
Challenges and Advances in Chimeric Antigen Receptor Therapy for Acute Myeloid Leukemia


Abstract
:Simple Summary
Abstract
1. Introduction
2. Overview of CARs
The Ideal CAR Target in AML
3. Barriers to Development of AML CARs
3.1. Lack of AML-Specific Antigens
3.2. Antigen Escape
3.3. AML-Induced Immunosuppressive Microenvironment
3.4. Issues with Quality of Autologous Cells
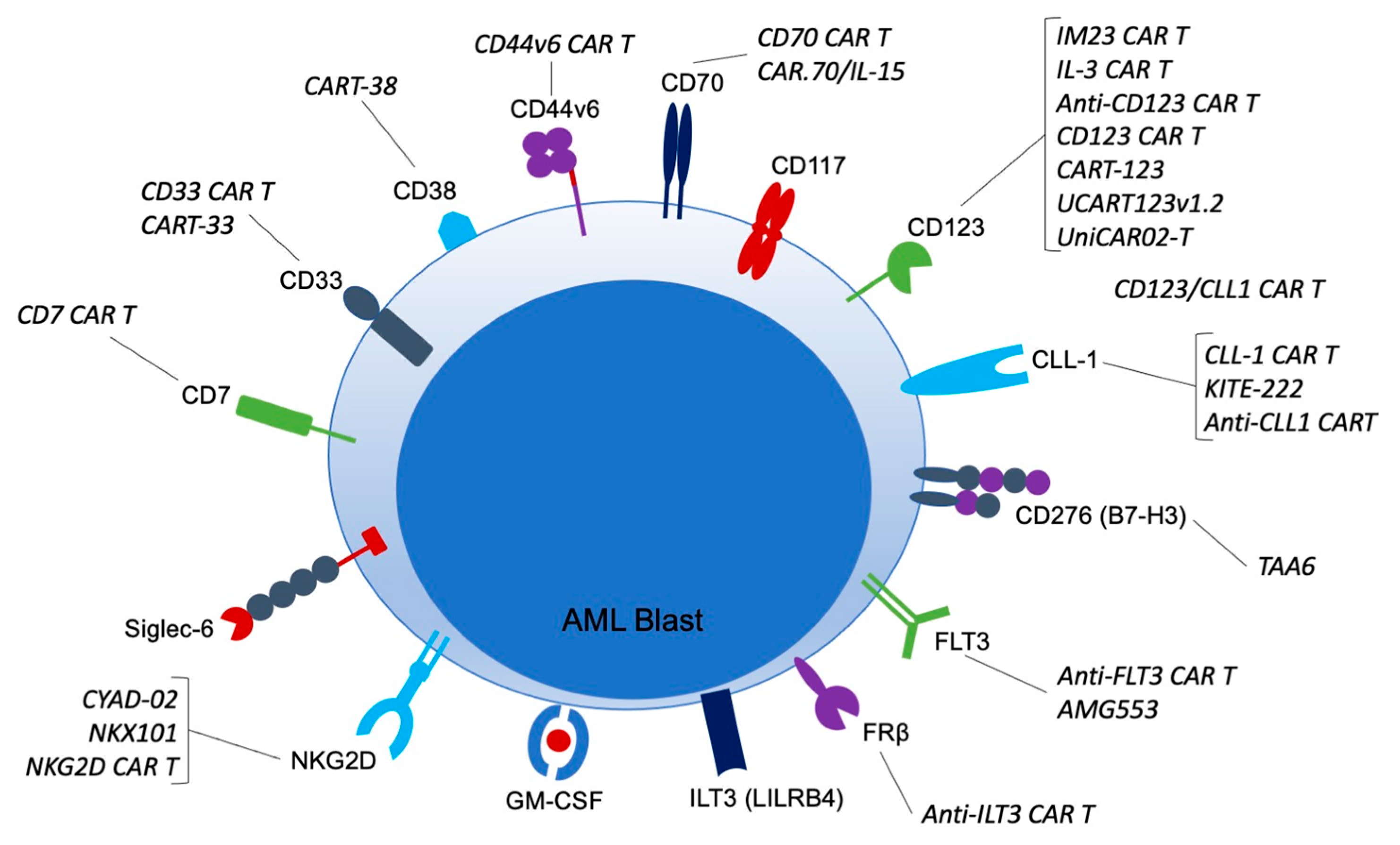
4. Experimental Targets under Investigation for AML CARs
4.1. CD7
4.2. CD33
4.3. CD38
4.4. CD44v6
4.5. CD70
4.6. CD117
4.7. CD123
4.8. CD276 (B7-H3)
4.9. CLL-1
4.10. FLT3
4.11. FRβ
4.12. GM-CSF (CD116/CD131)
4.13. ILT3 (LILRB4)
4.14. NKG2D
4.15. Siglec-6
4.16. TIM3
5. Potential Impact of CAR on the AML Treatment Landscape
5.1. Target Population for AML CARs
5.2. Bridging Treatment for AML CAR
5.3. Combining Targeted Inhibitors with CAR Products
5.4. Integrating AML CARs with Allogenic SCTs
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef]
- Zhylko, A.; Winiarska, M.; Graczyk-Jarzynka, A. The Great War of Today: Modifications of CAR-T Cells to Effectively Combat Malignancies. Cancers 2020, 12, 2030. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv. Cell Gene Ther. 2020, 3, 84. [Google Scholar] [CrossRef]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18, 125. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Rahman, H.S.; Al-Obaidi, Z.M.J.; Jalil, A.T.; Abdelbasset, W.K.; Suksatan, W.; Dorofeev, A.E.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; et al. Novel CAR T therapy is a ray of hope in the treatment of seriously ill AML patients. Stem Cell Res. Ther. 2021, 12, 465. [Google Scholar] [CrossRef] [PubMed]
- Biernacki, M.A.; Brault, M.; Bleakley, M. T-Cell Receptor–Based Immunotherapy for Hematologic Malignancies. Cancer J. 2019, 25, 179–190. [Google Scholar] [CrossRef]
- Daver, N.; Alotaibi, A.S.; Bücklein, V.; Subklewe, M. T-cell-based immunotherapy of acute myeloid leukemia: Current concepts and future developments. Leukemia 2021, 35, 1843–1863. [Google Scholar] [CrossRef] [PubMed]
- Zahid, A.; Siegler, E.L.; Kenderian, S.S. CART Cell Toxicities: New Insight into Mechanisms and Management. Clin. Hematol. Int. 2020, 2, 149–155. [Google Scholar] [CrossRef]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuis, A.G.; Egan, D.N.; Bar, M.; Schmitt, T.M.; McAfee, M.S.; Paulson, K.G.; Voillet, V.; Gottardo, R.; Ragnarsson, G.B.; Bleakley, M.; et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019, 25, 1064–1072. [Google Scholar] [CrossRef]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Hönemann, D.; et al. Persistence and Efficacy of Second Generation CAR T Cell Against the LeY Antigen in Acute Myeloid Leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Tabata, R.; Chi, S.; Yuda, J.; Minami, Y. Emerging Immunotherapy for Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 1944. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; on behalf of the Study Alliance Leukemia; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; Von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.F.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.Y.; Yu, K.-R.; Kenderian, S.S.; Ruella, M.; Chen, S.; Shin, T.-H.; Aljanahi, A.A.; Schreeder, D.; Klichinsky, M.; Shestova, O.; et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173, 1439–1453.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKoy, J.M.; Angelotta, C.; Bennett, C.L.; Tallman, M.S.; Wadleigh, M.; Evens, A.; Kuzel, T.M.; Trifilio, S.M.; Raisch, D.W.; Kell, J.; et al. Gemtuzumab ozogamicin-associated sinusoidal obstructive syndrome (SOS): An overview from the research on adverse drug events and reports (RADAR) project. Leuk. Res. 2007, 31, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiner, J.; Ono, Y.; Hofmann, S.; Schmitt, A.; Mehring, E.; Götz, M.; Guillaume, P.; Döhner, K.; Mytilineos, J.; Döhner, H.; et al. Mutated regions of nucleophosmin 1 elicit both CD4+ and CD8+ T-cell responses in patients with acute myeloid leukemia. Blood 2012, 120, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Condomines, M.; Van Der Stegen, S.J.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lu, X.-A.; Yang, J.; Zhang, G.; Li, J.; Song, L.; Su, Y.; Shi, Y.; Zhang, M.; He, J.; et al. Efficacy and safety of anti-CD19 CAR T-cell therapy in 110 patients with B-cell acute lymphoblastic leukemia with high-risk features. Blood Adv. 2020, 4, 2325–2338. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, J.; Zhang, X.; Lu, X.-A.; Xiong, M.; Zhang, J.; Zhou, X.; Qi, F.; He, T.; Ding, Y.; et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol. Ther. Oncolytics 2020, 18, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; He, T.; Wang, X.; Zheng, W.; Lin, N.; Tu, M.; Xie, Y.; Ping, L.; Zhang, C.; Liu, W.; et al. Parallel Comparison of 4-1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma. Mol. Ther. Oncolytics 2019, 15, 60–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, K.D.; Frey, N.; Nelson, A.M.; Schmidt, A.; Luger, S.; Isaacs, R.E.; Lacey, S.F.; Hexner, E.; Melenhorst, J.J.; June, C.H.; et al. Treating Relapsed/Refractory (RR) AML with Biodegradable Anti-CD123 CAR Modified T Cells; American Society of Hematology: Washington, DC, USA, 2017. [Google Scholar]
