021, 19, 9. [Google Scholar] [CrossRef]
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a Barrier to Immunotherapy in Pancreatic Adenocarcinoma. Clin. Transl. Med. 2019, 8, 10. [Google Scholar] [CrossRef]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted Hypoxia Reduction Restores T Cell Infiltration and Sensitizes Prostate Cancer to Immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms Regulating T-Cell Infiltration and Activity in Solid Tumors. Ann. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The Tumor Microenvironment as a Metabolic Barrier to Effector T Cells and Immunotherapy. eLife 2020, 9, e55185. [Google Scholar] [CrossRef]
- Gillies, R.J.; Schornack, P.A.; Secomb, T.W.; Raghunand, N. Causes and Effects of Heterogeneous Perfusion in Tumors. Neoplasia 1999, 1, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Padera, T.P.; Stoll, B.R.; Tooredman, J.B.; Capen, D.; di Tomaso, E.; Jain, R.K. Cancer Cells Compress Intratumour Vessels. Nature 2004, 427, 695. [Google Scholar] [CrossRef]
- Welti, J.; Loges, S.; Dimmeler, S.; Carmeliet, P. Recent Molecular Discoveries in Angiogenesis and Antiangiogenic Therapies in Cancer. J. Clin. Investig. 2013, 123, 3190–3200. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the Role of the Tumor Vasculature in Antitumor Immunity and Immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Kim, B.Y.S.; Chan, C.K.; Hahn, S.M.; Weissman, I.L.; Jiang, W. Improving Immune-Vascular Crosstalk for Cancer Immunotherapy. Nat. Rev. Immunol. 2018, 18, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, Q.; Zheng, Z.; Liu, S.; Meng, L.; Dong, L.; Jiang, X. Vascular Normalization in Immunotherapy: A Promising Mechanisms Combined with Radiotherapy. Biomed. Pharmacother. 2021, 139, 111607. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Bajpai, S.; Wong, C.C.; Chaturvedi, P.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Procollagen Lysyl Hydroxylase 2 Is Essential for Hypoxia-Induced Breast Cancer Metastasis. Mol. Cancer Res. 2013, 11, 456–466. [Google Scholar] [CrossRef] [Green Version]
- Xiong, G.; Deng, L.; Zhu, J.; Rychahou, P.G.; Xu, R. Prolyl-4-Hydroxylase α Subunit 2 Promotes Breast Cancer Progression and Metastasis by Regulating Collagen Deposition. BMC Cancer 2014, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisinger-Mathason, T.S.K.; Zhang, M.; Qiu, Q.; Skuli, N.; Nakazawa, M.S.; Karakasheva, T.; Mucaj, V.; Shay, J.E.S.; Stangenberg, L.; Sadri, N.; et al. Hypoxia-Dependent Modification of Collagen Networks Promotes Sarcoma Metastasis. Cancer Discov. 2013, 3, 1190–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofbauer, K.-H.; Gess, B.; Lohaus, C.; Meyer, H.E.; Katschinski, D.; Kurtz, A. Oxygen Tension Regulates the Expression of a Group of Procollagen Hydroxylases. Eur. J. Biochem. 2003, 270, 4515–4522. [Google Scholar]
- Aro, E.; Khatri, R.; Gerard-O’Riley, R.; Mangiavini, L.; Myllyharju, J.; Schipani, E. Hypoxia-Inducible Factor-1 (HIF-1) but Not HIF-2 Is Essential for Hypoxic Induction of Collagen Prolyl 4-Hydroxylases in Primary Newborn Mouse Epiphyseal Growth Plate Chondrocytes. J. Biol. Chem. 2012, 287, 37134–37144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.Y.; Jang, Y.S.; Min, S.Y.; Song, J.Y. Overexpression of MMP-9 and HIF-1α in Breast Cancer Cells under Hypoxic Conditions. J. Breast Cancer 2011, 14, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-β Signaling and Epithelial-Mesenchymal Transition in Cancer Progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Tanghetti, E.; Ria, R.; Dell’Era, P.; Urbinati, C.; Rusnati, M.; Ennas, M.G.; Presta, M. Biological Activity of Substrate-Bound Basic Fibroblast Growth Factor (FGF2): Recruitment of FGF Receptor-1 in Endothelial Cell Adhesion Contacts. Oncogene 2002, 21, 3889–3897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.-T.; Giaccia, A.J. Hypoxia-Induced Lysyl Oxidase Is a Critical Mediator of Bone Marrow Cell Recruitment to Form the Premetastatic Niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Pietras, K.; Ostman, A. Hallmarks of Cancer: Interactions with the Tumor Stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [PubMed]
- Schietke, R.; Warnecke, C.; Wacker, I.; Schödel, J.; Mole, D.R.; Campean, V.; Amann, K.; Goppelt-Struebe, M.; Behrens, J.; Eckardt, K.-U.; et al. The Lysyl Oxidases LOX and LOXL2 Are Necessary and Sufficient to Repress E-Cadherin in Hypoxia: Insights into Cellular Transformation Processes Mediated by HIF-1. J. Biol. Chem. 2010, 285, 6658–6669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.C.-L.; Gilkes, D.M.; Zhang, H.; Chen, J.; Wei, H.; Chaturvedi, P.; Fraley, S.I.; Wong, C.-M.; Khoo, U.-S.; Ng, I.O.-L.; et al. Hypoxia-Inducible Factor 1 Is a Master Regulator of Breast Cancer Metastatic Niche Formation. Proc. Natl. Acad. Sci. USA 2011, 108, 16369–16374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal Cells in Tumor Microenvironment and Breast Cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Gilkes, D.M.; Chaturvedi, P.; Bajpai, S.; Wong, C.C.; Wei, H.; Pitcairn, S.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Collagen Prolyl Hydroxylases Are Essential for Breast Cancer Metastasis. Cancer Res. 2013, 73, 3285–3296. [Google Scholar] [CrossRef] [Green Version]
- Wan, R.; Mo, Y.; Chien, S.; Li, Y.; Li, Y.; Tollerud, D.J.; Zhang, Q. The Role of Hypoxia Inducible Factor-1α in the Increased MMP-2 and MMP-9 Production by Human Monocytes Exposed to Nickel Nanoparticles. Nanotoxicology 2011, 5, 568–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revuelta-López, E.; Castellano, J.; Roura, S.; Gálvez-Montón, C.; Nasarre, L.; Benitez, S.; Bayes-Genis, A.; Badimon, L.; Llorente-Cortés, V. Hypoxia Induces Metalloproteinase-9 Activation and Human Vascular Smooth Muscle Cell Migration Through Low-Density Lipoprotein Receptor–Related Protein 1–Mediated Pyk2 Phosphorylation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2877–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-H.; Zhou, K.; Kim, D.-K.; Park, S.; Noh, J.; Kwon, Y.; Kim, D.; Song, N.W.; Lee, J.-B.; Suh, P.-G.; et al. Analysis of Interactions between the Epidermal Growth Factor Receptor and Soluble Ligands on the Basis of Single-Molecule Diffusivity in the Membrane of Living Cells. Angew. Chem. Int. Ed. 2015, 54, 7028–7032. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Shih, C.-H.; Tseng, C.-C.; Yu, C.-C.; Tsai, Y.-J.; Bien, M.-Y.; Chen, B.-C. CXCL12 Induces Connective Tissue Growth Factor Expression in Human Lung Fibroblasts through the Rac1/ERK, JNK, and AP-1 Pathways. PLoS ONE 2014, 9, e104746. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-L.; Cheng, W.-E.; Chen, S.-C.; Chen, C.-Y.; Shih, C.-M. The Effects of Hypoxia on the Expression of MMP-2, MMP-9 in Human Lung Adenocarcinoma A549 Cells. Eur. Respir. J. 2014, 44, P2699. [Google Scholar]
