
Efficacy, Safety, and Challenges of CAR T-Cells in the Treatment of Solid Tumors


Abstract
:Simple Summary
Abstract
1. Introduction
2. Status of CAR T-Cell Therapy in Hematological Malignancies
3. CAR T-Cell Therapy in Solid Tumors
3.1. CAR T-Cells Targeting Prostate TAA
3.2. CAR T-Cells Targeting MSLN (ATA2271)
3.3. Other Tumor Antigens Used for CAR T-Cells in Solid Tumors
3.3.1. MUC1
3.3.2. ICAM1
3.3.3. EGFR
3.3.4. ROR1
3.3.5. Trop2
3.3.6. TAG72
3.3.7. CA9
3.3.8. CD133
3.3.9. Integrin αvβ6
4. Lesson on Safety of CAR T-Cells in Solid Tumors from Hematologic Malignancies
4.1. Cytokine-Release Syndrome (CRS)
4.2. Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS)
4.3. Macrophage Activation Syndrome (MAS)
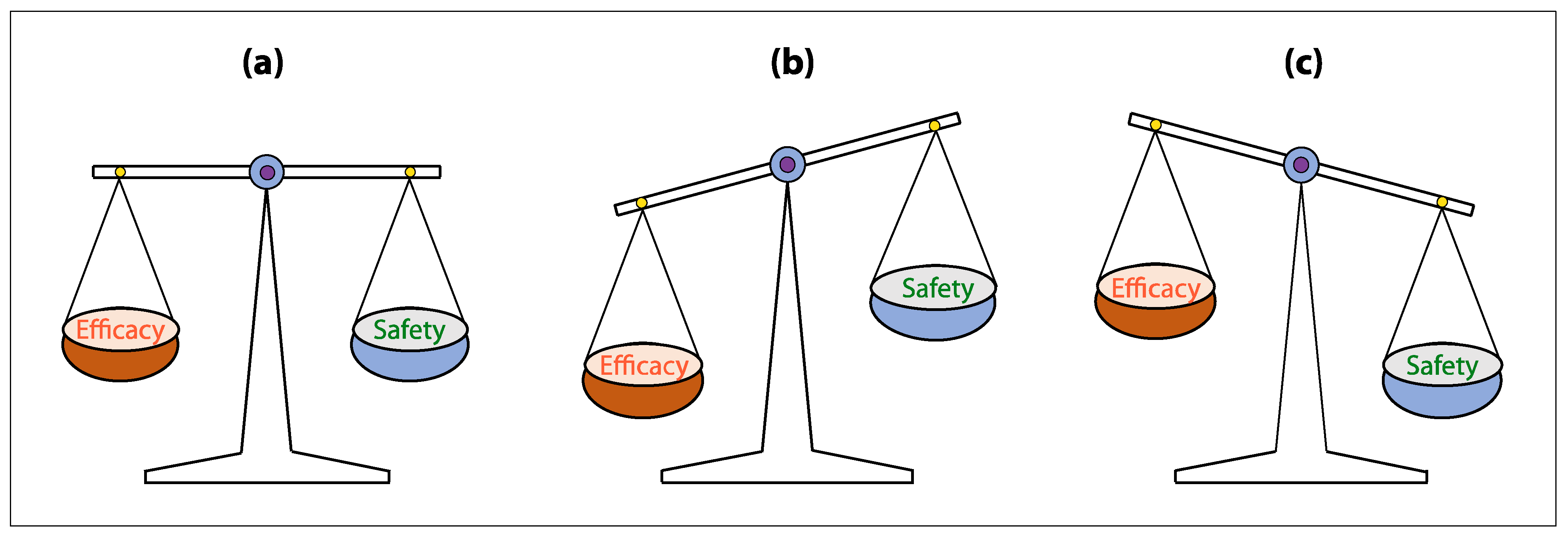
5. Challenges of CAR T-Cell Therapy in Solid Tumors
6. Improve the Effectiveness and Safety of CAR T-Cells
6.1. CD19 × CD22 CAR T-Cells
6.2. CD19 × BCMA CAR T-Cells
6.3. CD33 × CLL1 CAR T-Cells
6.4. HER2 × IL13Rα2 × EphA2 Trivalent CAR T-Cells
7. Discussion
8. Conclusions
9. Outlooks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| BCMA | B-cell maturation antigen |
| CAR | chimeric antigen receptor |
| CI | checkpoint inhibitor |
| CRS | cytokine release syndrome |
| CLL | chronic lymphocytic leukemia |
| DLBCL | diffuse large B-cell lymphoma |
| EFS | Event-free survival |
| FDA | Food and Drug Administration |
| HL | Hodgkin’s lymphoma |
| HLA | human leukocyte antigen |
| HSC | hematopoietic stem cells |
| ICANS | immune effector cell-associated neurotoxicity syndrome |
| IFN-γ | interferon-gamma |
| IL | interleukin |
| LSC | Leukemia stem cells |
| MAS | macrophage activation syndrome |
| MHC | major histocompatibility complex |
| MM | multiple myeloma |
| NHL | B-cell non-Hodgkin lymphoma |
| NK | natural killer |
| NKT | natural killer T |
| NS | neurologic symptoms |
| NSCLC | non-small-cell lung carcinoma |
| OS | objective response |
| PBMC | peripheral blood mononuclear cells |
| PD | progressive disease |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed death-ligand 1 |
| PFS | progression-free survival |
| PSMA | prostate-specific membrane antigen |
| R/R | relapsed and refractory |
| TIL | tumor infiltrating lymphocyte |
| TLS | tumor lysis syndrome |
| TMB | tumor mutational burden |
| TME | tumor microenvironment |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Jia, G.; Lu, L.; Bao, Y.; Ma, W. Factors affecting tumor responders and predictive biomarkers of toxicities in cancer patients treated with immune checkpoint inhibitors. Int. Immunopharmacol. 2020, 85, 106628. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell. Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Flego, M.; Frau, A.; Accardi, L.; Mallano, A.; Ascione, A.; Gellini, M.; Fanunza, E.; Vella, S.; Di Bonito, P.; Tramontano, E. Intracellular human antibody fragments recognizing the VP35 protein of Zaire Ebola filovirus inhibit the protein activity. BMC Biotechnol. 2019, 19, 64. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem. Cell. Res. 2021, 12, 81. [Google Scholar] [CrossRef]
- Ingegnere, T.; Mariotti, F.R.; Pelosi, A.; Quintarelli, C.; De Angelis, B.; Tumino, N.; Besi, F.; Cantoni, C.; Locatelli, F.; Vacca, P.; et al. Human CAR NK Cells: A New Non-viral Method Allowing High Efficient Transfection and Strong Tumor Cell Killing. Front. Immunol. 2019, 10, 957. [Google Scholar] [CrossRef] [Green Version]
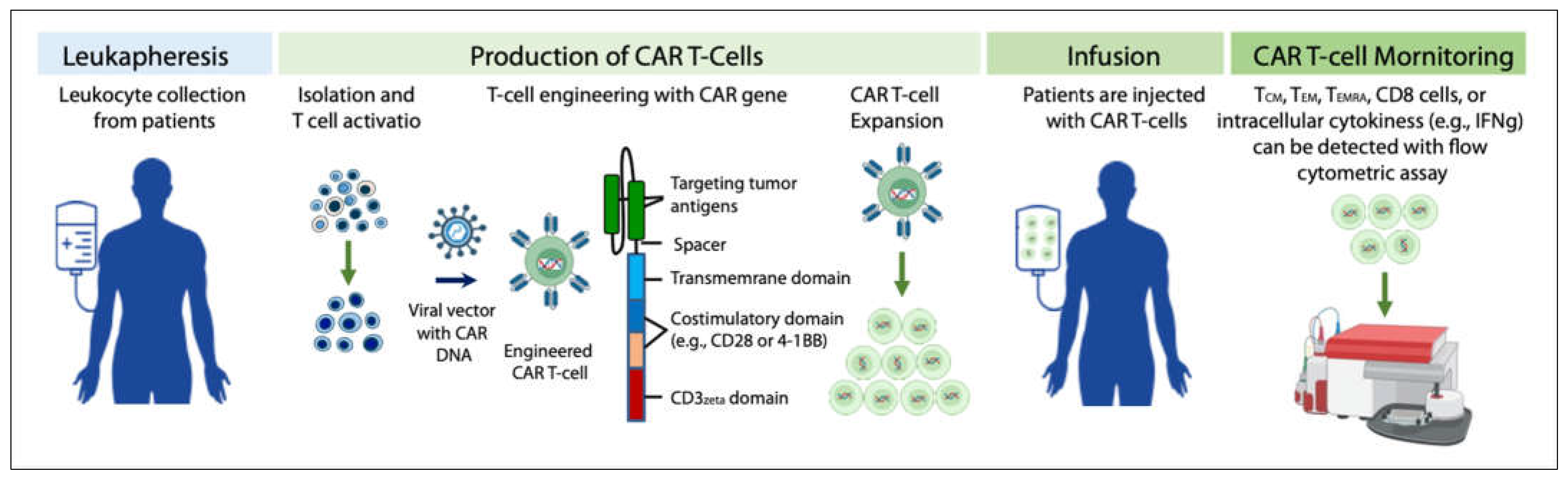
- Peinelt, A.; Bremm, M.; Kreyenberg, H.; Cappel, C.; Banisharif-Dehkordi, J.; Erben, S.; Rettinger, E.; Jarisch, A.; Meisel, R.; Schlegel, P.G.; et al. Monitoring of Circulating CAR T Cells: Validation of a Flow Cytometric Assay, Cellular Kinetics, and Phenotype Analysis Following Tisagenlecleucel. Front. Immunol. 2022, 13, 830773. [Google Scholar] [CrossRef]
- Neelapu, S.S. CAR-T efficacy: Is conditioning the key? Blood 2019, 133, 1799–1800. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Nastoupil, L.J.; Jain, M.D.; Feng, L.; Spiegel, J.Y.; Ghobadi, A.; Lin, Y.; Dahiya, S.; Lunning, M.; Lekakis, L.; Reagan, P.; et al. Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma CAR T Consortium. J. Clin. Oncol. 2020, 38, 3119–3128. [Google Scholar] [CrossRef]
- Gupta, A.; Gill, S. CAR-T cell persistence in the treatment of leukemia and lymphoma. Leuk. Lymphoma. 2021, 62, 2587–2599. [Google Scholar] [CrossRef] [PubMed]
- Sarikonda, G.; Mathieu, M.; Natalia, M.; Pahuja, A.; Xue, Q.; Pierog, P.L.; Trampont, P.C.; Decman, V.; Reynolds, S.; Hanafi, L.A.; et al. Best practices for the development, analytical validation and clinical implementation of flow cytometric methods for chimeric antigen receptor T cell analyses. Cytom. B Clin. Cytom. 2021, 100, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Bar-Ephraim, Y.E.; Koning, J.J.; Burniol Ruiz, E.; Konijn, T.; Mourits, V.P.; Lakeman, K.A.; Boon, L.; Bogels, M.; van Maanen, J.P.; Den Haan, J.M.M.; et al. CD62L Is a Functional and Phenotypic Marker for Circulating Innate Lymphoid Cell Precursors. J. Immunol. 2019, 202, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Yin, Y. Measuring Chimeric Antigen Receptor T Cells (CAR T Cells) Activation by Coupling Intracellular Cytokine Staining with Flow Cytometry. Methods. Mol. Biol. 2020, 2108, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menetrier-Caux, C.; Ray-Coquard, I.; Blay, J.Y.; Caux, C. Lymphopenia in Cancer Patients and its Effects on Response to Immunotherapy: An opportunity for combination with Cytokines? J. Immunother. Cancer 2019, 7, 85. [Google Scholar] [CrossRef] [Green Version]