- Alishah, K.; Birtel, M.; Masoumi, E.; Jafarzadeh, L.; Mirzaee, H.R.; Hadjati, J.; Voss, R.-H.; Diken, M.; Asad, S. CRISPR/Cas9-mediated TGFβRII disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells in vitro. J. Transl. Med. 2021, 19, 482. [Google Scholar] [CrossRef]
- Oluwole, O.O.; Davila, M.L. At The Bedside: Clinical review of chimeric antigen receptor (CAR) T cell therapy for B cell malignancies. J. Leukoc. Biol. 2016, 100, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Bordignon, C.; Bonini, C.; Verzeletti, S.; Nobili, N.; Maggioni, D.; Traversari, C.; Giavazzi, R.; Servida, P.; Zappone, E.; Benazzi, E.; et al. Transfer of the HSV-tk Gene into Donor Peripheral Blood Lymphocytes for In Vivo Modulation of Donor Anti-Tumor Immunity after Allogeneic Bone Marrow Transplantation. The San Raffaele Hospital, Milan, Italy. Hum. Gene Ther. 1995, 6, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Straathof, K.C.; Pule, M.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, P.; Li, Z.; He, Y.; Gan, W.; Jiang, H. Anti-CLL1 Chimeric Antigen Receptor T-Cell Therapy in Children with Relapsed/Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 3549–3555. [Google Scholar] [CrossRef] [PubMed]
- Petrov, J.C.; Wada, M.; Pinz, K.G.; Yan, L.E.; Chen, K.; Shuai, X.; Liu, H.; Chen, X.; Leung, L.-H.; Salman, H.; et al. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia 2018, 32, 1317–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, C.; Cheng, H.-Y.; Nguyen, D.; Dettling, D.; Yeung, Y.A.; Sutton, J.; Hamze, M.; Valton, J.; Smith, J.; Djuretic, I.; et al. Allogeneic FLT3 CAR T Cells with an Off-Switch Exhibit Potent Activity against AML and Can Be Depleted to Expedite Bone Marrow Recovery. Mol. Ther. 2020, 28, 2237–2251. [Google Scholar] [CrossRef]
- Tasian, S.K.; Kenderian, S.S.; Shen, F.; Ruella, M.; Shestova, O.; Kozlowski, M.; Li, Y.; Schrank-Hacker, A.; Morrissette, J.J.D.; Carroll, M.; et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 2017, 129, 2395–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszkiewicz, P.J.; Fräßle, S.P.; Srivastava, S.; Sommermeyer, D.; Hudecek, M.; Drexler, I.; Sadelain, M.; Liu, L.; Jensen, M.C.; Riddell, S.R.; et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Investig. 2016, 126, 4262–4272. [Google Scholar] [CrossRef] [PubMed]
- Warda, W.; Da Rocha, M.N.; Trad, R.; Haderbache, R.; Salma, Y.; Bouquet, L.; Roussel, X.; Nicod, C.; Deschamps, M.; Ferrand, C. Overcoming target epitope masking resistance that can occur on low-antigen-expresser AML blasts after IL-1RAP chimeric antigen receptor T cell therapy using the inducible caspase 9 suicide gene safety switch. Cancer Gene Ther. 2021, 28, 1365–1375. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; He, D.; Li, C.; Wang, H.; Yang, G. Development of Inducible CD19-CAR T Cells with a Tet-On System for Controlled Activity and Enhanced Clinical Safety. Int. J. Mol. Sci. 2018, 19, 3455. [Google Scholar] [CrossRef] [Green Version]
- Benmebarek, M.-R.; Cadilha, B.L.; Herrmann, M.; Lesch, S.; Schmitt, S.; Stoiber, S.; Darwich, A.; Augsberger, C.; Brauchle, B.; Rohrbacher, L.; et al. A modular and controllable T cell therapy platform for acute myeloid leukemia. Leukemia 2021, 35, 2243–2257. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef] [PubMed]
- Salman, H.; Pinz, K.G.; Wada, M.; Shuai, X.; Yan, L.E.; Petrov, J.C.; Ma, Y. Preclinical Targeting of Human Acute Myeloid Leukemia Using CD4-specific Chimeric Antigen Receptor (CAR) T Cells and NK Cells. J. Cancer 2019, 10, 4408–4419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salih, H.R.; Antropius, H.; Gieseke, F.; Lutz, S.Z.; Kanz, L.; Rammensee, H.-G.; Steinle, A. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood 2003, 102, 1389–1396. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Beksac, M. Natural Killer Cell-Mediated Cellular Therapy of Hematological Malignancies. Clin. Hematol. Int. 2019, 1, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Feng, Z.; Ma, J.; Ling, S.; Cao, Y.; Gurung, B.; Wu, Y.; Katona, B.W.; O’Dwyer, K.P.; Siegel, D.L.; et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood 2020, 135, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; De Silva, P.; Bron, D.; Willard-Gallo, K.; Sangiolo, D. Significance of TIM3 expression in cancer: From biology to the clinic. Semin. Oncol. 2019, 46, 372–379. [Google Scholar] [CrossRef]