- Miao, J.-W.; Liu, L.-J.; Huang, J. Interleukin-6-Induced Epithelial-Mesenchymal Transition through Signal Transducer and Activator of Transcription 3 in Human Cervical Carcinoma. Int. J. Oncol. 2014, 45, 165–176. [Google Scholar] [CrossRef]
- Masola, V.; Carraro, A.; Granata, S.; Signorini, L.; Bellin, G.; Violi, P.; Lupo, A.; Tedeschi, U.; Onisto, M.; Gambaro, G.; et al. In Vitro Effects of Interleukin (IL)-1 Beta Inhibition on the Epithelial-to-Mesenchymal Transition (EMT) of Renal Tubular and Hepatic Stellate Cells. J. Transl. Med. 2019, 17, 12. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Ye, Y.; Zhang, L.; Liu, P.; Yu, W.; Wei, F.; Ren, X.; Yu, J. IL-8, a Novel Messenger to Cross-Link Inflammation and Tumor EMT via Autocrine and Paracrine Pathways (Review). Int. J. Oncol. 2016, 48, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Palena, C.; Hamilton, D.H.; Fernando, R.I. Influence of IL-8 on the Epithelial-Mesenchymal Transition and the Tumor Microenvironment. Future Oncol. 2012, 8, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Sistigu, A.; Di Modugno, F.; Manic, G.; Nisticò, P. Deciphering the Loop of Epithelial-Mesenchymal Transition, Inflammatory Cytokines and Cancer Immunoediting. Cytokine Growth Factor Rev. 2017, 36, 67–77. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-Induced Epithelial to Mesenchymal Transition. Cell Res. 2009, 19, 156–172. [Google Scholar]
- Gonzalez, D.M.; Medici, D. Signaling Mechanisms of the Epithelial-Mesenchymal Transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-Mesenchymal Transition (EMT) Signature Is Inversely Associated with T-Cell Infiltration in Non-Small Cell Lung Cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Mu, Y.; Sa, N.; Wang, H.; Xu, W. Tumor Necrosis Factor α Induces Epithelial-Mesenchymal Transition and Promotes Metastasis via NF-κB Signaling Pathway-Mediated TWIST Expression in Hypopharyngeal Cancer. Oncol. Rep. 2014, 31, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-W.; Xia, W.; Huo, L.; Lim, S.-O.; Wu, Y.; Hsu, J.L.; Chao, C.-H.; Yamaguchi, H.; Yang, N.-K.; Ding, Q.; et al. Epithelial–Mesenchymal Transition Induced by TNF-α Requires NF-κB–Mediated Transcriptional Upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Ong, S.L.; Tran, L.M.; Jing, Z.; Liu, B.; Park, S.J.; Huang, Z.L.; Walser, T.C.; Heinrich, E.L.; Lee, G.; et al. Chronic IL-1β-Induced Inflammation Regulates Epithelial-to-Mesenchymal Transition Memory Phenotypes via Epigenetic Modifications in Non-Small Cell Lung Cancer. Sci. Rep. 2020, 10, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Zhang, C.; Pan, J.; Chen, L.; Qi, S.-T. Interleukin-6 Induces an Epithelial-mesenchymal Transition Phenotype in Human Adamantinomatous Craniopharyngioma Cells and Promotes Tumor Cell Migration. Mol. Med. Rep. 2017, 15, 4123–4131. [Google Scholar] [CrossRef] [Green Version]
- Farrell, J.; Kelly, C.; Rauch, J.; Kida, K.; García-Muñoz, A.; Monsefi, N.; Turriziani, B.; Doherty, C.; Mehta, J.P.; Matallanas, D.; et al. HGF Induces Epithelial-to-Mesenchymal Transition by Modulating the Mammalian hippo/MST2 and ISG15 Pathways. J. Proteome Res. 2014, 13, 2874–2886. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Song, S.; Yi, Z.; Zhang, M.; Li, J.; Yang, F.; Yin, H.; Yu, X.; Guan, C.; Liu, Y.; et al. HGF Induces EMT in Non-Small-Cell Lung Cancer through the hBVR Pathway. Eur. J. Pharmacol. 2017, 811, 180–190. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M.; Ziyadeh, F.N.; Yang, C.-Q.; Kalluri, R.; Müller, G.A.; Neilson, E.G. Role of Basic Fibroblast Growth Factor-2 in Epithelial-Mesenchymal Transformation. Kidney Int. 2002, 61, 1714–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savagner, P.; Vallés, A.M.; Jouanneau, J.; Yamada, K.M.; Thiery, J.P. Alternative Splicing in Fibroblast Growth Factor Receptor 2 Is Associated with Induced Epithelial-Mesenchymal Transition in Rat Bladder Carcinoma Cells. Mol. Biol. Cell 1994, 5, 851–862. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, Q.; Ishida, Y.; Hajjar, S.; Tang, X.; Shi, H.; Dang, C.V.; Le, A.D. EGF Induces Epithelial-Mesenchymal Transition and Cancer Stem-like Cell Properties in Human Oral Cancer Cells via Promoting Warburg Effect. Oncotarget 2017, 8, 9557–9571. [Google Scholar] [CrossRef] [Green Version]
- Shu, D.; Lovicu, F.J. EGF Potentiates TGFβ-Induced Epithelial-Mesenchymal Transition (EMT) in Lens Epithelial Cells by Enhancing EGFR Signaling. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1202. [Google Scholar]
- Wu, Q.; Hou, X.; Xia, J.; Qian, X.; Miele, L.; Sarkar, F.H.; Wang, Z. Emerging Roles of PDGF-D in EMT Progression during Tumorigenesis. Cancer Treat. Rev. 2013, 39, 640–646. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of Vascular Endothelial Growth Factor Gene Transcription by Hypoxia-Inducible Factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.D.; Hackett, S.F.; Hirota, K.; Oshima, Y.; Cai, Z.; Berg-Dixon, S.; Rowan, A.; Yan, Z.; Campochiaro, P.A.; Semenza, G.L. Cell Type-Specific Regulation of Angiogenic Growth Factor Gene Expression and Induction of Angiogenesis in Nonischemic Tissue by a Constitutively Active Form of Hypoxia-Inducible Factor 1. Circ. Res. 2003, 93, 1074–1081. [Google Scholar] [CrossRef] [Green Version]
- Hitchon, C.; Wong, K.; Ma, G.; Reed, J.; Lyttle, D.; El-Gabalawy, H. Hypoxia-Induced Production of Stromal Cell-Derived Factor 1 (CXCL12) and Vascular Endothelial Growth Factor by Synovial Fibroblasts. Arthritis Rheum. 2002, 46, 2587–2597. [Google Scholar] [CrossRef] [PubMed]