- Gutkin, P.M.; Kozak, M.M.; von Eyben, R.; Horst, K.C. Lymphopenia and clinical outcomes in patients with residual nodal disease after neoadjuvant chemotherapy for breast cancer. Cancer Causes Control 2020, 31, 1021–1026. [Google Scholar] [CrossRef]
- Grossman, S.A.; Ellsworth, S.; Campian, J.; Wild, A.T.; Herman, J.M.; Laheru, D.; Brock, M.; Balmanoukian, A.; Ye, X. Survival in Patients With Severe Lymphopenia Following Treatment With Radiation and Chemotherapy for Newly Diagnosed Solid Tumors. J. Natl. Compr. Canc. Netw. 2015, 13, 1225–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabrizio, V.A.; Boelens, J.J.; Mauguen, A.; Baggott, C.; Prabhu, S.; Egeler, E.; Mavroukakis, S.; Pacenta, H.; Phillips, C.L.; Rossoff, J.; et al. Optimal fludarabine lymphodepletion is associated with improved outcomes after CAR T-cell therapy. Blood Adv. 2022, 6, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, V.; Todd, L.A.; Goswami, A.; Stefanson, O.; Yang, Z.; Marincola, F. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 2021, 22, 10828. [Google Scholar] [CrossRef] [PubMed]
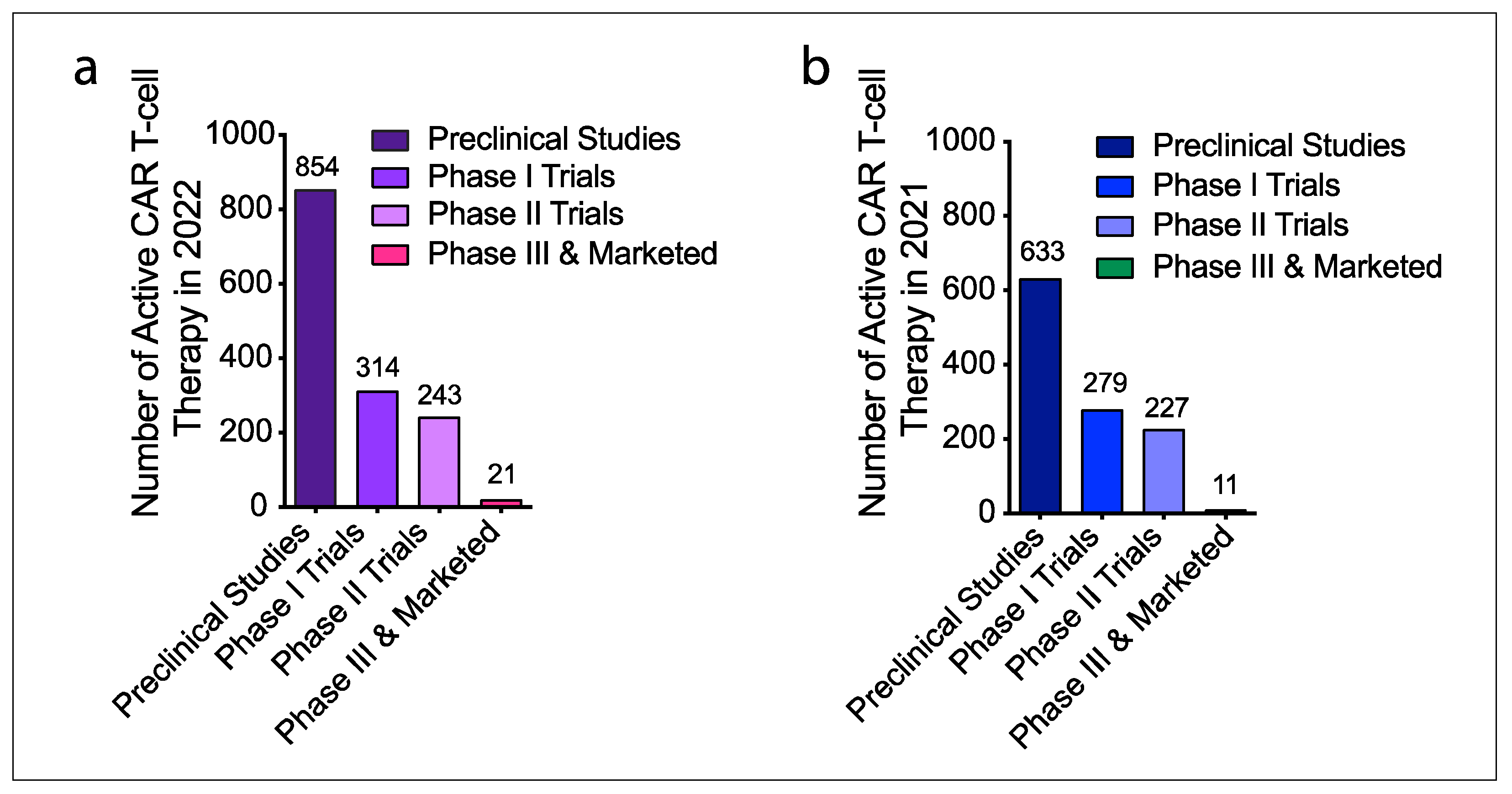
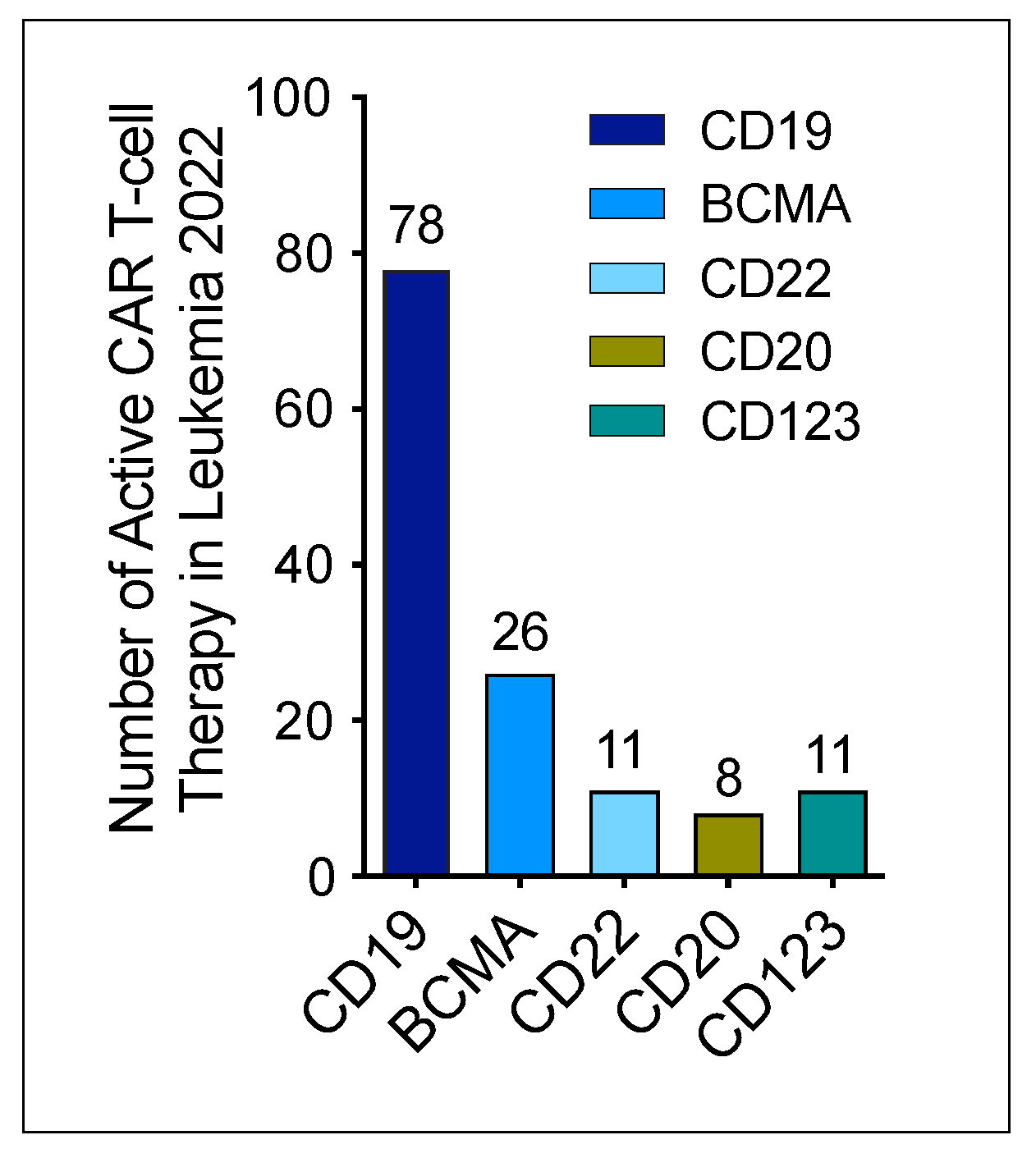
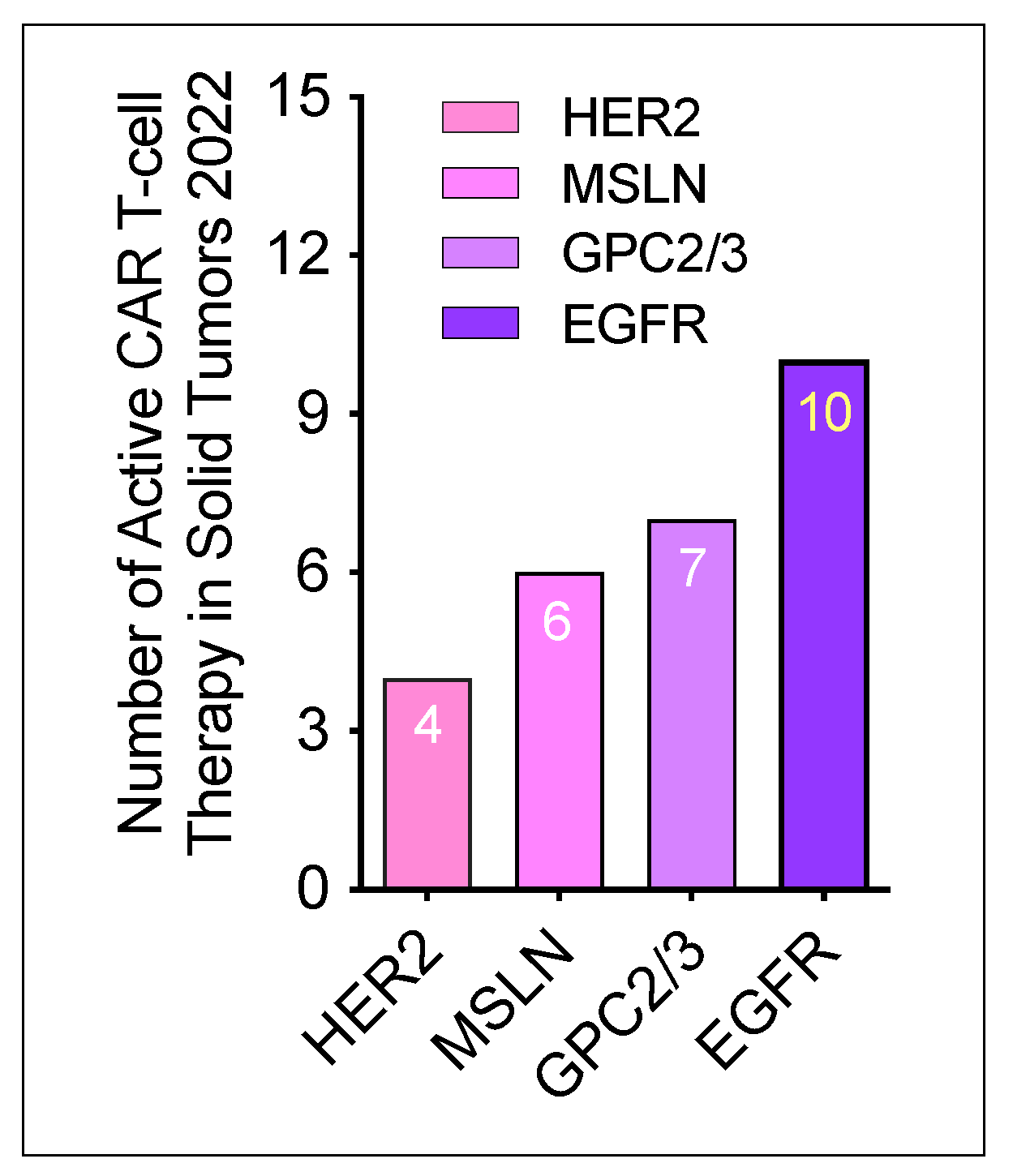
- Saez-Ibanez, A.R.; Upadhaya, S.; Partridge, T.; Shah, M.; Correa, D.; Campbell, J. Landscape of cancer cell therapies: Trends and real-world data. Nat. Rev. Drug Discov. 2022, 21, 631–632. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Kleber, M.; Ntanasis-Stathopoulos, I.; Terpos, E. BCMA in Multiple Myeloma-A Promising Key to Therapy. J. Clin. Med. 2021, 10, 4088. [Google Scholar] [CrossRef]
- Wang, J.N.; Gu, T.; Hu, Y.; Huang, H. Novel cellular immunotherapies for hematological malignancies: Recent updates from the 2021 ASH annual meeting. Exp. Hematol. Oncol. 2022, 11, 61. [Google Scholar] [CrossRef]
- Lu, J.; Jiang, G. The journey of CAR-T therapy in hematological malignancies. Mol. Cancer 2022, 21, 194. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Cai, H.; Li, C.; Deng, B.; Song, W.; Ling, Z.; Hu, G.; Yang, Y.; Niu, P.; Meng, G.; et al. A novel full-human CD22-CAR T cell therapy with potent activity against CD22(low) B-ALL. Blood Cancer J. 2021, 11, 71. [Google Scholar] [CrossRef]
- Shah, N.N.; Highfill, S.L.; Shalabi, H.; Yates, B.; Jin, J.; Wolters, P.L.; Ombrello, A.; Steinberg, S.M.; Martin, S.; Delbrook, C.; et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J. Clin. Oncol. 2020, 38, 1938–1950. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Zhang, Y.; Zhao, H.; Wang, Y.; Liu, Y.; Liang, B.; Wang, X.; Xu, H.; Cui, J.; Wu, W.; et al. CD19/CD22 Dual-Targeted CAR T-cell Therapy for Relapsed/Refractory Aggressive B-cell Lymphoma: A Safety and Efficacy Study. Cancer Immunol. Res. 2021, 9, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- van der Schans, J.J.; van de Donk, N.; Mutis, T. Dual Targeting to Overcome Current Challenges in Multiple Myeloma CAR T-Cell Treatment. Front. Oncol. 2020, 10, 1362. [Google Scholar] [CrossRef]
- Prinzing, B.; Zebley, C.C.; Petersen, C.T.; Fan, Y.; Anido, A.A.; Yi, Z.; Nguyen, P.; Houke, H.; Bell, M.; Haydar, D.; et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci. Transl. Med. 2021, 13, eabh0272. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Bi, E.; Ma, X.; Xiong, W.; Qian, J.; Ye, L.; Su, P.; Wang, Q.; Xiao, L.; Yang, M.; et al. Enhanced CAR-T activity against established tumors by polarizing human T cells to secrete interleukin-9. Nat. Commun. 2020, 11, 5902. [Google Scholar] [CrossRef]
- Koedam, J.; Wermke, M.; Ehninger, A.; Cartellieri, M.; Ehninger, G. Chimeric antigen receptor T-cell therapy in acute myeloid leukemia. Curr. Opin. Hematol. 2022, 29, 74–83. [Google Scholar] [CrossRef]
- El Khawanky, N.; Hughes, A.; Yu, W.; Myburgh, R.; Matschulla, T.; Taromi, S.; Aumann, K.; Clarson, J.; Vinnakota, J.M.; Shoumariyeh, K.; et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat. Commun. 2021, 12, 6436. [Google Scholar] [CrossRef]
- Wang, L. Clinical determinants of relapse following CAR-T therapy for hematologic malignancies: Coupling active strategies to overcome therapeutic limitations. Curr. Res. Transl. Med. 2022, 70, 103320. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Yu, W.L.; Hua, Z.C. Chimeric Antigen Receptor T-cell (CAR T) Therapy for Hematologic and Solid Malignancies: Efficacy and Safety-A Systematic Review with Meta-Analysis. Cancers 2019, 11, 47. [Google Scholar] [CrossRef]