- Walseng, E.; Köksal, H.; Sektioglu, I.M.; Fåne, A.; Skorstad, G.; Kvalheim, G.; Gaudernack, G.; Inderberg, E.M.; Wälchli, S. A TCR-based Chimeric Antigen Receptor. Sci. Rep. 2017, 7, 10713. [Google Scholar] [CrossRef]
- Bendle, G.M.; Linnemann, C.; Hooijkaas, A.I.; Bies, L.; De Witte, M.A.; Jorritsma, A.; Kaiser, A.D.M.; Pouw, N.; Debets, R.; Kieback, E.; et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med. 2010, 16, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Stenger, D.; Stief, T.A.; Kaeuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Vick, B.; Lotfi, R.; Wagner, B.; Grünewald, T.G.P.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020, 136, 1407–1418. [Google Scholar] [CrossRef]
- Poorebrahim, M.; Mohammadkhani, N.; Mahmoudi, R.; Gholizadeh, M.; Fakhr, E.; Cid-Arregui, A. TCR-like CARs and TCR-CARs targeting neoepitopes: An emerging potential. Cancer Gene Ther. 2021, 28, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Raskin, S.; Van Pelt, S.; Toner, K.; Balakrishnan, P.B.; Dave, H.; Bollard, C.M.; Yvon, E. Novel TCR-like CAR-T cells targeting an HLA∗0201-restricted SSX2 epitope display strong activity against acute myeloid leukemia. Mol. Ther. Methods Clin. Dev. 2021, 23, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Ivica, N.A.; Jia, B.; Li, Y.; Dong, H.; Liang, Y.; Brown, D.; Romee, R.; Chen, J. CAR-T cells targeting a nucleophosmin neoepitope exhibit potent specific activity in mouse models of acute myeloid leukaemia. Nat. Biomed. Eng. 2021, 5, 399–413. [Google Scholar] [CrossRef]
- Akahori, Y.; Wang, L.; Yoneyama, M.; Seo, N.; Okumura, S.; Miyahara, Y.; Amaishi, Y.; Okamoto, S.; Mineno, J.; Ikeda, H.; et al. Antitumor activity of CAR-T cells targeting the intracellular oncoprotein WT1 can be enhanced by vaccination. Blood 2018, 132, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Van Rhenen, A.; Moshaver, B.; Kelder, A.; Feller, N.; Nieuwint, A.W.M.; Zweegman, S.; Ossenkoppele, G.J.; Schuurhuis, G.J. Aberrant marker expression patterns on the CD34+CD38− stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia 2007, 21, 1700–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rhenen, A.; van Dongen, G.A.M.S.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Walsum, M.S.-V.; Zweegman, S.; Ossenkoppele, G.J.; Schuurhuis, G.J. The novel AML stem cell–associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007, 110, 2659–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Cao, Y.; Pinz, K.; Ma, Y.; Wada, M.; Chen, K.; Ma, G.; Shen, J.; Tse, C.O.; Su, Y.; et al. First-in-Human CLL1-CD33 Compound CAR T Cell Therapy Induces Complete Remission in Patients with Refractory Acute Myeloid Leukemia: Update on Phase 1 Clinical Trial. Blood 2018, 132, 901. [Google Scholar] [CrossRef]
- Yoshida, T.; Mihara, K.; Takei, Y.; Yanagihara, K.; Kubo, T.; Bhattacharyya, J.; Imai, C.; Mino, T.; Takihara, Y.; Ichinohe, T. All-trans retinoic acid enhances cytotoxic effect of T cells with an anti-CD38 chimeric antigen receptor in acute myeloid leukemia. Clin. Transl. Immunol. 2016, 5, e116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynn, R.C.; Poussin, M.; Kalota, A.; Feng, Y.; Low, P.; Dimitrov, D.S.; Powell, D.J. Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor–expressing T cells. Blood 2015, 125, 3466–3476. [Google Scholar] [CrossRef] [PubMed]
- Driouk, L.; Gicobi, J.K.; Kamihara, Y.; Rutherford, K.; Dranoff, G.; Ritz, J.; Baumeister, S.H.C. Chimeric Antigen Receptor T Cells Targeting NKG2D-Ligands Show Robust Efficacy Against Acute Myeloid Leukemia and T-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2020, 11, 580328. [Google Scholar] [CrossRef]
- El Khawanky, N.; Hughes, A.; Yu, W.; Myburgh, R.; Matschulla, T.; Taromi, S.; Aumann, K.; Clarson, J.; Vinnakota, J.M.; Shoumariyeh, K.; et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat. Commun. 2021, 12, 6436. [Google Scholar] [CrossRef]
- Li, S.; Xue, L.; Wang, M.; Qiang, P.; Xu, H.; Zhang, X.; Kang, W.; You, F.; Xu, H.; Wang, Y.; et al. Decitabine enhances cytotoxic effect of T cells with an anti-CD19 chimeric antigen receptor in treatment of lymphoma. OncoTargets Ther. 2019, 12, 5627–5638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epperly, R.; Gottschalk, S.; Velasquez, M.P. A Bump in the Road: How the Hostile AML Microenvironment Affects CAR T Cell Therapy. Front. Oncol. 2020, 10, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christopher, M.J.; Petti, A.A.; Rettig, M.; Miller, C.A.; Chendamarai, E.; Duncavage, E.J.; Klco, J.M.; Helton, N.M.; O’Laughlin, M.; Fronick, C.C.; et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N. Engl. J. Med. 2018, 379, 2330–2341. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanski, M.J.; Szajnik, M.; Czystowska-Kuźmicz, M.; Mandapathil, M.; Strauss, L.; Welsh, A.; Foon, K.A.; Whiteside, T.L.; Boyiadzis, M. Increased Frequency and Suppression by Regulatory T Cells in Patients with Acute Myelogenous Leukemia. Clin. Cancer Res. 2009, 15, 3325–3332. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Kaur, G.; Sankin, A.I.; Chen, F.; Guan, F.; Zang, X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J. Hematol. Oncol. 2019, 12, 59. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Li, A.M.; Hucks, G.E.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132 (Suppl. 1), 556. [Google Scholar] [CrossRef]