- Elvert, G.; Kappel, A.; Heidenreich, R.; Englmeier, U.; Lanz, S.; Acker, T.; Rauter, M.; Plate, K.; Sieweke, M.; Breier, G.; et al. Cooperative Interaction of Hypoxia-Inducible Factor-2alpha (HIF-2alpha) and Ets-1 in the Transcriptional Activation of Vascular Endothelial Growth Factor Receptor-2 (Flk-1). J. Biol. Chem. 2003, 278, 7520–7530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.-P.; Tournaire, R.; Pouyssegur, J. The Angiopoietin-2 Gene of Endothelial Cells Is up-Regulated in Hypoxia by a HIF Binding Site Located in Its First Intron and by the Central Factors GATA-2 and Ets-1. J. Cell. Physiol. 2008, 217, 809–818. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, P.; Gilkes, D.M.; Wong, C.C.L.; Kshitiz; Luo, W.; Zhang, H.; Wei, H.; Takano, N.; Schito, L.; Levchenko, A.; et al. Hypoxia-Inducible Factor-Dependent Breast Cancer-Mesenchymal Stem Cell Bidirectional Signaling Promotes Metastasis. J. Clin. Investig. 2013, 123, 189–205. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by Hypoxia-Inducible Factor-1. Interaction between H-Ras and Hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimura, I.; Nangaku, M.; Kanki, Y.; Tsutsumi, S.; Inoue, T.; Kohro, T.; Yamamoto, S.; Fujita, T.; Shimamura, T.; Suehiro, J.-I.; et al. Dynamic Change of Chromatin Conformation in Response to Hypoxia Enhances the Expression of GLUT3 (SLC2A3) by Cooperative Interaction of Hypoxia-Inducible Factor 1 and KDM3A. Mol. Cell. Biol. 2012, 32, 3018–3032. [Google Scholar] [CrossRef] [Green Version]
- Wood, I.S.; Wang, B.; Lorente-Cebrián, S.; Trayhurn, P. Hypoxia Increases Expression of Selective Facilitative Glucose Transporters (GLUT) and 2-Deoxy-D-Glucose Uptake in Human Adipocytes. Biochem. Biophys. Res. Commun. 2007, 361, 468–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-C.; Lin, S.-C.; Wu, M.-H.; Tsai, S.-J. Induction of Pyruvate Dehydrogenase Kinase 1 by Hypoxia Alters Cellular Metabolism and Inhibits Apoptosis in Endometriotic Stromal Cells. Reprod. Sci. 2019, 26, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Jiang, B.-H.; Leung, S.W.; Passantino, R.; Concordet, J.-P.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-Inducible Factor 1*. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional Regulation of Genes Encoding Glycolytic Enzymes by Hypoxia-Inducible Factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Granja, S.; Martinho, O.; Honavar, M.; Pojo, M.; Costa, B.M.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; et al. Hypoxia-Mediated Upregulation of MCT1 Expression Supports the Glycolytic Phenotype of Glioblastomas. Oncotarget 2016, 7, 46335–46353. [Google Scholar] [CrossRef] [Green Version]
- Jamali, S.; Klier, M.; Ames, S.; Barros, L.F.; McKenna, R.; Deitmer, J.W.; Becker, H.M. Hypoxia-Induced Carbonic Anhydrase IX Facilitates Lactate Flux in Human Breast Cancer Cells by Non-Catalytic Function. Sci. Rep. 2015, 5, 13605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraswati, S.; Guo, Y.; Atkinson, J.; Young, P.P. Prolonged Hypoxia Induces Monocarboxylate Transporter-4 Expression in Mesenchymal Stem Cells Resulting in a Secretome That Is Deleterious to Cardiovascular Repair. Stem Cells 2015, 33, 1333–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia in Cancer Cell Metabolism and pH Regulation. Essays Biochem. 2007, 43, 165–178. [Google Scholar] [PubMed] [Green Version]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour Hypoxia Induces a Metabolic Shift Causing Acidosis: A Common Feature in Cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [Green Version]
- Svastová, E.; Hulíková, A.; Rafajová, M.; Zat’ovicová, M.; Gibadulinová, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia Activates the Capacity of Tumor-Associated Carbonic Anhydrase IX to Acidify Extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Bobulescu, I.A.; Di Sole, F.; Moe, O.W. Na+/H+ Exchangers: Physiology and Link to Hypertension and Organ Ischemia. Curr. Opin. Nephrol. Hypertens. 2005, 14, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedlakova, O.; Svastova, E.; Takacova, M.; Kopacek, J.; Pastorek, J.; Pastorekova, S. Carbonic Anhydrase IX, a Hypoxia-Induced Catalytic Component of the pH Regulating Machinery in Tumors. Front. Physiol. 2014, 4, 400. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.A.; Ganesan, R.; Reynolds, G.; Gross, L.; Stevens, A.; Pastorek, J.; Murray, P.G.; Perunovic, B.; Anwar, M.S.; Billingham, L.; et al. Hypoxia-Regulated Carbonic Anhydrase IX Expression Is Associated with Poor Survival in Patients with Invasive Breast Cancer. Br. J. Cancer 2007, 96, 104–109. [Google Scholar] [CrossRef]
- Parks, S.K.; Pouyssegur, J. The Na+/HCO3− Co-Transporter SLC4A4 Plays a Role in Growth and Migration of Colon and Breast Cancer Cells. J. Cell. Physiol. 2015, 230, 1954–1963. [Google Scholar] [CrossRef]
- Ye, Z.; Yue, L.; Shi, J.; Shao, M.; Wu, T. Role of IDO and TDO in Cancers and Related Diseases and the Therapeutic Implications. J. Cancer 2019, 10, 2771–2782. [Google Scholar] [CrossRef] [Green Version]
- Xiang, L.; Mou, J.; Shao, B.; Wei, Y.; Liang, H.; Takano, N.; Semenza, G.L.; Xie, G. Glutaminase 1 Expression in Colorectal Cancer Cells Is Induced by Hypoxia and Required for Tumor Growth, Invasion, and Metastatic Colonization. Cell Death Dis. 2019, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Chimote, A.A.; Kuras, Z.; Conforti, L. Disruption of kv1.3 Channel Forward Vesicular Trafficking by Hypoxia in Human T Lymphocytes. J. Biol. Chem. 2012, 287, 2055–2067. [Google Scholar] [CrossRef] [Green Version]
- Conforti, L.; Petrovic, M.; Mohammad, D.; Lee, S.; Ma, Q.; Barone, S.; Filipovich, A.H. Hypoxia Regulates Expression and Activity of Kv1.3 Channels in T Lymphocytes: A Possible Role in T Cell Proliferation. J. Immunol. 2003, 170, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Ernstoff, M.S.; Hernandez, C.; Atkins, M.; Zabaleta, J.; Sierra, R.; Ochoa, A.C. Arginase I-Producing Myeloid-Derived Suppressor Cells in Renal Cell Carcinoma Are a Subpopulation of Activated Granulocytes. Cancer Res. 2009, 69, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, P.C.; Ochoa, A.C. Arginine Regulation by Myeloid Derived Suppressor Cells and Tolerance in Cancer: Mechanisms and Therapeutic Perspectives. Immunol. Rev. 2008, 222, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan Catabolism in Cancer: Beyond IDO and Tryptophan Depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef]