- Gumber, D.; Wang, L.D. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; LaBelle, J.L.; Bishop, M.R. Current Status of CAR T Cell Therapy for Leukemias. Curr. Treat. Options. Oncol. 2021, 22, 62. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, X.A.; Yang, J.; Zhang, G.; Li, J.; Song, L.; Su, Y.; Shi, Y.; Zhang, M.; He, J.; et al. Efficacy and safety of anti-CD19 CAR T-cell therapy in 110 patients with B-cell acute lymphoblastic leukemia with high-risk features. Blood Adv. 2020, 4, 2325–2338. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V. CD19 CAR T cells for adults with relapsed or refractory acute lymphoblastic leukaemia. Lancet 2021, 398, 466–467. [Google Scholar] [CrossRef] [PubMed]
- Juluri, K.R.; Wu, Q.V.; Voutsinas, J.; Hou, J.; Hirayama, A.V.; Mullane, E.; Miles, N.; Maloney, D.G.; Turtle, C.J.; Bar, M.; et al. Severe cytokine release syndrome is associated with hematologic toxicity following CD19 CAR T-cell therapy. Blood Adv. 2022, 6, 2055–2068. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; Moon, E.; Albelda, S.M. Chimeric antigen receptor T-cell therapy for solid tumors. Mol. Oncolytics. 2016, 3, 16006. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Jenkins, Y.; Zabkiewicz, J.; Ottmann, O.; Jones, N. Tinkering under the Hood: Metabolic Optimisation of CAR-T Cell Therapy. Antibodies 2021, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Pianou, N.K.; Stavrou, P.Z.; Vlontzou, E.; Rondogianni, P.; Exarhos, D.N.; Datseris, I.E. More advantages in detecting bone and soft tissue metastases from prostate cancer using (18)F-PSMA PET/CT. Hell. J. Nucl. Med. 2019, 22, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Kessel, K.; Bernemann, C.; Bogemann, M.; Rahbar, K. Evolving Castration Resistance and Prostate Specific Membrane Antigen Expression: Implications for Patient Management. Cancers 2021, 13, 3556. [Google Scholar] [CrossRef]
- Chang, S.S. Overview of prostate-specific membrane antigen. Rev. Urol. 2004, 6 (Suppl. 10), S13–S18. [Google Scholar]
- Van de Wiele, C.; Sathekge, M.; de Spiegeleer, B.; De Jonghe, P.J.; Debruyne, P.R.; Borms, M.; Beels, L.; Maes, A. PSMA expression on neovasculature of solid tumors. Histol. Histopathol. 2020, 35, 919–927. [Google Scholar] [CrossRef]
- Hillerdal, V.; Essand, M. Chimeric antigen receptor-engineered T cells for the treatment of metastatic prostate cancer. BioDrugs 2015, 29, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Weimin, S.; Abula, A.; Qianghong, D.; Wenguang, W. Chimeric cytokine receptor enhancing PSMA-CAR-T cell-mediated prostate cancer regression. Cancer Biol. Ther. 2020, 21, 570–580. [Google Scholar] [CrossRef]
- Junghans, R.P.; Ma, Q.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate 2016, 76, 1257–1270. [Google Scholar] [CrossRef]
- Slovin, S.F.; Wang, X.; Hullings, M.; Arauz, G.; Bartido, S.; Lewis, J.S.; Schöder, H.; Zanzonico, P.; Scher, H.I.; Riviere, I. Chimeric antigen receptor (CAR+) modified T cells targeting prostate specific membrane antigen (PSMA) in patients (pts) with castrate metastatic prostate cancer (CMPC). J. Clin. Oncol. 2013, 31, 72. [Google Scholar] [CrossRef]
- Gu, Z.; Thomas, G.; Yamashiro, J.; Shintaku, I.P.; Dorey, F.; Raitano, A.; Witte, O.N.; Said, J.W.; Loda, M.; Reiter, R.E. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene 2000, 19, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Feng, F.; Wang, K.; Men, C.; Lin, C.; Liu, Q.; Yang, D.; Gao, Z. The therapeutic efficacy of I131-PSCA-mAb in orthotopic mouse models of prostate cancer. Eur. J. Med. Res. 2013, 18, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannull, J.; Diener, P.A.; Prikler, L.; Furstenberger, G.; Cerny, T.; Schmid, U.; Ackermann, D.K.; Groettrup, M. Prostate stem cell antigen is a promising candidate for immunotherapy of advanced prostate cancer. Cancer Res. 2000, 60, 5522–5528. [Google Scholar] [PubMed]
- Morello, A.; Sadelain, M.; Adusumilli, P.S. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016, 6, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Klampatsa, A.; Dimou, V.; Albelda, S.M. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert. Opin. Biol. Ther. 2021, 21, 473–486. [Google Scholar] [CrossRef]
- Lv, J.; Zhao, R.; Wu, D.; Zheng, D.; Wu, Z.; Shi, J.; Wei, X.; Wu, Q.; Long, Y.; Lin, S.; et al. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J. Hematol. Oncol. 2019, 12, 18. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, S.; Gagelmann, P.; Gorbokon, N.; Lennartz, M.; Menz, A.; Luebke, A.M.; Kluth, M.; Hube-Magg, C.; Blessin, N.C.; Fraune, C.; et al. Mesothelin Expression in Human Tumors: A Tissue Microarray Study on 12,679 Tumors. Biomedicines 2021, 9, 397. [Google Scholar] [CrossRef]
- Kachala, S.S.; Bograd, A.J.; Villena-Vargas, J.; Suzuki, K.; Servais, E.L.; Kadota, K.; Chou, J.; Sima, C.S.; Vertes, E.; Rusch, V.W.; et al. Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clin. Cancer Res. 2014, 20, 1020–1028. [Google Scholar] [CrossRef] [Green Version]
- Schoutrop, E.; El-Serafi, I.; Poiret, T.; Zhao, Y.; Gultekin, O.; He, R.; Moyano-Galceran, L.; Carlson, J.W.; Lehti, K.; Hassan, M.; et al. Mesothelin-Specific CAR T Cells Target Ovarian Cancer. Cancer Res. 2021, 81, 3022–3035. [Google Scholar] [CrossRef]