- Mardiana, S.; Gill, S. CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W. Allogeneic CAR T cell therapies for leukemia. Am. J. Hematol. 2019, 94, S50–S54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, C.; Jozwik, A.; Pepper, A.; Benjamin, R. Allogeneic CAR-T Cells: More than Ease of Access? Cells 2018, 7, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torikai, H.; Reik, A.; Liu, P.-Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovitch, B.; et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef] [PubMed]
- Loff, S.; Dietrich, J.; Meyer, J.-E.; Riewaldt, J.; Spehr, J.; von Bonin, M.; Gründer, C.; Swayampakula, M.; Franke, K.; Feldmann, A.; et al. Rapidly Switchable Universal CAR-T Cells for Treatment of CD123-Positive Leukemia. Mol. Ther. Oncolytics 2020, 17, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Wermke, M.; Kraus, S.; Ehninger, A.; Bargou, R.C.; Goebeler, M.-E.; Middeke, J.M.; Kreissig, C.; von Bonin, M.; Koedam, J.; Pehl, M.; et al. Proof of concept for a rapidly switchable universal CAR-T platform with UniCAR-T-CD123 in relapsed/refractory AML. Blood 2021, 137, 3145–3148. [Google Scholar] [CrossRef]
- Lu, H.; Zhao, X.; Li, Z.; Hu, Y.; Wang, H. From CAR-T Cells to CAR-NK Cells: A Developing Immunotherapy Method for Hematological Malignancies. Front. Oncol. 2021, 11, 3151. [Google Scholar] [CrossRef] [PubMed]
- Handgretinger, R.; Schilbach, K. The potential role of γδ T cells after allogeneic HCT for leukemia. Blood 2018, 131, 1063–1072. [Google Scholar] [CrossRef] [Green Version]
- Rozenbaum, M.; Meir, A.; Aharony, Y.; Itzhaki, O.; Schachter, J.; Bank, I.; Jacoby, E.; Besser, M.J. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front. Immunol. 2020, 11, 1347. [Google Scholar] [CrossRef] [PubMed]
- Cummins, K.; Gill, S. Will CAR T cell therapy have a role in AML? Promises and pitfalls. Semin. Hematol. 2019, 56, 155–163. [Google Scholar] [CrossRef]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Silva, D.; Atilla, E.; Atilla, P.A.; Mo, F.; Tashiro, H.; Srinivasan, M.; Lulla, P.; Rouce, R.H.; Cabral, J.M.; Ramos, C.A.; et al. CD7 CAR T Cells for the Therapy of Acute Myeloid Leukemia. Mol. Ther. 2019, 27, 272–280. [Google Scholar] [CrossRef]
- Laszlo, G.S.; Estey, E.H.; Walter, R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014, 28, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.I. How close are we to CAR T-cell therapy for AML? Best Pract. Res. Clin. Haematol. 2019, 32, 101104. [Google Scholar] [CrossRef]
- De Propris, M.S.; Raponi, S.; Diverio, D.; Milani, M.L.; Meloni, G.; Falini, B.; Foa, R.; Guarini, A. High CD33 expression levels in acute myeloid leukemia cells carrying the nucleophosmin (NPM1) mutation. Haematologica 2011, 96, 1548–1551. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.-S.; Wang, Y.; Lv, H.-Y.; Han, Q.-W.; Fan, H.; Guo, B.; Wang, L.-L.; Han, W.-D. Treatment of CD33-directed Chimeric Antigen Receptor-modified T Cells in One Patient with Relapsed and Refractory Acute Myeloid Leukemia. Mol. Ther. 2015, 23, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tambaro, F.P.; Singh, H.; Jones, E.; Rytting, M.; Mahadeo, K.M.; Thompson, P.; Daver, N.; DiNardo, C.; Kadia, T.; Garcia-Manero, G.; et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia 2021, 35, 3282–3286. [Google Scholar] [CrossRef]
- Cui, Q.; Qian, C.; Xu, N.; Kang, L.; Dai, H.; Cui, W.; Song, B.; Yin, J.; Li, Z.; Zhu, X.; et al. CD38-directed CAR-T cell therapy: A novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J. Hematol. Oncol. 2021, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Casucci, M.; Nicolis di Robilant, B.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef]
- Riether, C.; Pabst, T.; Höpner, S.; Bacher, U.; Hinterbrandner, M.; Banz, Y.; Müller, R.; Manz, M.G.; Gharib, W.H.; Francisco, D.; et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat. Med. 2020, 26, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Sauer, T.; Parikh, K.; Sharma, S.; Omer, B.; Sedloev, D.N.; Chen, Q.; Angenendt, L.; Schliemann, C.; Schmitt, M.; Müller-Tidow, C.; et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood 2021, 138, 318–330. [Google Scholar] [CrossRef]