- Schmidt, S.K.; Ebel, S.; Keil, E.; Woite, C.; Ernst, J.F.; Benzin, A.E.; Rupp, J.; Däubener, W. Regulation of IDO Activity by Oxygen Supply: Inhibitory Effects on Antimicrobial and Immunoregulatory Functions. PLoS ONE 2013, 8, e63301. [Google Scholar] [CrossRef] [Green Version]
- Mohapatra, S.R.; Sadik, A.; Tykocinski, L.-O.; Dietze, J.; Poschet, G.; Heiland, I.; Opitz, C.A. Hypoxia Inducible Factor 1α Inhibits the Expression of Immunosuppressive Tryptophan-2,3-Dioxygenase in Glioblastoma. Front. Immunol. 2019, 10, 2762. [Google Scholar] [CrossRef]
- Sener, Z.; Cederkvist, F.H.; Volchenkov, R.; Holen, H.L.; Skålhegg, B.S. T Helper Cell Activation and Expansion Is Sensitive to Glutaminase Inhibition under Both Hypoxic and Normoxic Conditions. PLoS ONE 2016, 11, e0160291. [Google Scholar] [CrossRef] [Green Version]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha Induces the Recruitment of Bone Marrow-Derived Vascular Modulatory Cells to Regulate Tumor Angiogenesis and Invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Wan, S.; Sun, L.; Hu, J.; Fang, D.; Zhao, R.; Yuan, S.; Zhang, L. Chemokine C-C Motif Receptor 5 and C-C Motif Ligand 5 Promote Cancer Cell Migration under Hypoxia. Cancer Sci. 2012, 103, 904–912. [Google Scholar] [CrossRef]
- Schioppa, T.; Uranchimeg, B.; Saccani, A.; Biswas, S.K.; Doni, A.; Rapisarda, A.; Bernasconi, S.; Saccani, S.; Nebuloni, M.; Vago, L.; et al. Regulation of the Chemokine Receptor CXCR4 by Hypoxia. J. Exp. Med. 2003, 198, 1391–1402. [Google Scholar] [CrossRef] [Green Version]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding Macrophage Entry into Hypoxic Tumor Areas by Sema3A/Nrp1 Signaling Blockade Inhibits Angiogenesis and Restores Antitumor Immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Dineen, S.P.; Lynn, K.D.; Holloway, S.E.; Miller, A.F.; Sullivan, J.P.; Shames, D.S.; Beck, A.W.; Barnett, C.C.; Fleming, J.B.; Brekken, R.A. Vascular Endothelial Growth Factor Receptor 2 Mediates Macrophage Infiltration into Orthotopic Pancreatic Tumors in Mice. Cancer Res. 2008, 68, 4340–4346. [Google Scholar] [CrossRef] [Green Version]
- Grimshaw, M.J.; Naylor, S.; Balkwill, F.R. Endothelin-2 Is a Hypoxia-Induced Autocrine Survival Factor for Breast Tumor Cells. Mol. Cancer Ther. 2002, 1, 1273–1281. [Google Scholar]
- Grimshaw, M.J. Endothelins and Hypoxia-Inducible Factor in Cancer. Endocr. Relat. Cancer 2007, 14, 233–244. [Google Scholar] [CrossRef]
- Leek, R.D.; Hunt, N.C.; Landers, R.J.; Lewis, C.E.; Royds, J.A.; Harris, A.L. Macrophage Infiltration Is Associated with VEGF and EGFR Expression in Breast Cancer. J. Pathol. 2000, 190, 430–436. [Google Scholar] [CrossRef]
- Poth, J.M.; Brodsky, K.; Ehrentraut, H.; Grenz, A.; Eltzschig, H.K. Transcriptional Control of Adenosine Signaling by Hypoxia-Inducible Transcription Factors during Ischemic or Inflammatory Disease. J. Mol. Med. 2013, 91, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synnestvedt, K.; Furuta, G.T.; Comerford, K.M.; Louis, N.; Karhausen, J.; Eltzschig, H.K.; Hansen, K.R.; Thompson, L.F.; Colgan, S.P. Ecto-5′-Nucleotidase (CD73) Regulation by Hypoxia-Inducible Factor-1 Mediates Permeability Changes in Intestinal Epithelia. J. Clin. Investig. 2002, 110, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 Regulates CD47 Expression in Breast Cancer Cells to Promote Evasion of Phagocytosis and Maintenance of Cancer Stem Cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-Y.; Ye, Z.-H.; Huang, M.-Y.; Lu, J.-J. Regulation of CD47 Expression in Cancer Cells. Transl. Oncol. 2020, 13, 100862. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhu, Y.; Wang, X.; Gong, J.; Hu, C.; Guo, B.; Zhu, B.; Li, Y. Temporal Regulation of HIF-1 and NF-κB in Hypoxic Hepatocarcinoma Cells. Oncotarget 2015, 6, 9409–9419. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; Trzyna, W.C.; McClintock, D.S.; Schumacker, P.T. Role of Oxidants in NF-Kappa B Activation and TNF-Alpha Gene Transcription Induced by Hypoxia and Endotoxin. J. Immunol. 2000, 165, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB Links Innate Immunity to the Hypoxic Response through Transcriptional Regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Belaiba, R.S.; Bonello, S.; Zähringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Görlach, A. Hypoxia up-Regulates Hypoxia-Inducible Factor-1alpha Transcription by Involving Phosphatidylinositol 3-Kinase and Nuclear Factor kappaB in Pulmonary Artery Smooth Muscle Cells. Mol. Biol. Cell 2007, 18, 4691–4697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlus, M.R.; Wang, L.; Hu, C.-J. STAT3 and HIF1α Cooperatively Activate HIF1 Target Genes in MDA-MB-231 and RCC4 Cells. Oncogene 2014, 33, 1670–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.-H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.-W.; Ye, S.-K.; et al. STAT3 Is a Potential Modulator of HIF-1-Mediated VEGF Expression in Human Renal Carcinoma Cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef]
- Yaguchi, T.; Goto, Y.; Kido, K.; Mochimaru, H.; Sakurai, T.; Tsukamoto, N.; Kudo-Saito, C.; Fujita, T.; Sumimoto, H.; Kawakami, Y. Immune Suppression and Resistance Mediated by Constitutive Activation of Wnt/β-Catenin Signaling in Human Melanoma Cells. J. Immunol. 2012, 189, 2110–2117. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-Intrinsic β-Catenin Signalling Prevents Anti-Tumour Immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 Signaling Pathway in Hypoxia-ischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef] [Green Version]