- Raghav, K.; Liu, S.; Overman, M.J.; Willett, A.F.; Knafl, M.; Fu, S.C.; Malpica, A.; Prasad, S.; Royal, R.E.; Scally, C.P.; et al. Efficacy, Safety, and Biomarker Analysis of Combined PD-L1 (Atezolizumab) and VEGF (Bevacizumab) Blockade in Advanced Malignant Peritoneal Mesothelioma. Cancer Discov. 2021, 11, 2738–2747. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A randomised, controlled, open-label, phase 3 trial. Lancet 2016, 387, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4, e126908. [Google Scholar] [CrossRef] [Green Version]
- Adusumilli, P.S.; Zauderer, M.G.; Riviere, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef] [PubMed]
- Saltos, A.; Khalil, F.; Smith, M.; Li, J.; Schell, M.; Antonia, S.J.; Gray, J.E. Clinical associations of mucin 1 in human lung cancer and precancerous lesions. Oncotarget 2018, 9, 35666–35675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, J.; Tang, M.; Li, G.; Chen, L.; Wang, Y.; Huang, Y. Expression of Mucin Family Proteins in Non-Small-Cell Lung Cancer and its Role in Evaluation of Prognosis. J. Oncol. 2022, 2022, 4181658. [Google Scholar] [CrossRef]
- Patel, U.; Abernathy, J.; Savani, B.N.; Oluwole, O.; Sengsayadeth, S.; Dholaria, B. CAR T cell therapy in solid tumors: A review of current clinical trials. EJHaem 2022, 3, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends. Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdanifar, M.; Roy, L.D.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front. Immunol. 2019, 10, 1149. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, N.; Li, Z.; Chang, A.; Chen, Y.; Zhao, T.; Li, Y.; Wang, X.; Zhang, W.; Wang, Z.; et al. Tumour-derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat. Commun. 2017, 8, 14035. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K.; Campbell, S.C.; Alexander, J.P.; Steinbach, F.; Edinger, M.G.; Tubbs, R.R.; Novick, A.C.; Klein, E.A. Molecular regulation of intercellular adhesion molecule 1 (ICAM-1) expression in renal cell carcinoma. Urol. Res. 1997, 25, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Taftaf, R.; Liu, X.; Singh, S.; Jia, Y.; Dashzeveg, N.K.; Hoffmann, A.D.; El-Shennawy, L.; Ramos, E.K.; Adorno-Cruz, V.; Schuster, E.J.; et al. ICAM1 initiates CTC cluster formation and trans-endothelial migration in lung metastasis of breast cancer. Nat. Commun. 2021, 12, 4867. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, Z.; Kuang, Y.; Wu, Z.; Zhao, S.; Zhang, Z.; Li, H.; Zheng, M.; Zhang, N.; Long, C.; et al. Intercellular Adhesion Molecule-1 as Target for CAR-T-Cell Therapy of Triple-Negative Breast. Cancer Front. Immunol. 2020, 11, 573823. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.J.; Zhou, D.D.; Yang, X.X.; Cui, B.; Tan, F.W.; Wang, J.; Li, K.; Shang, S.; Zhang, C.; Lv, X.X.; et al. TRIB3-EGFR interaction promotes lung cancer progression and defines a therapeutic target. Nat. Commun. 2020, 11, 3660. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Kumarakulasinghe, N.B.; Huang, Y.Q.; Ang, Y.L.E.; Choo, J.R.; Goh, B.C.; Soo, R.A. Third generation EGFR TKIs: Current data and future directions. Mol. Cancer 2018, 17, 29. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Chong, C.R.; Janne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhou, Y.; Huang, K.H.; Li, Y.; Fang, X.; An, L.; Wang, F.; Chen, Q.; Zhang, Y.; Shi, A.; et al. EGFR-specific CAR-T cells trigger cell lysis in EGFR-positive TNBC. Aging 2019, 11, 11054–11072. [Google Scholar] [CrossRef]
- Santos, E.D.S.; Nogueira, K.A.B.; Fernandes, L.C.C.; Martins, J.R.P.; Reis, A.V.F.; Neto, J.B.V.; Junior, I.; Pessoa, C.; Petrilli, R.; Eloy, J.O. EGFR targeting for cancer therapy: Pharmacology and immunoconjugates with drugs and nanoparticles. Int. J. Pharm. 2021, 592, 120082. [Google Scholar] [CrossRef]
- Diaz-Serrano, A.; Gella, P.; Jimenez, E.; Zugazagoitia, J.; Paz-Ares Rodriguez, L. Targeting EGFR in Lung Cancer: Current Standards and Developments. Drugs 2018, 78, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; De Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Huang, Y.; Jiang, D.Q.; Cui, L.Z.; He, Z.; Wang, C.; Zhang, Z.W.; Zhu, H.L.; Ding, Y.M.; Li, L.F.; et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell. Death. Dis. 2018, 9, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, Z.; Ding, Y.; Fang, Y.; Wang, P.; Chu, W.; Jin, Z.; Yang, X.; Wang, J.; Lou, J.; et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J. Cancer Res. Clin. Oncol. 2021, 147, 3725–3734. [Google Scholar] [CrossRef]
- Schiavone, G.; Epistolio, S.; Martin, V.; Molinari, F.; Barizzi, J.; Mazzucchelli, L.; Frattini, M.; Wannesson, L. Functional and clinical significance of ROR1 in lung adenocarcinoma. BMC Cancer 2020, 20, 1085, Erratum in BMC Cancer 2020, 20, 1196. [Google Scholar]
- Karachaliou, N.; Gimenez-Capitan, A.; Drozdowskyj, A.; Viteri, S.; Moran, T.; Carcereny, E.; Massuti, B.; Vergnenegre, A.; de Marinis, F.; Molina, M.A.; et al. ROR1 as a novel therapeutic target for EGFR-mutant non-small-cell lung cancer patients with the EGFR T790M mutation. Transl. Lung. Cancer Res. 2014, 3, 122–130. [Google Scholar] [CrossRef]