- Myburgh, R.; Kiefer, J.D.; Russkamp, N.F.; Magnani, C.F.; Nuñez, N.; Simonis, A.; Pfister, S.; Wilk, C.M.; McHugh, D.; Friemel, J.; et al. Anti-human CD117 CAR T-cells efficiently eliminate healthy and malignant CD117-expressing hematopoietic cells. Leukemia 2020, 34, 2688–2703. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lin, J.; Gao, L.; Deng, A.; Lu, X.; Li, Y.; Wang, L.; Yu, L. High Expression of c-kit mRNA Predicts Unfavorable Outcome in Adult Patients with t(8;21) Acute Myeloid Leukemia. PLoS ONE 2015, 10, e0124241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergez, F.; Green, A.S.; Tamburini, J.; Sarry, J.-E.; Gaillard, B.; Cornillet-Lefebvre, P.; Pannetier, M.; Neyret, A.; Chapuis, N.; Ifrah, N.; et al. High levels of CD34+CD38low/−CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: A Groupe Ouest-Est des Leucémies Aiguës et Maladies du Sang (GOELAMS) study. Haematologica 2011, 96, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Mardiros, A.; Forman, S.J.; Budde, L.E. T cells expressing CD123 chimeric antigen receptors for treatment of acute myeloid leukemia. Curr. Opin. Hematol. 2015, 22, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Pizzitola, I.; Afonso, F.D.A.; Rouault-Pierre, K.; Lassailly, F.; Tettamanti, S.; Spinelli, O.; Biondi, A.; Biagi, E.; Bonnet, D. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia 2014, 28, 1596–1605. [Google Scholar] [CrossRef]
- Budde, L.; Song, J.Y.; Kim, Y.; Blanchard, S.; Wagner, B.J.; Stein, A.S.; Weng, L.; Del Real, M.; Hernandez, R.; Marcucci, E.; et al. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-Specific CAR T Cells: A First-in-Human Clinical Trial. Blood 2017, 130, 811. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, C.; Liu, Z.; Yang, M.; Tang, X.; Wang, Y.; Zheng, M.; Huang, J.; Zhong, K.; Zhao, S.; et al. B7-H3-Targeted CAR-T Cells Exhibit Potent Antitumor Effects on Hematologic and Solid Tumors. Mol. Ther. Oncolytics 2020, 17, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Tang, X.; Zhang, Z.; Gu, L.; Wei, H.; Zhao, S.; Zhong, K.; Mu, M.; Huang, C.; Jiang, C.; et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 2020, 10, 7622–7634. [Google Scholar] [CrossRef]
- Lichtman, E.I.; Du, H.; Shou, P.; Song, F.; Suzuki, K.; Ahn, S.; Li, G.; Ferrone, S.; Su, L.; Savoldo, B.; et al. Preclinical Evaluation of B7-H3–specific Chimeric Antigen Receptor T Cells for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 3141–3153. [Google Scholar] [CrossRef]
- Bakker, A.B.H.; Oudenrijn, S.V.D.; Bakker, A.Q.; Feller, N.; Van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.C.; Visser, T.J.; Bijl, N.; Geuijen, C.A.W.; et al. C-Type Lectin-Like Molecule-1. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [CrossRef] [Green Version]
- Bill, M.; Niekerk, P.B.V.K.; Woll, P.S.; Herborg, L.L.; Roug, A.S.; Hokland, P.; Nederby, L. Mapping the CLEC 12A expression on myeloid progenitors in normal bone marrow; implications for understanding CLEC 12A-related cancer stem cell biology. J. Cell. Mol. Med. 2018, 22, 2311–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting CLL-1 for acute myeloid leukemia therapy. J. Hematol. Oncol. 2019, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- De Togni, E.; Kim, M.Y.; Cooper, M.L.; Ritchey, J.; O’Neal, J.; Niswonger, J.; DiPersio, J.F. Chimeric Antigen Receptor T Cells Specific for CLL-1 for Treatment of Acute Myeloid Leukemia. Blood 2018, 132, 2205. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Xiao, W.; Li, W.; Wang, L.; Yang, S.; Wang, W.; Xu, L.; Liao, S.; Liu, W.; et al. CAR-T cells targeting CLL-1 as an approach to treat acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Gan, W.-T.; Hao, W.-G.; Wang, P.-F.; Li, Z.-Y.; Chang, L.-J. Successful Anti-CLL1 CAR T-Cell Therapy in Secondary Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 685. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacher, U.; Haferlach, C.; Kern, W.; Haferlach, T.; Schnittger, S. Prognostic relevance of FLT3-TKD mutations in AML: The combination matters—An analysis of 3082 patients. Blood 2008, 111, 2527–2537. [Google Scholar] [CrossRef]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3−ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.D.; Sauter, C.T.; Ishii, K.; Nguyen, S.M.; Shen, F.; Tasian, S.K.; Chen, W.; Dimitrov, D.S.; Fry, T.J. Preclinical Development of FLT3-Redirected Chimeric Antigen Receptor T Cell Immunotherapy for Acute Myeloid Leukemia. Blood 2016, 128, 1072. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Li, S.; Liu, J.; Xing, Y.; Xing, H.; Tian, Z.; Tang, K.; Rao, Q.; Wang, M.; et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J. Hematol. Oncol. 2018, 11, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiorova, V.; Mollaev, M.D.; Vikhreva, P.; Kulakovskaya, E.; Pershin, D.; Chudakov, D.M.; Kibardin, A.; Maschan, M.A.; Larin, S. Natural Flt3Lg-Based Chimeric Antigen Receptor (Flt3-CAR) T Cells Successfully Target Flt3 on AML Cell Lines. Vaccines 2021, 9, 1238. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, S.; Shestova, O.; Santiago, F.; Shrestha, E.; Liang, R.; Jehuda, R.B.; Sandler, V.; Gill, S. Anti-FLT3 CAR T Cells in Acute Myeloid Leukemia (Abst). Blood 2021, 138, 1703. [Google Scholar] [CrossRef]