- Sethumadhavan, S.; Silva, M.; Philbrook, P.; Nguyen, T.; Hatfield, S.M.; Ohta, A.; Sitkovsky, M.V. Hypoxia and Hypoxia-Inducible Factor (HIF) Downregulate Antigen-Presenting MHC Class I Molecules Limiting Tumor Cell Recognition by T Cells. PLoS ONE 2017, 12, e0187314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct Regulation of GAS6/AXL Signaling by HIF Promotes Renal Metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Wang, Y.; Singh, I.; Bell, R.D.; Deane, R.; Zhong, Z.; Sagare, A.; Winkler, E.A.; Zlokovic, B.V. Protein S Controls Hypoxic/ischemic Blood-Brain Barrier Disruption through the TAM Receptor Tyro3 and Sphingosine 1-Phosphate Receptor. Blood 2010, 115, 4963–4972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.-Z.; Wang, W.; Xian, N.; Wu, B. Inhibition of TYRO3/Akt Signaling Participates in Hypoxic Injury in Hippocampal Neurons. Neural Regen. Res. 2016, 11, 752. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Wang, J.; Shiozawa, Y.; McGee, S.; Kim, J.; Jung, Y.; Joseph, J.; Berry, J.E.; Havens, A.; Pienta, K.J.; et al. Hypoxia Stabilizes GAS6/Axl Signaling in Metastatic Prostate Cancer. Mol. Cancer Res. 2012, 10, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 Is a Novel Direct Target of HIF-1α, and Its Blockade under Hypoxia Enhanced MDSC-Mediated T Cell Activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Zapata, C.; Avendaño-Ortiz, J.; Hernandez-Jimenez, E.; Toledano, V.; Casas-Martin, J.; Varela-Serrano, A.; Torres, M.; Almendros, I.; Casitas, R.; Fernández-Navarro, I.; et al. Hypoxia-Induced PD-L1/PD-1 Crosstalk Impairs T-Cell Function in Sleep Apnoea. Eur. Respir. J. 2017, 50, 1700833. [Google Scholar] [CrossRef]
- Wen, Q.; Han, T.; Wang, Z.; Jiang, S. Role and Mechanism of Programmed Death-Ligand 1 in Hypoxia-Induced Liver Cancer Immune Escape. Oncol. Lett. 2020, 19, 2595–2601. [Google Scholar] [CrossRef] [Green Version]
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Sauvage, D.; Van Moer, K.; Viry, E.; Bocci, I.; Hasmim, M.; Bosseler, M.; Berchem, G.; et al. Impact of Hypoxic Tumor Microenvironment and Tumor Cell Plasticity on the Expression of Immune Checkpoints. Cancer Lett. 2019, 458, 13–20. [Google Scholar] [CrossRef]
- Corpechot, C.; Barbu, V.; Wendum, D.; Kinnman, N.; Rey, C.; Poupon, R.; Housset, C.; Rosmorduc, O. Hypoxia-Induced VEGF and Collagen I Expressions Are Associated with Angiogenesis and Fibrogenesis in Experimental Cirrhosis. Hepatology 2002, 35, 1010–1021. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the Extracellular Matrix: Drivers of Tumour Metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Gilkes, D.M.; Bajpai, S.; Chaturvedi, P.; Wirtz, D.; Semenza, G.L. Hypoxia-Inducible Factor 1 (HIF-1) Promotes Extracellular Matrix Remodeling under Hypoxic Conditions by Inducing P4HA1, P4HA2, and PLOD2 Expression in Fibroblasts. J. Biol. Chem. 2013, 288, 10819–10829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, T.B.; Hjortdal, J.; Priyadarsini, S.; Karamichos, D. Acute Hypoxia Influences Collagen and Matrix Metalloproteinase Expression by Human Keratoconus Cells In Vitro. PLoS ONE 2017, 12, e0176017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, A.T.; Bürgers, H.F.; Rabie, T.; Marti, H.H. Matrix Metalloproteinase-9 Mediates Hypoxia-Induced Vascular Leakage in the Brain via Tight Junction Rearrangement. J. Cereb. Blood Flow Metab. 2010, 30, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Zhou, Y.; Wang, L.; Zhang, J.; Wu, H.; Xiong, J.; Zhang, J.; Tian, Y.; Wang, C.; Wu, H. Transcriptional Upregulation of MT2-MMP in Response to Hypoxia Is Promoted by HIF-1α in Cancer Cells. Mol. Carcinog. 2011, 50, 770–780. [Google Scholar] [CrossRef]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.-L.; Carretta, M.; Kalvisa, A.; Siersbæk, M.S.; Simões, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen Density Regulates the Activity of Tumor-Infiltrating T Cells. J Immunother Cancer 2019, 7, 68. [Google Scholar] [CrossRef] [Green Version]
- Casey, T.M.; Eneman, J.; Crocker, A.; White, J.; Tessitore, J.; Stanley, M.; Harlow, S.; Bunn, J.Y.; Weaver, D.; Muss, H.; et al. Cancer Associated Fibroblasts Stimulated by Transforming Growth Factor beta1 (TGF-Beta 1) Increase Invasion Rate of Tumor Cells: A Population Study. Breast Cancer Res. Treat. 2008, 110, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Tang, C.-M.; Banerjee, S.; Delgado, A.L.; Yebra, M.; Davis, J.; Sicklick, J.K. TGF-β1-Mediated Transition of Resident Fibroblasts to Cancer-Associated Fibroblasts Promotes Cancer Metastasis in Gastrointestinal Stromal Tumor. Oncogenesis 2021, 10, 13. [Google Scholar] [CrossRef]
- McMahon, S.; Charbonneau, M.; Grandmont, S.; Richard, D.E.; Dubois, C.M. Transforming Growth Factor beta1 Induces Hypoxia-Inducible Factor-1 Stabilization through Selective Inhibition of PHD2 Expression. J. Biol. Chem. 2006, 281, 24171–24181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingyuan, X.; Qianqian, P.; Shengquan, X.; Chenyi, Y.; Rui, L.; Yichen, S.; Jinghong, X. Hypoxia-Inducible Factor-1α Activates Transforming Growth Factor-β1/Smad Signaling and Increases Collagen Deposition in Dermal Fibroblasts. Oncotarget 2018, 9, 3188–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in Cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Yang, P.; Hu, Y.; Zhou, Q. The CXCL12-CXCR4 Signaling Axis Plays a Key Role in Cancer Metastasis and Is a Potential Target for Developing Novel Therapeutics against Metastatic Cancer. Curr. Med. Chem. 2020, 27, 5543–5561. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 Induces an Epithelial–mesenchymal Transition Phenotype in Human Breast Cancer Cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [Green Version]
- Borthwick, L.A. The IL-1 Cytokine Family and Its Role in Inflammation and Fibrosis in the Lung. Semin. Immunopathol. 2016, 38, 517–534. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and Inflammation: Inseparable Actors of Cancer Progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef]