- Wallstabe, L.; Gottlich, C.; Nelke, L.C.; Kuhnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 2019, 4, e126345. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Zhu, H.; Zhang, S.; Yong, H.; Wang, W.; Zhou, Y.; Wang, B.; Wen, J.; Qiu, Z.; Ding, G.; et al. Trop2 is overexpressed in gastric cancer and predicts poor prognosis. Oncotarget 2016, 7, 6136–6145. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wu, W.; Togashi, Y.; Taira Nihira, N.; Johmura, Y.; Zhu, D.; Nakanishi, M.; Miyoshi, Y.; Ohta, T. Alteration of Trop-2 expression in breast cancer cells by clinically used therapeutic agents and acquired tamoxifen resistance. Breast Cancer 2022, 29, 1076–1087. [Google Scholar] [CrossRef]
- Zhao, W.; Kuai, X.; Zhou, X.; Jia, L.; Wang, J.; Yang, X.; Tian, Z.; Wang, X.; Lv, Q.; Wang, B.; et al. Trop2 is a potential biomarker for the promotion of EMT in human breast cancer. Oncol. Rep. 2018, 40, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Shvartsur, A.; Bonavida, B. Trop2 and its overexpression in cancers: Regulation and clinical/therapeutic implications. Genes Cancer 2015, 6, 84–105. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Jia, L.; Zhang, M.; Huang, X.; Qian, P.; Tang, Q.; Zhu, J.; Feng, Z. The killing effect of novel bi-specific Trop2/PD-L1 CAR-T cell targeted gastric cancer. Am. J. Cancer Res. 2019, 9, 1846–1856. [Google Scholar] [PubMed]
- Ponnusamy, M.P.; Venkatraman, G.; Singh, A.P.; Chauhan, S.C.; Johansson, S.L.; Jain, M.; Smith, L.; Davis, J.S.; Remmenga, S.W.; Batra, S.K. Expression of TAG-72 in ovarian cancer and its correlation with tumor stage and patient prognosis. Cancer Lett. 2007, 251, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Murad, J.P.; Kozlowska, A.K.; Lee, H.J.; Ramamurthy, M.; Chang, W.C.; Yazaki, P.; Colcher, D.; Shively, J.; Cristea, M.; Forman, S.J.; et al. Effective Targeting of TAG72(+) Peritoneal Ovarian Tumors via Regional Delivery of CAR-Engineered T Cells. Front. Immunol. 2018, 9, 2268. [Google Scholar] [CrossRef] [PubMed]
- Shu, R.; Evtimov, V.J.; Hammett, M.V.; Nguyen, N.N.; Zhuang, J.; Hudson, P.J.; Howard, M.C.; Pupovac, A.; Trounson, A.O.; Boyd, R.L. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol. Ther. Oncolytics 2021, 20, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Uemura, H.; Hirao, Y.; Yoshida, K.; Saga, S.; Yoshikawa, K. Radiation hybrid mapping of the human MN/CA9 locus to chromosome band 9p12-p13. Genomics 1998, 53, 118–119. [Google Scholar] [CrossRef]
- Courcier, J.; de la Taille, A.; Nourieh, M.; Leguerney, I.; Lassau, N.; Ingels, A. Carbonic Anhydrase IX in Renal Cell Carcinoma, Implications for Disease Management. Int. J. Mol. Sci. 2020, 21, 7146. [Google Scholar] [CrossRef]
- Genega, E.M.; Ghebremichael, M.; Najarian, R.; Fu, Y.; Wang, Y.; Argani, P.; Grisanzio, C.; Signoretti, S. Carbonic anhydrase IX expression in renal neoplasms: Correlation with tumor type and grade. Am. J. Clin. Pathol. 2010, 134, 873–879. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Liao, G.; Li, Y.; Zhou, S.; Zou, H.; Fernando, S. Prognostic value of carbonic anhydrase IX immunohistochemical expression in renal cell carcinoma: A meta-analysis of the literature. PLoS ONE 2014, 9, e114096. [Google Scholar] [CrossRef]
- Hsin, M.C.; Hsieh, Y.H.; Hsiao, Y.H.; Chen, P.N.; Wang, P.H.; Yang, S.F. Carbonic Anhydrase IX Promotes Human Cervical Cancer Cell Motility by Regulating PFKFB4 Expression. Cancers 2021, 13, 1174. [Google Scholar] [CrossRef]
- Chiche, J.; Brahimi-Horn, M.C.; Pouyssegur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, R.; Yang, H.; Lu, L.; Lin, L. Tiliroside as a CAXII inhibitor suppresses liver cancer development and modulates E2Fs/Caspase-3 axis. Sci. Rep. 2021, 11, 8626. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ding, J.; Lu, M.; Liu, H.; Miao, Y.; Li, L.; Wang, G.; Zheng, J.; Pei, D.; Zhang, Q. CAIX-specific CAR-T Cells and Sunitinib Show Synergistic Effects Against Metastatic Renal Cancer Models. J. Immunother. 2020, 43, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gong, P.; Li, J.; Fu, Y.; Zhou, Z.; Liu, L. Role of CD133 in human embryonic stem cell proliferation and teratoma formation. Stem. Cell. Res. Ther. 2020, 11, 208. [Google Scholar] [CrossRef]
- Ehteram, H.; Aslanbeigi, F.; Ghoochani Khorasani, E.; Tolouee, M.; Haddad Kashani, H. Expression and Prognostic Significance of Stem Cell Marker CD133 in Survival Rate of Patients with Colon Cancer. Oncol. Ther. 2022, 10, 451–461. [Google Scholar] [CrossRef]
- Nomura, A.; Banerjee, S.; Chugh, R.; Dudeja, V.; Yamamoto, M.; Vickers, S.M.; Saluja, A.K. CD133 initiates tumors, induces epithelial-mesenchymal transition and increases metastasis in pancreatic cancer. Oncotarget 2015, 6, 8313–8322. [Google Scholar] [CrossRef] [Green Version]
- Tsunekuni, K.; Konno, M.; Haraguchi, N.; Koseki, J.; Asai, A.; Matsuoka, K.; Kobunai, T.; Takechi, T.; Doki, Y.; Mori, M.; et al. CD44/CD133-Positive Colorectal Cancer Stem Cells are Sensitive to Trifluridine Exposure. Sci. Rep. 2019, 9, 14861. [Google Scholar] [CrossRef] [Green Version]