- Kandalaft, L.E.; Powell, D.J.; Coukos, G. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. J. Transl. Med. 2012, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.F.; Wang, H.; Behm, F.G.; Mathew, P.; Wu, M.; Booth, R.; Ratnam, M. Folate receptor type beta is a neutrophilic lineage marker and is differentially expressed in myeloid leuke-mia. Cancer 1999, 85, 348–357. [Google Scholar] [CrossRef]
- Lynn, R.C.; Feng, Y.; Schutsky, K.; Poussin, M.; Kalota, A.; Dimitrov, D.S.; Powell, D.J., Jr. High-affinity FRβ-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia 2016, 30, 1355–1364. [Google Scholar] [CrossRef]
- Hasegawa, A.; Saito, S.; Narimatsu, S.; Nakano, S.; Nagai, M.; Ohnota, H.; Inada, Y.; Morokawa, H.; Nakashima, I.; Morita, D.; et al. Mutated GM-CSF-based CAR-T cells targeting CD116/CD131 complexes exhibit enhanced anti-tumor effects against acute myeloid leukaemia. Clin. Transl. Immunol. 2021, 10, e1282. [Google Scholar] [CrossRef] [PubMed]
- Lanza, F.; Castagnari, B.; Rigolin, G.M.; Moretti, S.; Latorraca, A.; Ferrari, L.; Bardi, A.; Castoldi, G. Flow cytometry measurement of GM-CSF receptors in acute leukemic blasts, and normal hemopoietic cells. Leukemia 1997, 11, 1700–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccioni, R.; Diverio, D.; Riti, V.; Buffolino, S.; Mariani, G.; Boe, A.; Cedrone, M.; Ottone, T.; Foà, R.; Testa, U. Interleukin (IL)-3/granulocyte macrophage-colony stimulating factor/IL-5 receptor alpha and beta chains are preferentially expressed in acute myeloid leukaemias with mutated FMS-related tyrosine kinase 3 receptor. Br. J. Haematol. 2009, 144, 376–387. [Google Scholar] [CrossRef]
- Nair, R.; Salinas-Illarena, A.; Baldauf, H.-M. New strategies to treat AML: Novel insights into AML survival pathways and combination therapies. Leukemia 2021, 35, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Gui, X.; Kim, J.; Xie, L.; Chen, W.; Li, Z.; He, L.; Chen, Y.; Chen, H.; Luo, W.; et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature 2018, 562, 605–609. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Chen, H.; Deng, M.; Gui, X.; Wu, G.; Chen, W.; Li, Z.; Zhang, N.; An, Z.; Zhang, C.C. A Novel Anti-LILRB4 CAR-T Cell for the Treatment of Monocytic AML. Mol. Ther. 2018, 26, 2487–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Niu, T. Natural killer cell-based immunotherapy for acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 167. [Google Scholar] [CrossRef]
- Diermayr, S.; Himmelreich, H.; Müller-Durovic, B.; Mathys-Schneeberger, A.; Siegler, U.; Langenkamp, U.; Hofsteenge, J.; Gratwohl, A.; Tichelli, A.; Paluszewska, M.; et al. NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood 2008, 111, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef]
- Jetani, H.; Navarro-Bailón, A.; Maucher, M.; Frenz, S.; Verbruggen, C.; Yeguas, A.; Vidriales, M.B.; González, M.; Saborido, J.R.; Kraus, S.; et al. Siglec-6 is a novel target for CAR T-cell therapy in acute myeloid leukemia. Blood 2021, 138, 1830–1842. [Google Scholar] [CrossRef]
- Li, C.; Chen, X.; Yu, X.; Zhu, Y.; Ma, C.; Xia, R.; Ma, J.; Gu, C.; Ye, L.; Wu, D. Tim-3 is highly expressed in T cells in acute myeloid leukemia and associated with clinicopathological prognos-tic stratification. Int. J. Clin. Exp. Pathol. 2014, 7, 6880–6888. [Google Scholar] [PubMed]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients with Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndrome (HR-MDS): Updated Results from a Phase 1b Study. Blood 2020, 136, 657. [Google Scholar] [CrossRef]
- Lee, W.-H.S.; Ye, Z.; Cheung, A.M.; Goh, Y.S.; Oh, H.L.J.; Rajarethinam, R.; Yeo, S.P.; Soh, M.K.; Chan, E.H.L.; Tan, L.K.; et al. Effective Killing of Acute Myeloid Leukemia by TIM-3 Targeted Chimeric Antigen Receptor T Cells. Mol. Cancer Ther. 2021, 20, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wei, Y.; Cao, Y.; Xiao, X.; Li, Q.; Lyu, H.; Jiang, Y.; Zhang, H.; Li, X.; Jiang, Y.; et al. CD19 CAR-T cell treatment conferred sustained remission in B-ALL patients with minimal residual disease. Cancer Immunol. Immunother. 2021, 70, 3501–3511. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, X.-S.; Liu, Y.-R.; Xu, L.-P.; Zhang, X.-H.; Chen, H.; Chen, Y.-H.; Wang, F.-R.; Han, W.; Sun, Y.-Q.; et al. Association of Persistent Minimal Residual Disease with Poor Outcomes of Patients with Acute Myeloid Leukemia Undergoing Allogeneic Hematopoietic Stem Cell Transplantation. Chin. Med. J. 2018, 131, 2808–2816. [Google Scholar] [CrossRef] [PubMed]