- Said, N.A.B.M.; Williams, E.D. Growth Factors in Induction of Epithelial-Mesenchymal Transition and Metastasis. Cells Tissues Organs 2011, 193, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Saci, A.; Szabo, P.M.; Chasalow, S.D.; Castillo-Martin, M.; Domingo-Domenech, J.; Siefker-Radtke, A.; Sharma, P.; Sfakianos, J.P.; Gong, Y.; et al. EMT- and Stroma-Related Gene Expression and Resistance to PD-1 Blockade in Urothelial Cancer. Nat. Commun. 2018, 9, 3503. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Miao, W.; Jiang, X.; Cao, J.; Wang, B.; Wang, Y.; Yu, J.; Wang, X.; Liu, H. The Epithelial to Mesenchymal Transition Related Gene Calumenin Is an Adverse Prognostic Factor of Bladder Cancer Correlated with Tumor Microenvironment Remodeling, Gene Mutation, and Ferroptosis. Front. Oncol. 2021, 11, 683951. [Google Scholar] [CrossRef]
- Lou, Y.; Diao, L.; Cuentas, E.R.P.; Denning, W.L.; Chen, L.; Fan, Y.H.; Byers, L.A.; Wang, J.; Papadimitrakopoulou, V.A.; Behrens, C.; et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 3630–3642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiler, R.; Ashab, H.A.D.; Erho, N.; van Rhijn, B.W.G.; Winters, B.; Douglas, J.; Van Kessel, K.E.; Fransen van de Putte, E.E.; Sommerlad, M.; Wang, N.Q.; et al. Impact of Molecular Subtypes in Muscle-Invasive Bladder Cancer on Predicting Response and Survival after Neoadjuvant Chemotherapy. Eur. Urol. 2017, 72, 544–554. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Easley, C.A., 4th; Ramalho-Santos, J.; Van Houten, B.; Schatten, G. Energy Metabolism in Human Pluripotent Stem Cells and Their Differentiated Counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Wu, H.; Dai, C.; Pan, Q.; Ding, Z.; Hu, D.; Ji, B.; Luo, Y.; Hu, X. Beyond Warburg Effect--Dual Metabolic Nature of Cancer Cells. Sci. Rep. 2014, 4, 4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ye, C.; Chen, C.; Xiong, H.; Xie, B.; Zhou, J.; Chen, Y.; Zheng, S.; Wang, L. Glucose Transporter GLUT1 Expression and Clinical Outcome in Solid Tumors: A Systematic Review and Meta-Analysis. Oncotarget 2017, 8, 16875–16886. [Google Scholar] [CrossRef] [Green Version]
- Kierans, S.J.; Taylor, C.T. Regulation of Glycolysis by the Hypoxia-Inducible Factor (HIF): Implications for Cellular Physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Lum, J.J.; Bui, T.; Gruber, M.; Gordan, J.D.; DeBerardinis, R.J.; Covello, K.L.; Simon, M.C.; Thompson, C.B. The Transcription Factor HIF-1alpha Plays a Critical Role in the Growth Factor-Dependent Regulation of Both Aerobic and Anaerobic Glycolysis. Genes Dev. 2007, 21, 1037–1049. [Google Scholar] [CrossRef] [Green Version]
- Erra Díaz, F.; Dantas, E.; Geffner, J. Unravelling the Interplay between Extracellular Acidosis and Immune Cells. Mediat. Inflamm. 2018, 2018, 1218297. [Google Scholar] [CrossRef]
- Kellum, J.A.; Song, M.; Li, J. Science Review: Extracellular Acidosis and the Immune Response: Clinical and Physiologic Implications. Crit. Care 2004, 8, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshkin, S.J.; Cardone, R.A.; Harguindey, S. Na+-H+ Exchanger, pH Regulation and Cancer. Recent Pat. Anticancer Drug Discov. 2013, 8, 85–99. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-Tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-Arginine Availability Regulates T-Lymphocyte Cell-Cycle Progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, L.W.; Ashcroft, M. Exploring the Molecular Interface between Hypoxia-Inducible Factor Signalling and Mitochondria. Cell. Mol. Life Sci. 2019, 76, 1759–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and mTORC1 Kinase Activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.-B.; Taylor, P.M.; Cantrell, D.A. Control of Amino-Acid Transport by Antigen Receptors Coordinates the Metabolic Reprogramming Essential for T Cell Differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef]
- Chiu, D.K.-C.; Tse, A.P.-W.; Xu, I.M.-J.; Di Cui, J.; Lai, R.K.-H.; Li, L.L.; Koh, H.-Y.; Tsang, F.H.-C.; Wei, L.L.; Wong, C.-M.; et al. Hypoxia Inducible Factor HIF-1 Promotes Myeloid-Derived Suppressor Cells Accumulation through ENTPD2/CD39L1 in Hepatocellular Carcinoma. Nat. Commun. 2017, 8, 517. [Google Scholar] [CrossRef] [Green Version]
- Abou Khouzam, R.; Brodaczewska, K.; Filipiak, A.; Zeinelabdin, N.A.; Buart, S.; Szczylik, C.; Kieda, C.; Chouaib, S. Tumor Hypoxia Regulates Immune Escape/Invasion: Influence on Angiogenesis and Potential Impact of Hypoxic Biomarkers on Cancer Therapies. Front. Immunol. 2020, 11, 613114. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. Biology of the TAM Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef] [PubMed]
- Michaels, A.D.; Newhook, T.E.; Adair, S.J.; Morioka, S.; Goudreau, B.J.; Nagdas, S.; Mullen, M.G.; Persily, J.B.; Bullock, T.N.J.; Slingluff, C.L., Jr.; et al. CD47 Blockade as an Adjuvant Immunotherapy for Resectable Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 1415–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Pantoja, D.R.; Terabe, M.; Ghosh, A.; Ridnour, L.A.; DeGraff, W.G.; Wink, D.A.; Berzofsky, J.A.; Roberts, D.D. CD47 in the Tumor Microenvironment Limits Cooperation between Antitumor T-Cell Immunity and Radiotherapy. Cancer Res. 2014, 74, 6771–6783. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Chang, T.; Singh, S.P.; Lim, L.; Mannan, P.; Garfield, S.H.; Pendrak, M.L.; Soto-Pantoja, D.R.; Rosenberg, A.Z.; Jin, S.; et al. CD47 Signaling Regulates the Immunosuppressive Activity of VEGF in T Cells. J. Immunol. 2014, 193, 3914–3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy Induces Enrichment of CD47+/CD73+/PDL1+ Immune Evasive Triple-Negative Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front. Immunol. 2018, 9, 1591. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.M.; Matosevic, S. Immunometabolic Dysfunction of Natural Killer Cells Mediated by the Hypoxia-CD73 Axis in Solid Tumors. Front Mol. Biosci. 2019, 6, 60. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Ahmad, S.; Glover, L.; Miller, S.M.; Shannon, J.M.; Guo, X.; Franklin, W.A.; Bridges, J.P.; Schaack, J.B.; Colgan, S.P.; et al. Adenosine A2A Receptor Is a Unique Angiogenic Target of HIF-2α in Pulmonary Endothelial Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10684–10689. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Lu, H.; Samanta, D.; Salman, S.; Lu, Y.; Semenza, G.L. Hypoxia-Inducible Factor 1-Dependent Expression of Adenosine Receptor 2B Promotes Breast Cancer Stem Cell Enrichment. Proc. Natl. Acad. Sci. USA 2018, 115, E9640–E9648. [Google Scholar] [CrossRef] [Green Version]