- Brugnoli, F.; Grassilli, S.; Al-Qassab, Y.; Capitani, S.; Bertagnolo, V. CD133 in Breast Cancer Cells: More than a Stem Cell Marker. J. Oncol. 2019, 2019, 7512632. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Yang, H.; Ling, C.; Lu, L. Neurospora crassa is a potential source of anti-cancer agents against breast cancer. Breast Cancer 2022, 29, 1032–1041. [Google Scholar] [CrossRef]
- Bueno, C.; Velasco-Hernandez, T.; Gutierrez-Aguera, F.; Zanetti, S.R.; Baroni, M.L.; Sanchez-Martinez, D.; Molina, O.; Closa, A.; Agraz-Doblas, A.; Marin, P.; et al. CD133-directed CAR T-cells for MLL leukemia: On-target, off-tumor myeloablative toxicity. Leukemia 2019, 33, 2090–2125. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reader, C.S.; Vallath, S.; Steele, C.W.; Haider, S.; Brentnall, A.; Desai, A.; Moore, K.M.; Jamieson, N.B.; Chang, D.; Bailey, P.; et al. The integrin alphavbeta6 drives pancreatic cancer through diverse mechanisms and represents an effective target for therapy. J. Pathol. 2019, 249, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Qian, L.; Cui, J. Focused evaluation of the roles of macrophages in chimeric antigen receptor (CAR) T cell therapy associated cytokine release syndrome. Cancer Biol. Med. 2021, 19, 333–342. [Google Scholar] [CrossRef]
- Brown, M.P.; Ebert, L.M.; Gargett, T. Clinical chimeric antigen receptor-T cell therapy: A new and promising treatment modality for glioblastoma. Clin. Transl. Immunol. 2019, 8, e1050. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Titov, A.; Petukhov, A.; Staliarova, A.; Motorin, D.; Bulatov, E.; Shuvalov, O.; Soond, S.M.; Piacentini, M.; Melino, G.; Zaritskey, A.; et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018, 9, 897. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Zhang, Z.; Ren, Z.; Li, Y. Reactions Related to CAR-T Cell Therapy. Front. Immunol. 2021, 12, 663201. [Google Scholar] [CrossRef]
- Lerkvaleekul, B.; Vilaiyuk, S. Macrophage activation syndrome: Early diagnosis is key. Open Access Rheumatol. 2018, 10, 117–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Rojas, R.M.; Gomez-Centurion, I.; Bailen, R.; Bastos, M.; Diaz-Crespo, F.; Carbonell, D.; Correa-Rocha, R.; Pion, M.; Munoz, C.; Sancho, M.; et al. Hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS) following treatment with tisagenlecleucel. Clin. Case. Rep. 2022, 10, e05209. [Google Scholar] [CrossRef]
- Drent, E.; Themeli, M.; Poels, R.; de Jong-Korlaar, R.; Yuan, H.; de Bruijn, J.; Martens, A.C.M.; Zweegman, S.; van de Donk, N.; Groen, R.W.J.; et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef]
- Donnadieu, E.; Dupre, L.; Pinho, L.G.; Cotta-de-Almeida, V. Surmounting the obstacles that impede effective CAR T cell trafficking to solid tumors. J. Leukoc. Biol. 2020, 108, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Andrea, A.E.; Chiron, A.; Mallah, S.; Bessoles, S.; Sarrabayrouse, G.; Hacein-Bey-Abina, S. Advances in CAR-T Cell Genetic Engineering Strategies to Overcome Hurdles in Solid Tumors Treatment. Front. Immunol. 2022, 13, 830292. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.; Zhan, X.; Zhang, D.; Jain, R.; Wang, K.W.; Choi, J.H.; Misawa, T.; Su, L.; Quan, J.; Hildebrand, S.; et al. SLFN2 protection of tRNAs from stress-induced cleavage is essential for T cell-mediated immunity. Science 2021, 372, eaba4220. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Bai, Y.; Wang, Z. Elevated T cell activation score is associated with improved survival of breast cancer. Breast Cancer Res. Treat. 2017, 164, 689–696. [Google Scholar] [CrossRef]
- Shao, S.; Risch, E.; Burner, D.; Lu, L.; Minev, B.; Ma, W. IFNgamma enhances cytotoxic efficiency of the cytotoxic T lymphocytes against human glioma cells. Int. Immunopharmacol. 2017, 47, 159–165. [Google Scholar] [CrossRef]
- Philipp, N.; Kazerani, M.; Nicholls, A.; Vick, B.; Wulf, J.; Straub, T.; Scheurer, M.; Muth, A.; Hanel, G.; Nixdorf, D.; et al. T-cell exhaustion induced by continuous bispecific molecule exposure is ameliorated by treatment-free intervals. Blood 2022, 140, 1104–1118. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, Y.; Hu, Y.; Mei, H. T Cell Exhaustion and CAR-T Immunotherapy in Hematological Malignancies. Biomed. Res. Int. 2021, 2021, 6616391. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Woodhouse, S.; Zhao, Z.; Arya, R.; Govek, K.; Kim, D.; Lundh, S.; Baysoy, A.; Sun, H.; Deng, Y.; et al. Single-cell antigen-specific landscape of CAR T infusion product identifies determinants of CD19-positive relapse in patients with ALL. Sci. Adv. 2022, 8, eabj2820. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.; Oluwole, O.O.; Savani, B.; Majhail, N.S.; Hill, B.T.; Locke, F.L. Understanding and Managing Large B Cell Lymphoma Relapses after Chimeric Antigen Receptor T Cell Therapy. Biol. Blood Marrow. Transpl. 2019, 25, e344–e351. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, N.; Smith, M. Custom CARs: Leveraging the Adaptability of Allogeneic CAR Therapies to Address Current Challenges in Relapsed/Refractory DLBCL. Front. Immunol. 2022, 13, 887866. [Google Scholar] [CrossRef] [PubMed]