- Anthias, C.; Dignan, F.L.; Morilla, R.; Morilla, A.; Ethell, M.E.; Potter, M.N.; Shaw, B.E. Pre-transplant MRD predicts outcome following reduced-intensity and myeloablative allogeneic hemopoietic SCT in AML. Bone Marrow Transplant. 2014, 49, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leahy, A.B.; Newman, H.; Li, Y.; Liu, H.; Myers, R.; DiNofia, A.; Dolan, J.G.; Callahan, C.; Baniewicz, D.; Devine, K.; et al. CD19-targeted chimeric antigen receptor T-cell therapy for CNS relapsed or refractory acute lymphocytic leukaemia: A post-hoc analysis of pooled data from five clinical trials. Lancet Haematol. 2021, 8, e711–e722. [Google Scholar] [CrossRef]
- Perica, K.; Flynn, J.; Curran, K.J.; Rivere, I.; Wang, X.; Senechal, B.; Halton, E.; Diamonte, C.; Pineda, J.; Bernal, Y.; et al. Impact of bridging chemotherapy on clinical outcome of CD19 CAR T therapy in adult acute lymphoblastic leukemia. Leukemia 2021, 35, 3268–3271. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Khan, D.H.; Hurren, R.; Xu, M.; Na, Y.; Kang, H.; Mirali, S.; Wang, X.; Gronda, M.V.; Jitkova, Y.; et al. Venetoclax enhances T cell-mediated anti-leukemic activity by increasing ROS production. Blood 2021, 138, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, L.; Ni, M.; Neuber, B.; Wang, S.; Gong, W.; Sauer, T.; Sellner, L.; Schubert, M.-L.; Hückelhoven-Krauss, A.; et al. Pre-sensitization of Malignant B Cells Through Venetoclax Significantly Improves the Cytotoxic Efficacy of CD19.CAR-T Cells. Front. Immunol. 2020, 11, 3233. [Google Scholar] [CrossRef]
- You, L.; Han, Q.; Zhu, L.; Zhu, Y.; Bao, C.; Yang, C.; Lei, W.; Qian, W. Decitabine-Mediated Epigenetic Reprograming Enhances Anti-leukemia Efficacy of CD123-Targeted Chimeric Antigen Receptor T-Cells. Front. Immunol. 2020, 11, 1787. [Google Scholar] [CrossRef] [PubMed]
- Dholaria, B.; Savani, B.N.; Hamilton, B.K.; Oran, B.; Liu, H.D.; Tallman, M.S.; Ciurea, S.O.; Holtzman, N.G.; Ii, G.L.P.; Devine, S.M.; et al. Hematopoietic Cell Transplantation in the Treatment of Newly Diagnosed Adult Acute Myeloid Leukemia: An Evidence-Based Review from the American Society of Transplantation and Cellular Therapy. Transplant. Cell. Ther. 2021, 27, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.J.; Battiwalla, M. Relapse after allogeneic stem cell transplantation. Expert Rev. Hematol. 2010, 3, 429–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dholaria, B.; Savani, B.N.; Huang, X.; Nagler, A.; Perales, M.; Mohty, M. The evolving role of allogeneic haematopoietic cell transplantation in the era of chimaeric antigen receptor T-cell therapy. Br. J. Haematol. 2021, 193, 1060–1075. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Highfill, S.L.; Shalabi, H.; Yates, B.; Jin, J.; Wolters, P.L.; Ombrello, A.; Steinberg, S.M.; Martin, S.; Delbrook, C.; et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J. Clin. Oncol. 2020, 38, 1938–1950. [Google Scholar] [CrossRef]
- Li, L.; Liu, J.; Xu, M.; Yu, H.; Lv, C.; Cao, F.; Wang, Z.; Fu, Y.; Zhang, M.; Meng, H.; et al. Treatment response, survival, safety, and predictive factors to chimeric antigen receptor T cell therapy in Chinese relapsed or refractory B cell acute lymphoblast leukemia patients. Cell Death Dis. 2020, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L.M.; Baggott, R.C.; Prabhu, S.; Pacenta, H.; Phillips, C.L.; Rossoff, J.; Stefanski, M.H.; Talano, J.-A.; Moskop, A.; Margossian, S.P.; et al. Disease Burden Impacts Outcomes in Pediatric and Young Adult B-Cell Acute Lymphoblastic Leukemia after Commercial Tisagenlecleucel: Results from the Pediatric Real World CAR Consortium (PRWCC). Blood 2020, 136, 14–15. [Google Scholar] [CrossRef]
- Zhang, R.; He, Y.; Yang, D.; Wang, Y.; Feng, S.; Wang, J.; Han, M.-Z. Donor-Derived Second Generation of CD19 CAR-T Cell Therapy for Relapsed B-Cell Acute Lymphoblastic Leukemia after Allogenic Stem Cell Transplantation. Blood 2018, 132, 2179. [Google Scholar] [CrossRef]
- Jain, T.; Sauter, C.S.; Shah, G.L.; Maloy, M.A.; Chan, J.; Scordo, M.; Avecilla, S.T.; Batlevi, Y.; Dahi, P.B.; Batlevi, C.W.; et al. Safety and feasibility of chimeric antigen receptor T cell therapy after allogeneic hematopoietic cell transplantation in relapsed/ refractory B cell non-Hodgkin lymphoma. Leukemia 2019, 33, 2540–2544. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Zhang, J.; Zhang, X.; Wu, X.; Zhou, L.; Bao, X.; Han, Y.; Miao, M.; Li, C.; Fu, C.; et al. Donor-derived anti-CD19 CAR T cells compared with donor lymphocyte infusion for recurrent B-ALL after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2021, 56, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Kebriaei, P.; Ciurea, S.O.; Huls, B.M.H.; Singh, H.; Olivares, B.S.; Su, B.S.; Figliola, B.M.J.; Kumar, P.; Jena, B.; Forget, M.-A.; et al. Pre-Emptive Donor Lymphocyte Infusion with CD19-Directed, CAR-Modified T Cells Infused after Allogeneic Hematopoietic Cell Transplantation for Patients with Advanced CD19+ Malignancies. Blood 2015, 126, 862. [Google Scholar] [CrossRef]