- Steingold, J.M.; Hatfield, S.M. Targeting Hypoxia-A2A Adenosinergic Immunosuppression of Antitumor T Cells During Cancer Immunotherapy. Front. Immunol. 2020, 11, 570041. [Google Scholar] [CrossRef]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Belikoff, B.; Schreiber, T.H.; Sethumadhavan, S.; Abbott, R.; Philbrook, P.; Thayer, M.; Shujia, D.; et al. Systemic Oxygenation Weakens the Hypoxia and Hypoxia Inducible Factor 1α-Dependent and Extracellular Adenosine-Mediated Tumor Protection. J. Mol. Med. 2014, 92, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Sitkovsky, M. A2A Adenosine Receptor Antagonists to Weaken the Hypoxia-HIF-1α Driven Immunosuppression and Improve Immunotherapies of Cancer. Curr. Opin. Pharmacol. 2016, 29, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Murthy, A.; Gerber, S.A.; Koch, C.J.; Lord, E.M. Intratumoral Hypoxia Reduces IFN-γ-Mediated Immunity and MHC Class I Induction in a Preclinical Tumor Model. Immunohorizons 2019, 3, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Mayer, A. Hypoxia in Cancer: Significance and Impact on Clinical Outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.C.; Lebedev, A.; Aten, E.; Madsen, K.; Marciano, L.; Kolb, H.C. The Clinical Importance of Assessing Tumor Hypoxia: Relationship of Tumor Hypoxia to Prognosis and Therapeutic Opportunities. Antioxid. Redox Signal. 2014, 21, 1516–1554. [Google Scholar] [CrossRef]
- Foehrenbacher, A.; Secomb, T.W.; Wilson, W.R.; Hicks, K.O. Design of Optimized Hypoxia-Activated Prodrugs Using Pharmacokinetic/pharmacodynamic Modeling. Front. Oncol. 2013, 3, 314. [Google Scholar] [CrossRef] [Green Version]
- Phillips, R.M. Targeting the Hypoxic Fraction of Tumours Using Hypoxia-Activated Prodrugs. Cancer Chemother. Pharmacol. 2016, 77, 441–457. [Google Scholar] [CrossRef] [Green Version]
- Hamis, S.; Kohandel, M.; Dubois, L.J.; Yaromina, A.; Lambin, P.; Powathil, G.G. Combining Hypoxia-Activated Prodrugs and Radiotherapy in Silico: Impact of Treatment Scheduling and the Intra-Tumoural Oxygen Landscape. PLoS Comput. Biol. 2020, 16, e1008041. [Google Scholar] [CrossRef]
- Spiegelberg, L.; Houben, R.; Niemans, R.; de Ruysscher, D.; Yaromina, A.; Theys, J.; Guise, C.P.; Smaill, J.B.; Patterson, A.V.; Lambin, P.; et al. Hypoxia-Activated Prodrugs and (lack of) Clinical Progress: The Need for Hypoxia-Based Biomarker Patient Selection in Phase III Clinical Trials. Clin. Transl. Radiat. Oncol. 2019, 15, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, P.R. Let There Be Oxygen and T Cells. J. Clin. Investig. 2018, 128, 4761–4763. [Google Scholar] [CrossRef] [PubMed]
- Damgaci, S.; Enriquez-Navas, P.M.; Pilon-Thomas, S.; Guvenis, A.; Gillies, R.J.; Ibrahim-Hashim, A. Immunotherapy on Acid: Opportunities and Challenges. Eur. J. Clin. Nutr. 2020, 74, 3–6. [Google Scholar] [CrossRef]
- Ding, X.; Yu, J.; Hu, M. The Relationship between Expression of PD-L1 and HIF-1α in Glioma Cells under Hypoxia. J. Clin. Orthod. 2021, 39, e14043. [Google Scholar]
- Choi, W.S.W.; Boland, J.; Lin, J. Hypoxia-Inducible Factor-2α as a Novel Target in Renal Cell Carcinoma. J Kidney Cancer VHL 2021, 8, 1–7. [Google Scholar] [CrossRef]
- Tatli Dogan, H.; Kiran, M.; Bilgin, B.; Kiliçarslan, A.; Sendur, M.A.N.; Yalçin, B.; Ardiçoglu, A.; Atmaca, A.F.; Gumuskaya, B. Prognostic Significance of the Programmed Death Ligand 1 Expression in Clear Cell Renal Cell Carcinoma and Correlation with the Tumor Microenvironment and Hypoxia-Inducible Factor Expression. Diagn. Pathol. 2018, 13, 60. [Google Scholar] [CrossRef]
- Magnussen, A.L.; Mills, I.G. Vascular Normalisation as the Stepping Stone into Tumour Microenvironment Transformation. Br. J. Cancer 2021, 125, 324–336. [Google Scholar] [CrossRef]
- Goel, S.; Wong, A.H.-K.; Jain, R.K. Vascular Normalization as a Therapeutic Strategy for Malignant and Nonmalignant Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006486. [Google Scholar] [CrossRef]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular Normalization as an Emerging Strategy to Enhance Cancer Immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular Normalizing Doses of Antiangiogenic Treatment Reprogram the Immunosuppressive Tumor Microenvironment and Enhance Immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewhirst, M.W.; Mowery, Y.M.; Mitchell, J.B.; Cherukuri, M.K.; Secomb, T.W. Rationale for Hypoxia Assessment and Amelioration for Precision Therapy and Immunotherapy Studies. J. Clin. Investig. 2019, 129, 489–491. [Google Scholar] [CrossRef]
- He, H.; Liao, Q.; Zhao, C.; Zhu, C.; Feng, M.; Liu, Z.; Jiang, L.; Zhang, L.; Ding, X.; Yuan, M.; et al. Conditioned CAR-T Cells by Hypoxia-Inducible Transcription Amplification (HiTA) System Significantly Enhances Systemic Safety and Retains Antitumor Efficacy. J. Immunother. Cancer 2021, 9, e002755. [Google Scholar] [CrossRef] [PubMed]
- Kosti, P.; Opzoomer, J.W.; Larios-Martinez, K.I.; Henley-Smith, R.; Scudamore, C.L.; Okesola, M.; Taher, M.Y.M.; Davies, D.M.; Muliaditan, T.; Larcombe-Young, D.; et al. Hypoxia-Sensing CAR T Cells Provide Safety and Efficacy in Treating Solid Tumors. Cell. Rep. Med. 2021, 2, 100227. [Google Scholar] [CrossRef]
- Gropper, Y.; Feferman, T.; Shalit, T.; Salame, T.-M.; Porat, Z.; Shakhar, G. Culturing CTLs under Hypoxic Conditions Enhances Their Cytolysis and Improves Their Anti-Tumor Function. Cell Rep. 2017, 20, 2547–2555. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, C.C.; Kojima, H.; Lukashev, D.; Armstrong, J.; Farber, M.; Apasov, S.G.; Sitkovsky, M.V. Differential Effects of Physiologically Relevant Hypoxic Conditions on T Lymphocyte Development and Effector Functions. J. Immunol. 2001, 167, 6140–6149. [Google Scholar] [CrossRef]
- Vuillefroy de Silly, R.; Ducimetière, L.; Yacoub Maroun, C.; Dietrich, P.-Y.; Derouazi, M.; Walker, P.R. Phenotypic Switch of CD8+ T Cells Reactivated under Hypoxia toward IL-10 Secreting, Poorly Proliferative Effector Cells. Eur. J. Immunol. 2015, 45, 2263–2275. [Google Scholar] [CrossRef] [Green Version]