- Cherkassky, L.; Hou, Z.; Amador-Molina, A.; Adusumilli, P.S. Regional CAR T cell therapy: An ignition key for systemic immunity in solid tumors. Cancer Cell. 2022, 40, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Han, B.S.; Ji, S.; Woo, S.; Lee, J.H.; Sin, J.I. Regulation of the translation activity of antigen-specific mRNA is responsible for antigen loss and tumor immune escape in a HER2-expressing tumor model. Sci. Rep. 2019, 9, 2855. [Google Scholar] [CrossRef] [Green Version]
- Lemoine, J.; Ruella, M.; Houot, R. Born to survive: How cancer cells resist CAR T cell therapy. J. Hematol. Oncol. 2021, 14, 199. [Google Scholar] [CrossRef]
- Feng, Z.; He, X.; Zhang, X.; Wu, Y.; Xing, B.; Knowles, A.; Shan, Q.; Miller, S.; Hojnacki, T.; Ma, J.; et al. Potent suppression of neuroendocrine tumors and gastrointestinal cancers by CDH17CAR T cells without toxicity to normal tissues. Nat. Cancer 2022, 3, 581–594. [Google Scholar] [CrossRef]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology 2018, 20, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Cordoba, S.; Onuoha, S.; Thomas, S.; Pignataro, D.S.; Hough, R.; Ghorashian, S.; Vora, A.; Bonney, D.; Veys, P.; Rao, K.; et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: A phase 1 trial. Nat. Med. 2021, 27, 1797–1805. [Google Scholar] [CrossRef]
- Park, J.H.; Geyer, M.B.; Brentjens, R.J. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: Interpreting clinical outcomes to date. Blood 2016, 127, 3312–3320. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cao, J.; Gu, W.; Shi, M.; Lan, J.; Yan, Z.; Jin, L.; Xia, J.; Ma, S.; Liu, Y.; et al. Long-Term Follow-Up of Combination of B-Cell Maturation Antigen and CD19 Chimeric Antigen Receptor T Cells in Multiple Myeloma. J. Clin. Oncol. 2022, 40, 2246–2256. [Google Scholar] [CrossRef]
- Jiang, L.; Li, X.P.; Dai, Y.T.; Chen, B.; Weng, X.Q.; Xiong, S.M.; Zhang, M.; Huang, J.Y.; Chen, Z.; Chen, S.J. Multidimensional study of the heterogeneity of leukemia cells in t(8;21) acute myelogenous leukemia identifies the subtype with poor outcome. Proc. Natl. Acad. Sci. USA 2020, 117, 20117–20126. [Google Scholar] [CrossRef]
- Liu, J.; Tong, J.; Yang, H. Targeting CD33 for acute myeloid leukemia therapy. BMC Cancer 2022, 22, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gan, W.T.; Hao, W.G.; Wang, P.F.; Li, Z.Y.; Chang, L.J. Successful Anti-CLL1 CAR T-Cell Therapy in Secondary Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 685. [Google Scholar] [CrossRef] [PubMed]
- Rath, J.A.; Arber, C. Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy. Cells 2020, 9, 1485. [Google Scholar] [CrossRef]
- Edeline, J.; Houot, R.; Marabelle, A.; Alcantara, M. CAR-T cells and BiTEs in solid tumors: Challenges and perspectives. J. Hematol. Oncol. 2021, 14, 65. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Cao, W.; Wang, F.; Xie, X.; Li, Y.; Wang, X.; Guo, R.; Jiang, Z.; Guo, R. Single-Cell Analysis of Target Antigens of CAR-T Reveals a Potential Landscape of “On-Target, Off-Tumor Toxicity”. Front. Immunol. 2021, 12, 799206. [Google Scholar] [CrossRef]
- Li, W.; Amei, A.; Bui, F.; Norouzifar, S.; Lu, L.; Wang, Z. Impact of Neoantigen Expression and T-Cell Activation on Breast Cancer Survival. Cancers 2021, 13, 2879. [Google Scholar] [CrossRef]
- Felsberg, J.; Hentschel, B.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Malzkorn, B.; Kamp, M.; Sabel, M.; Simon, M.; Westphal, M.; et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin. Cancer Res. 2017, 23, 6846–6855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durgin, J.S.; Henderson, F., Jr.; Nasrallah, M.P.; Mohan, S.; Wang, S.; Lacey, S.F.; Melenhorst, J.J.; Desai, A.S.; Lee, J.Y.K.; Maus, M.V.; et al. Case Report: Prolonged Survival Following EGFRvIII CAR T Cell Treatment for Recurrent Glioblastoma. Front. Oncol. 2021, 11, 669071. [Google Scholar] [CrossRef] [PubMed]
- Supimon, K.; Sangsuwannukul, T.; Sujjitjoon, J.; Phanthaphol, N.; Chieochansin, T.; Poungvarin, N.; Wongkham, S.; Junking, M.; Yenchitsomanus, P.T. Anti-mucin 1 chimeric antigen receptor T cells for adoptive T cell therapy of cholangiocarcinoma. Sci. Rep. 2021, 11, 6276. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yang, X.; Fan, J.; Du, X. Claudin 6: Therapeutic prospects for tumours, and mechanisms of expression and regulation (Review). Mol. Med. Rep. 2021, 24, 677. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Jin, Q.; Quan, C. CLDN6: From Traditional Barrier Function to Emerging Roles in Cancers. Int. J. Mol. Sci. 2021, 22, 13416. [Google Scholar] [CrossRef]
- Haanen, J.B.; Mackensen, A.; Koenecke, C.; Alsdorf, W.; Wagner-Drouet, E.; Heudobler, D.; Borchmann, P.; Bokemeyer, C.; Klobuch, S.; Desuki, A.; et al. A Phase I trial to evaluate safety and efficacy of CLDN6 CAR-T cells and CARVac-mediated in vivo expansion in patients with CLDN6-positive advanced solid tumors. Cancer Res. 2022, 82, CT002. [Google Scholar] [CrossRef]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: Results of two phase 1 studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- DiNofia, A.M.; Grupp, S.A. Will allogeneic CAR T cells for CD19(+) malignancies take autologous CAR T cells ‘off the shelf’? Nat. Rev. Clin. Oncol. 2021, 18, 195–196. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Barsan, V.; Mackall, C. Prospects and challenges for use of CAR T cell therapies in solid tumors. Expert. Opin. Biol. Ther. 2020, 20, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.; Rosenblatt, J.; Raje, N.; Cook, D.; Gaballa, M.; Emmanuel-Alejandro, E.; Cornwell, C.; Banerjee, K.; Rotte, A.; Heery, C.; et al. 620O CART-ddBCMA for multiple myeloma: Interim results from phase I study. Ann. Oncol. 2022, 33, S828. [Google Scholar] [CrossRef]