- Yao, S.; Jianlin, C.; Yarong, L.; Botao, L.; Qinghan, W.; Hongliang, F.; Lu, Z.; Hongmei, N.; Pin, W.; Hu, C.; et al. Donor-Derived CD123-Targeted CAR T Cell Serves as a RIC Regimen for Haploidentical Transplantation in a Patient With FUS-ERG+ AML. Front. Oncol. 2019, 9, 1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Target Antigen | Name of Drug | CAR Cell Type | Phase | https://clinicaltrials.gov/ (accessed on 21 December 2021) ID | Age (years) | Country |
|---|---|---|---|---|---|---|
| CD7 | CD7 CAR-T | T | I/II | NCT04762485 | 12 to 65 | China |
| CD7 | CD7 CAR-T | T | I/II | NCT04033302 | Up to 75 | China |
| CD19 | CAR-T CD19 | T | II/III | NCT04257175 | 18 and up | Israel |
| CD33 | CD33 CAR-T | T | I/II | NCT04835519 | 1 to 70 | China |
| CD33 | CD33CART | T | I/II | NCT03971799 | 1 to 35 | USA |
| CD33 | CART-33 | T | I | NCT02799680 | 50 and up | China |
| CD33 | PRGN-3006 | T | I | NCT03927261 | 18 and up | USA |
| CD33/CLL1 | Dual CD33-CLL1 CAR-T | T | I | NCT05016063 | 18 to 70 | China |
| CD38 | CART-38 | T | I/II | NCT04351022 | 6 to 65 | China |
| CD44v6 | CD44v6 CAR-T | T | I/II | NCT04097301 | 1 to 75 | Italy |
| CD70 | CD70 CAR-T | T | I | NCT04662294 | All | China |
| CD70 | CAR.70/IL-15 | NK | I/II | NCT05092451 | 18 and up | USA |
| CD123 | IM23 CAR-T | T | I | NCT03585517 | 3 to 80 | China |
| CD123 | IL-3 CAR-T | T | I | NCT04599543 | All | China |
| CD123 | Anti-CD123 CAR-T | T | I | NCT04014881 | 18 to 70 | China |
| CD123 | CD123-CAR-T | T | I | NCT04318678 | Up to 21 | USA |
| CD123 | CART-123 | T | I/II | NCT03556982 | 14 to 75 | China |
| CD123 | CD123 CAR-T | T | I/II | NCT04272125 | 3 to 75 | China |
| CD123 | CD123 CAR-T | T | I/II | NCT04265963 | 2 to 75 | China |
| CD123 | UCART123v1.2 | T | I | NCT03190278 | 18 to 65 | USA |
| CD123 | UniCAR02-T | T | I | NCT04230265 | 18 and up | Germany |
| CD123 | CD123-CAR-CD28-CD3z-EGFRt | T | I | NCT02159495 | 12 and up | USA |
| CD123 | CART123 | T | I | NCT04678336 | 1 to 29 | USA |
| CD123 | CART123 | T | I | NCT03766126 | 18 and up | USA |
| CD123/CLL-1 | CD123/CLL1 CAR-T | T | II/III | NCT03631576 | Up to 70 | China |
| CD276 | TAA6 | T | I | NCT04692948 | 18 to 70 | China |
| CLL-1 | CLL-1 CAR-T | T | I | NCT04219163 | Up to 75 | USA |
| CLL-1 | KITE-222 | T | I | NCT04789408 | 18 and up | USA |
| CLL1 | Anti-CLL1 CART | T | I/II | NCT04884984 | 6 to 65 | China |
| CLL1 | Anti-CLL1 CART | T | I | NCT04923919 | 2 to 75 | China |
| FLT3 | Anti-FLT3 CAR-T | T | I/II | NCT05023707 | 16 to 65 | China |
| FLT3 | AMG553 | T | I | NCT03904069 | 12 and up | USA |
| FLT3 | TAA05 | T | NCT05017883 | 18 to 70 | China | |
| ILT3 | Anti-ILT3 CAR-T | T | I | NCT04803929 | 18 to 70 | China |
| NKG2D | CYAD-02 | NK | I | NCT04167696 | 18 and up | USA |
| NKG2D | NKX101 | NK | I | NCT04623944 | 18 and up | USA |
| NKG2D | NKG2D CAR-T | NK | I | NCT04658004 | 3 to 70 | China |
| Target Name | Normal Tissue Distribution | Expression on LSCs | Phase of Development |
|---|---|---|---|
| CD7 | activated T-cells, NK cells, lymphoid and myeloid progenitors | Yes | Phase I/II clinical trial |
| CD33 | myeloid cells, myeloid progenitors, Kupffer cells | Yes | Phase I/II clinical trial |
| CD38 | plasma cells, NK cells, B-cells, HSCs (low) | No | Phase I/II clinical trial |
| CD44v6 | activated T-cells, monocytes, keratinocytes | No | Phase I/II clinical trial |
| CD70 | dendritic cells, B-cells | Yes | Phase I/II clinical trial |
| CD117 | HSCs, myeloid progenitors, erythroid progenitors | No | Preclinical |
| CD123 | myeloid cells, myeloid progenitors | Yes | Phase II/III clinical trial |
| CD276 (B7-H3) | Antigen presenting cells, HSCs (low) | No | Phase I clinical trial |
| CLL-1 | myeloid cells, myeloid progenitors | Yes | Phase I/II clinical trial |
| FLT3 | HSCs | No | Phase I clinical trial |
| FRb | myeloid cells, HSCs (low) | No | Preclinical |
| GM-CSF (CD116) | myeloid cells | No | Preclinical |
| ILT3 (LILRB4) | myeloid antigen presenting cells | No | Phase I/II clinical trial |
| NKG2D | NK cells, γδ T-cells, CD8+ T-cells | No | Phase I/II clinical trial |
| Siglec-6 | B-cells, mast cells, placenta | Yes | Preclinical |
| TIM3 | T-cells, myeloid cells, NK cells | Yes | Preclinical |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marvin-Peek, J.; Savani, B.N.; Olalekan, O.O.; Dholaria, B. Challenges and Advances in Chimeric Antigen Receptor Therapy for Acute Myeloid Leukemia. Cancers 2022, 14, 497. https://doi.org/10.3390/cancers14030497
Marvin-Peek J, Savani BN, Olalekan OO, Dholaria B. Challenges and Advances in Chimeric Antigen Receptor Therapy for Acute Myeloid Leukemia. Cancers. 2022; 14(3):497. https://doi.org/10.3390/cancers14030497
Chicago/Turabian StyleMarvin-Peek, Jennifer, Bipin N. Savani, Oluwole O. Olalekan, and Bhagirathbhai Dholaria. 2022. "Challenges and Advances in Chimeric Antigen Receptor Therapy for Acute Myeloid Leukemia" Cancers 14, no. 3: 497. https://doi.org/10.3390/cancers14030497