- Roman, J.; Rangasamy, T.; Guo, J.; Sugunan, S.; Meednu, N.; Packirisamy, G.; Shimoda, L.A.; Golding, A.; Semenza, G.; Georas, S.N. T-Cell Activation under Hypoxic Conditions Enhances IFN-Gamma Secretion. Am. J. Respir. Cell Mol. Biol. 2010, 42, 123–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veliça, P.; Cunha, P.P.; Vojnovic, N.; Foskolou, I.P.; Bargiela, D.; Gojkovic, M.; Rundqvist, H.; Johnson, R.S. Modified Hypoxia-Inducible Factor Expression in CD8+ T Cells Increases Antitumor Efficacy. Cancer Immunol. Res. 2021, 9, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Berahovich, R.; Liu, X.; Zhou, H.; Tsadik, E.; Xu, S.; Golubovskaya, V.; Wu, L. Hypoxia Selectively Impairs CAR-T Cells In Vitro. Cancers 2019, 11, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuillefroy de Silly, R.; Dietrich, P.-Y.; Walker, P.R. Hypoxia and Antitumor CD8+ T Cells: An Incompatible Alliance? Oncoimmunology 2016, 5, e1232236. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, Y.; Negishi, Y.; Shimizu, M.; Takahashi, M.; Ichikawa, M.; Takahashi, H. Effects of Extracellular pH and Hypoxia on the Function and Development of Antigen-Specific Cytotoxic T Lymphocytes. Immunol. Lett. 2015, 167, 72–86. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
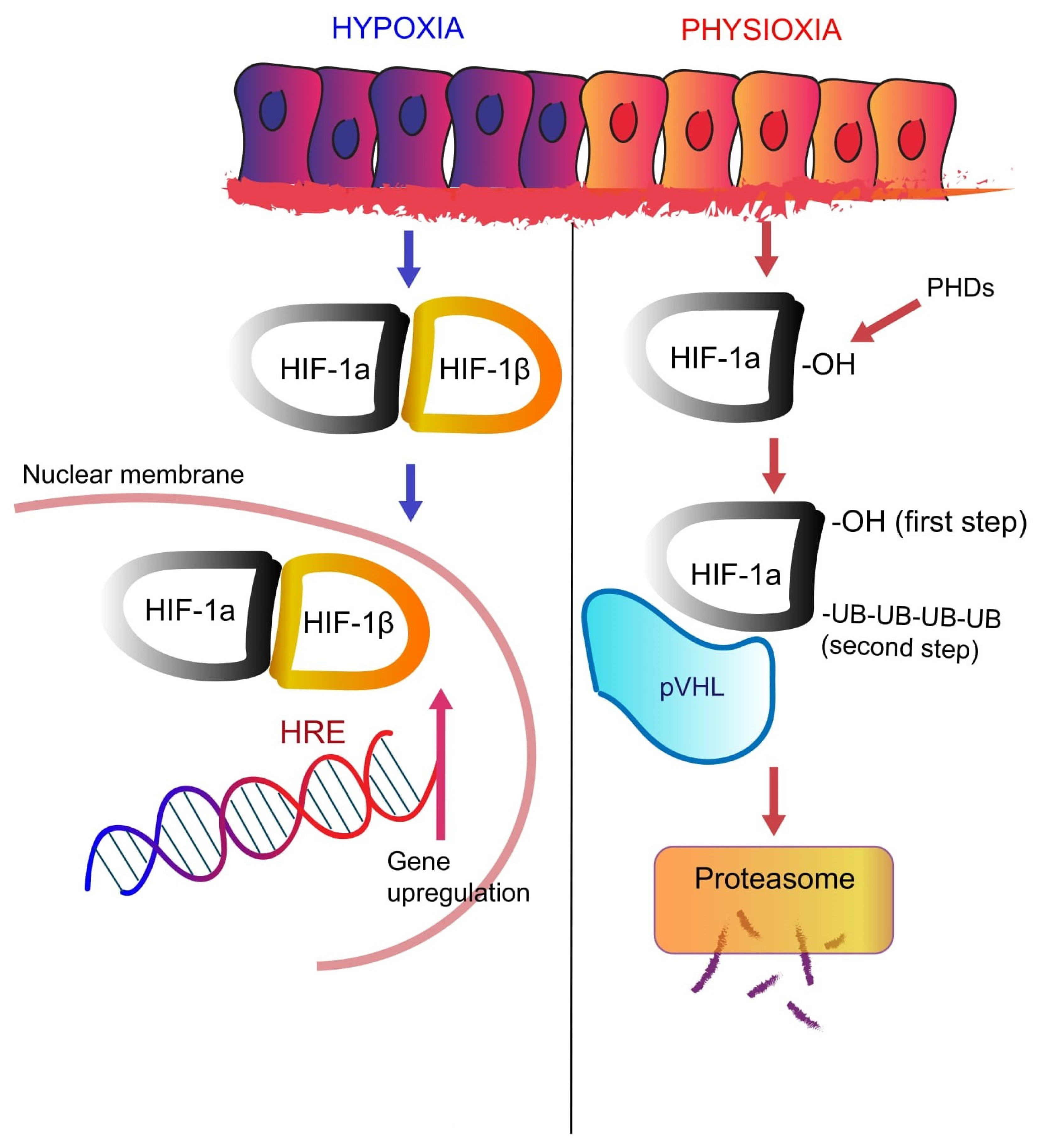{kind=link}
{kind=link}
| Signaling | Cancer | Response | Reference |
|---|---|---|---|
| ERα-mediated estrogen signaling | Ovarian | ↑ HIF-1α protein/signaling | [180] |
| Thyroid | ↑ HIF-1α protein | [183] | |
| Breast | ↑ HIF-1α protein/signaling | [184] | |
| Breast | ↓ HIF-2α mRNA/protein | [186] | |
| Breast | ↑ HIF-1α mRNA | [192] | |
| GPER-mediated estrogen signaling | Breast, CAFs | ↑ HIF-1α mRNA/protein/signaling | [155] |
| EGF/EGFR signaling | Prostate | ↑ HIF-1α protein/signaling | [199] |
| NSCLC | ↑ HIF-1α signaling | [200] | |
| Breast | ↑ HIF-1α protein/signaling | [201,203] | |
| Colorectal | ↑ HIF-1α mRNA/protein | [202] | |
| Heregulin | Breast | ↑ HIF-1α protein/signaling | [174] |
| IGF-I | Colon | ↑ HIF-1α protein/signaling | [175] |
| NSCLC, HNSCC | ↑ HIF-1α protein/signaling | [219] | |
| Kaposi sarcoma | ↑ HIF-1α and HIF-2α protein/signaling | [220] | |
| Breast | ↑ HIF-1α and HIF-2α protein/signaling | [221,222,223] | |
| bFGF | Breast | ↑ HIF-1α protein/signaling | [176,230,231] |
| Physical Barriers | Impediments to Direct Contact Between T-Cells and Cancer Cells | Reference |
|---|---|---|
| Stromal fibrosis | Epidermal growth factor (EGF), platelet-derived growth factor (PDGF), fibroblast growth factor 2 (FGF2), CXCL12, TGF-β, zinc finger E-box binding homeobox 1 and 2 (ZEB1, ZEB2) proteins, Snail, Slug, Twist, Goosecoid, FOXC2, LOX, PLOD1, PLOD2, P4HA1, P4HA2, MMP2, and MMP9 | [253,254,255,256,257,258,259,260,261,262,263,264,265,266,267,268,269,270,271] |
| Epithelial-mesenchymal transition (EMT) | Tumor necrosis factor α (TNF-α), TGF-β, interleukin 1 (IL-1), interleukin-6 (IL-6) and interleukin-8 (IL-8), hepatocyte growth factor (HGF), basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), platelet-derived growth factor (PDGF) | [272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287,288,289,290,291] |
| Vascular access | VEGF-family, angiopoietin-2 (Ang-2), transforming growth factor beta (TGF-β), platelet-derived growth factor B (PDGFB), placental growth factor (PGF), connective tissue growth factor (CTGF), stem cell factor (SCF), stromal cell-derived factor 1 (CXCL12), leptin, endoglin, nitric oxide synthase 2, haemoxygenase-1, endothelin-1 (ET-1), VEGF receptor-2, endothelial receptor tyrosine kinase (Tie-2) | [83,234,292,293,294,295,296,297,298] |
| Functional Barriers | Biological or metabolic interactions between cancer, stromal and immune cells limiting migration, function, and/or survival of T-cells | |
| Metabolic barriers | Warburg effect: enzymes glucose transporters (GLUTs 1–3), pyruvate dehydrogenase kinase 1 (PDK1), lactic dehydrogenase A (LDHA) and pyruvate kinase M2 subtype (PKM2), mono-carboxylate transporters (MCTs) | [299,300,301,302,303,304,305,306,307,308] |
| Acidification of the TME: carbonic anhydrases (CAs), Na+/H+ exchanger (NHE1), bicarbonate transporters (SLC4A4) | [90,309,310,311,312,313,314,315] | |
| Amino acids depletion and ionic misbalance: indoleamine 2,3 dioxygenase (IDO), Glutaminase 1 (GLS1), Kv1.3 | [316,317,318,319,320,321,322,323,324,325,326] | |
| Soluble factors and “Don’t eat-me” signals | Myeloid cells recruitment and activity: CCL5, CXCL12, CXCR4, VEGF, Sema3A, CCL28, endothelin 1 and 2, TGF-β | [327,328,329,330,331,332,333,334] |
| Adenosine signaling: CD39, CD73 | [335,336] | |
| “Don’t eat me” signals: CD47/signal regulatory protein (SIRP)-α axis | [337,338] | |
| Tumor cell-intrinsic signaling | Pathways involved in immune escape: extended PI3K pathway signaling, β-catenin/signaling, STAT-3 activation, MAPK signaling, p53 signaling | [199,339,340,341,342,343,344,345,346,347] |
| Downregulation of molecules necessary for effector immune cells recognition: major histocompatibility class-I (MHC-I) | [348] | |
| TAM receptor tyrosine kinases: Tyro3, Axl and Mertk | [349,350,351,352] | |
| Dynamic barriers | Interactions between cancer and T-cells resulting in limited function | |
| Checkpoint/ligand interactions: upregulation of CTLA-4, PD-L1, HLA-G | [353,354,355,356] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lappano, R.; Todd, L.A.; Stanic, M.; Cai, Q.; Maggiolini, M.; Marincola, F.; Pietrobon, V. Multifaceted Interplay between Hormones, Growth Factors and Hypoxia in the Tumor Microenvironment. Cancers 2022, 14, 539. https://doi.org/10.3390/cancers14030539
Lappano R, Todd LA, Stanic M, Cai Q, Maggiolini M, Marincola F, Pietrobon V. Multifaceted Interplay between Hormones, Growth Factors and Hypoxia in the Tumor Microenvironment. Cancers. 2022; 14(3):539. https://doi.org/10.3390/cancers14030539
Chicago/Turabian StyleLappano, Rosamaria, Lauren A. Todd, Mia Stanic, Qi Cai, Marcello Maggiolini, Francesco Marincola, and Violena Pietrobon. 2022. "Multifaceted Interplay between Hormones, Growth Factors and Hypoxia in the Tumor Microenvironment" Cancers 14, no. 3: 539. https://doi.org/10.3390/cancers14030539