- Martinez Bedoya, D.; Dutoit, V.; Migliorini, D. Allogeneic CAR T Cells: An Alternative to Overcome Challenges of CAR T Cell Therapy in Glioblastoma. Front. Immunol. 2021, 12, 640082. [Google Scholar] [CrossRef] [PubMed]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- Lanna, A.; Vaz, B.; D’Ambra, C.; Valvo, S.; Vuotto, C.; Chiurchiu, V.; Devine, O.; Sanchez, M.; Borsellino, G.; Akbar, A.N.; et al. An intercellular transfer of telomeres rescues T cells from senescence and promotes long-term immunological memory. Nat. Cell. Biol. 2022, 24, 1461–1474. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generic Name (Rx) | Brand Name | Target Antigen | Targeted Diseases | Indications (Patient Population) | Manufacturers | Time FDA Approved | Cost of One-Time Infusion |
|---|---|---|---|---|---|---|---|
| Tisagenlecleucel | Kymriah | CD19 | B-cell acute lymphoblastic leukemia (ALL) | Children and young adults with R/R B-cell ALL | Novartis | 30 August 2017 | USD 475,000 |
| B-cell non-Hodgkin lymphoma (NHL) | Adults with R/R B cell NHL | ||||||
| R/R follicular lymphoma (FL) | Adults with R/R FL | 28 May 2022 | |||||
| Axicabtagene | Yescarta | CD19 | B-cell non-Hodgkin lymphoma (NHL) | Adults with R/R B cell NHL | Kite Pharma | 18 October 2017 | USD 373,000 |
| Follicular lymphoma (FL) | Adults with R/R FL | ||||||
| Brexucabtagene | Tecartus | CD19 | Mantle cell lymphoma (MCL) | Adults with R/R MCL | Kite Pharma | 1 October 2021 | USD 373,000 |
| B-cell acute lymphoblastic leukemia (ALL) | Adults with R/R B-ALL | ||||||
| Lisocabtagene maraleucel | Breyanzi | CD19 | B-cell non-Hodgkin lymphoma (NHL) | Adults with R/R large B cell NHL | Bristol Myers Squibb | 5 February 2021 | USD 432,055 |
| Axicabtagene ciloleucel | Yescarta | CD19 | R/R follicular lymphoma (FL) | Adults with DLBCL | Kite Pharma | 5 March 2021 1 April 2022 | USD 373,000 |
| Ciltacabtagene autoleucel | Carvykti | BCMA | Multiple Myeloma (MM) | Adults with R/R MM | Janssen Biotech | 28 February 2022 | USD 465,000 |
| Idecabtagene vicleucel | Abecma | Bristol Myers Squibb | 27 March 2021 | USD 419,500 |
| Antigen | Cancer | Phase | Identifier (ID) |
| EGFR | Lung, colorectal, ovarian, pancreatic, renal cancers | Phase 1/2 | NCT01869166 |
| HER2 | Central nervous system tumor, pediatric glioma | Phase 1 | NCT03500991 |
| EGFR806 | Central nervous system tumor, pediatric glioma | Phase 1 | NCT03638167 |
| Mesothelin | Ovarian, cervical, pancreatic, lung | Phase 1/2 | NCT01583686 |
| PSCA | Lung | Phase 1 | NCT03198052 |
| MUC1 | Advanced solid tumors, lung | Phase 1/2 | NCT03179007, NCT03525782 |
| Claudin 18.2 | Advanced solid tumor | Phase 1 | NCT03874897 |
| EpCAM | Colon, pancreatic, prostate, gastric, liver | Phase 1/2 | NCT03013712 |
| GD2 | Brain | Phase 1 | NCT04099797 |
| VEGFR2 | Melanoma, brain | Phase 1 | NCT01218867 |
| AFP | Hepatocellular carcinoma liver cancer | Phase 1 | NCT03349255 |
| Nectin4/FAP | Nectin4-positive advanced malignant solid tumor | Phase 1 | NCT03932565 |
| CEA | Lung, colorectal, gastric, breast, pancreatic cancer | Phase 1 | NCT02349724 |
| Lewis Y | Advanced cancer | Phase 1 | NCT03851146 |
| Glypican-3 | Liver | Phase 1 | NCT02932956 |
| EGFRvIII | Glioblastoma and brain tumor | Phase 1 | NCT01454596 |
| IL-13Rα2 | Glioblastoma | Phase 1 | NCT02208362 |
| CD171 | Neuroblastoma | Phase 1 | NCT02311621 |
| MUC16 (CA-125) | Ovarian | Phase 1 | NCT 02498912 |
| PSMA | Prostate | Phase 1 | NCT01140373 |
| AFP | Hepatocellular carcinoma, liver | Phase 1 | NCT03349255 |
| AXL | Renal | Phase 1 | NCT03393936 |
| CD20 | Melanoma | Phase 1 | NCT03893019 |
| CD80/86 | Lung | Phase 1 | NCT03060343 |
| c-MET | Breast, hepatocellular | Phase 1 | NCT03060356, NCT03672305 |
| DLL-3 | Lung | Phase 1 | NCT03392064 |
| DR5 | Hepatoma | Phase 1 | NCT03638206 |
| EphA2 | Glioma | Phase 1 | NCT02575261 |
| TAG72 | Ovarian | Phase 1 | NCT05225363 |
| gp100 | Melanoma | Phase 1 | NCT03649529 |
| MAGE-A1/3/4 | Lung | Phase 1 | NCT03356808, NCT03535246 |
| LMP1 | Nasopharyngeal | Phase 1 | NCT02980315 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Lu, L.; Ma, W. Efficacy, Safety, and Challenges of CAR T-Cells in the Treatment of Solid Tumors. Cancers 2022, 14, 5983. https://doi.org/10.3390/cancers14235983
Chen Q, Lu L, Ma W. Efficacy, Safety, and Challenges of CAR T-Cells in the Treatment of Solid Tumors. Cancers. 2022; 14(23):5983. https://doi.org/10.3390/cancers14235983
Chicago/Turabian StyleChen, Qiuqiang, Lingeng Lu, and Wenxue Ma. 2022. "Efficacy, Safety, and Challenges of CAR T-Cells in the Treatment of Solid Tumors" Cancers 14, no. 23: 5983. https://doi.org/10.3390/cancers14235983