
ACAP1 Deficiency Predicts Inferior Immunotherapy Response in Solid Tumors

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. ACAP1 Expression in Tissues and Cell Lines
2.2. TCGA Datasets
2.3. Single-Cell Sequencing Datasets
2.4. Immunotherapy Datasets
2.5. Other Datasets
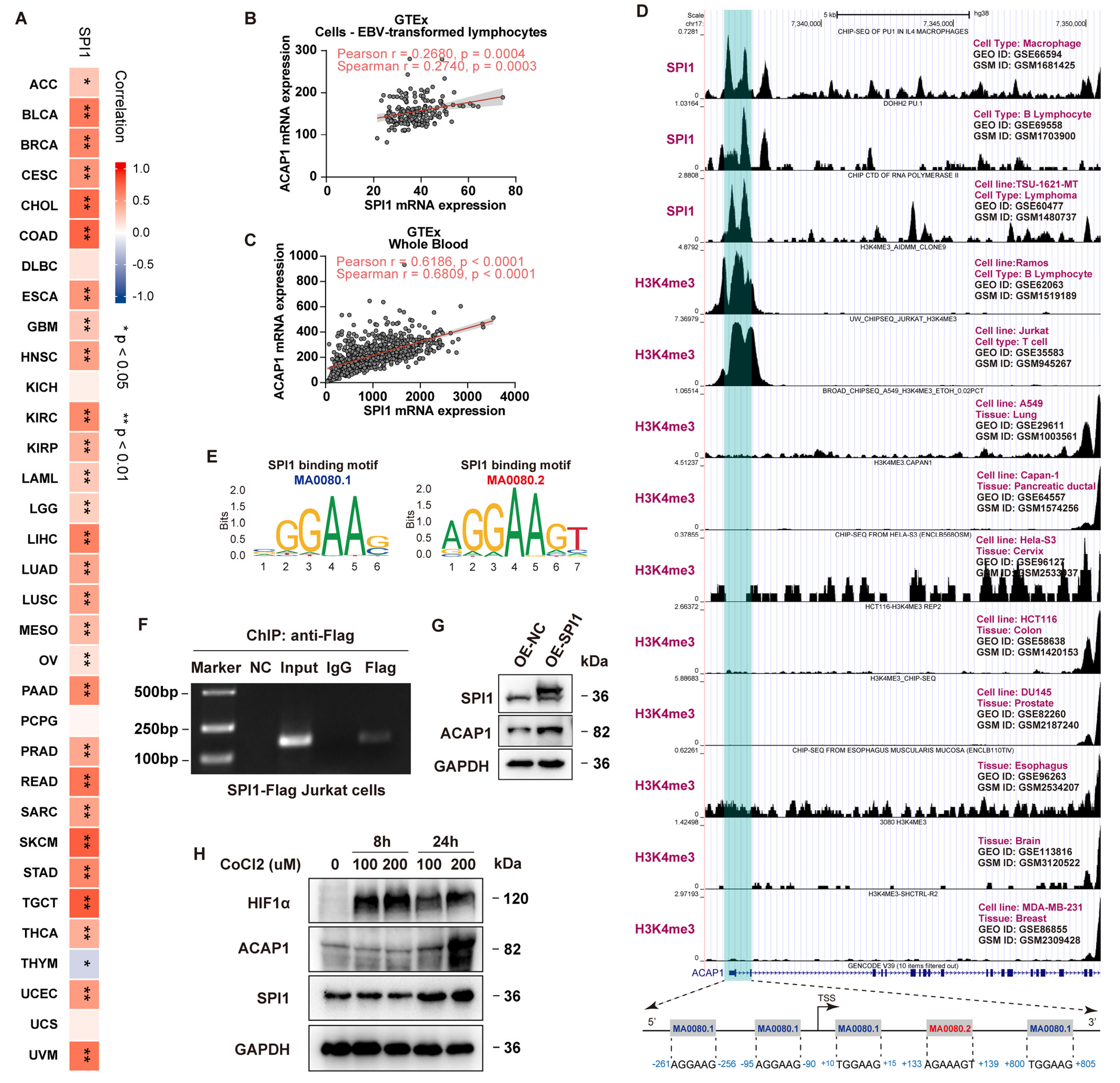
2.6. ChIP-Sequencing Analysis and JASPAR Analysis
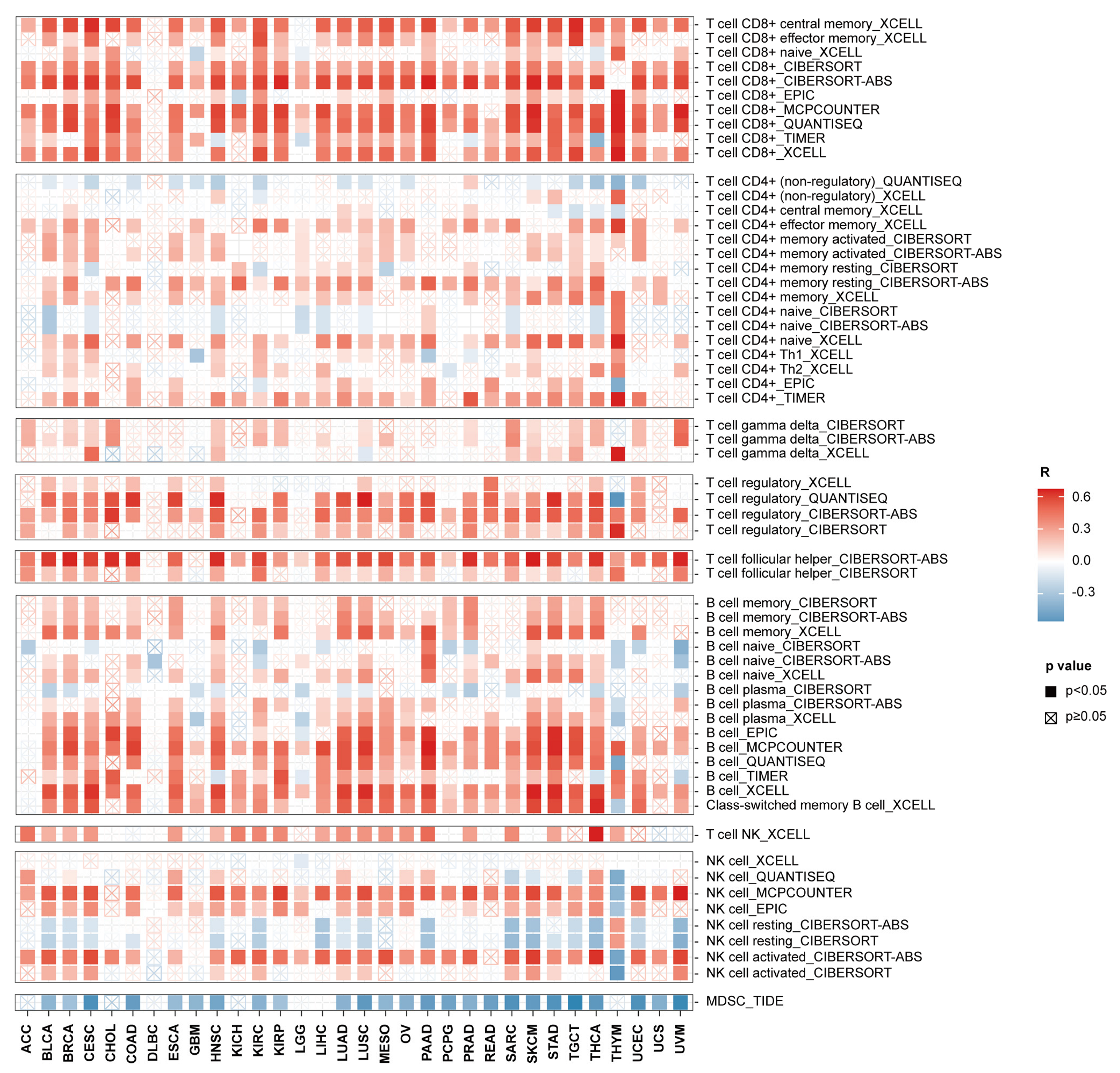
2.7. Calculation of Immune Cell Infiltration
2.8. Survival Analysis and Gene Set Enrichment Analysis (GSEA)
2.9. Cell Culture and Lentivirus Transfection
2.10. Cell Treatment
2.11. ChIP-PCR
2.12. qRT–PCR
2.13. Western Blotting
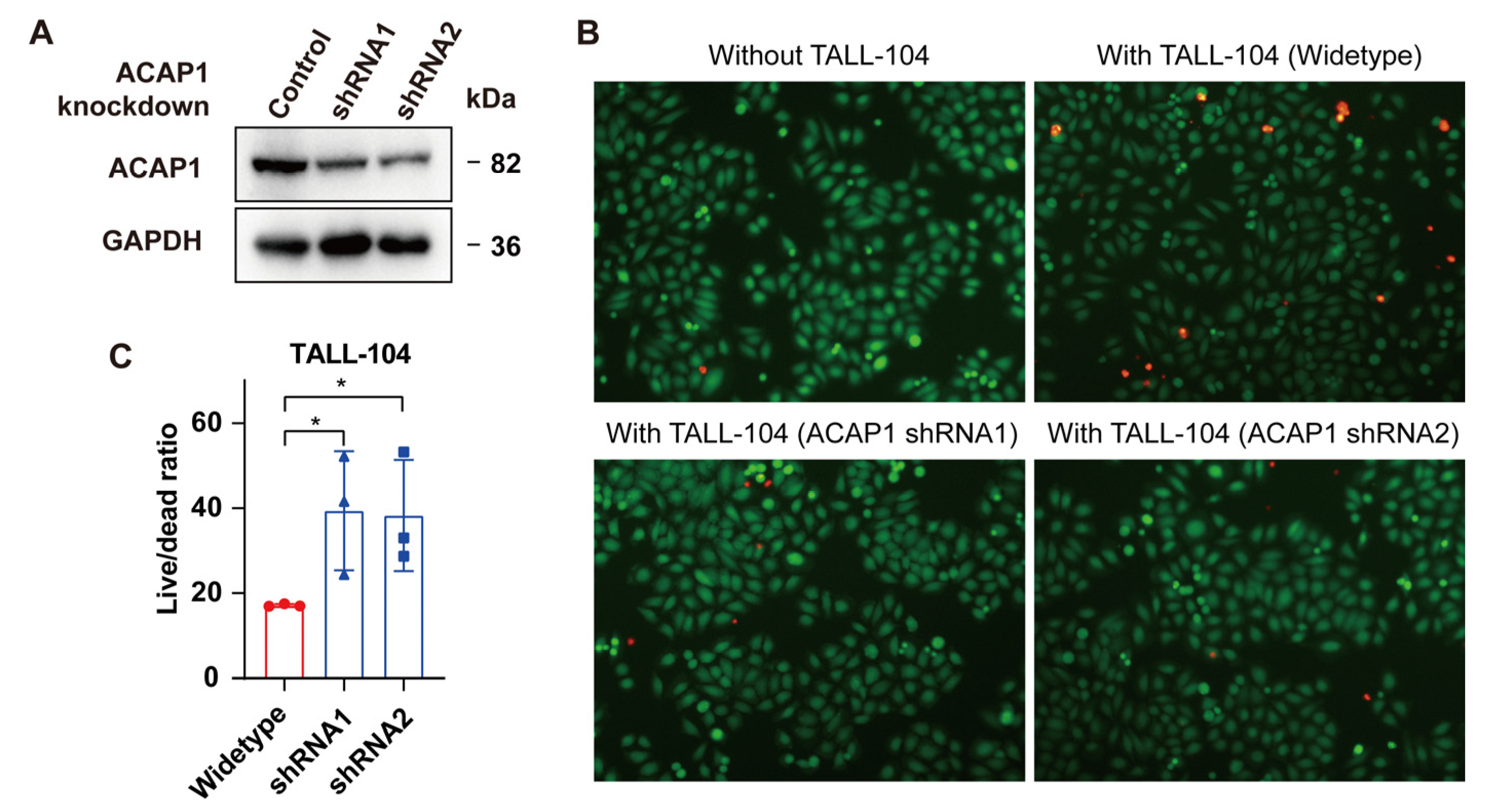
2.14. T-Cell Co-Culture Killing Assay
2.15. Statistical Analysis
3. Results
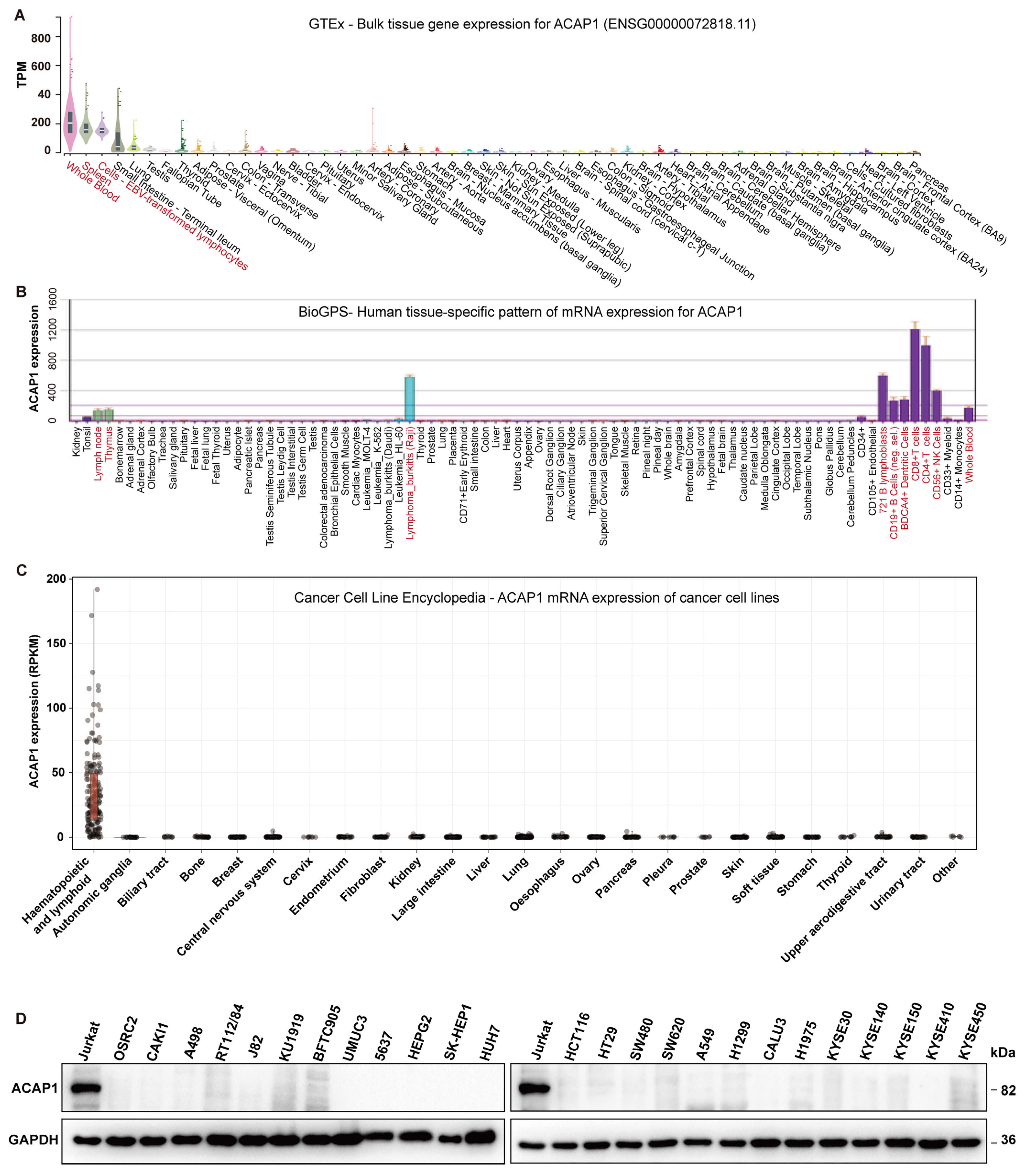
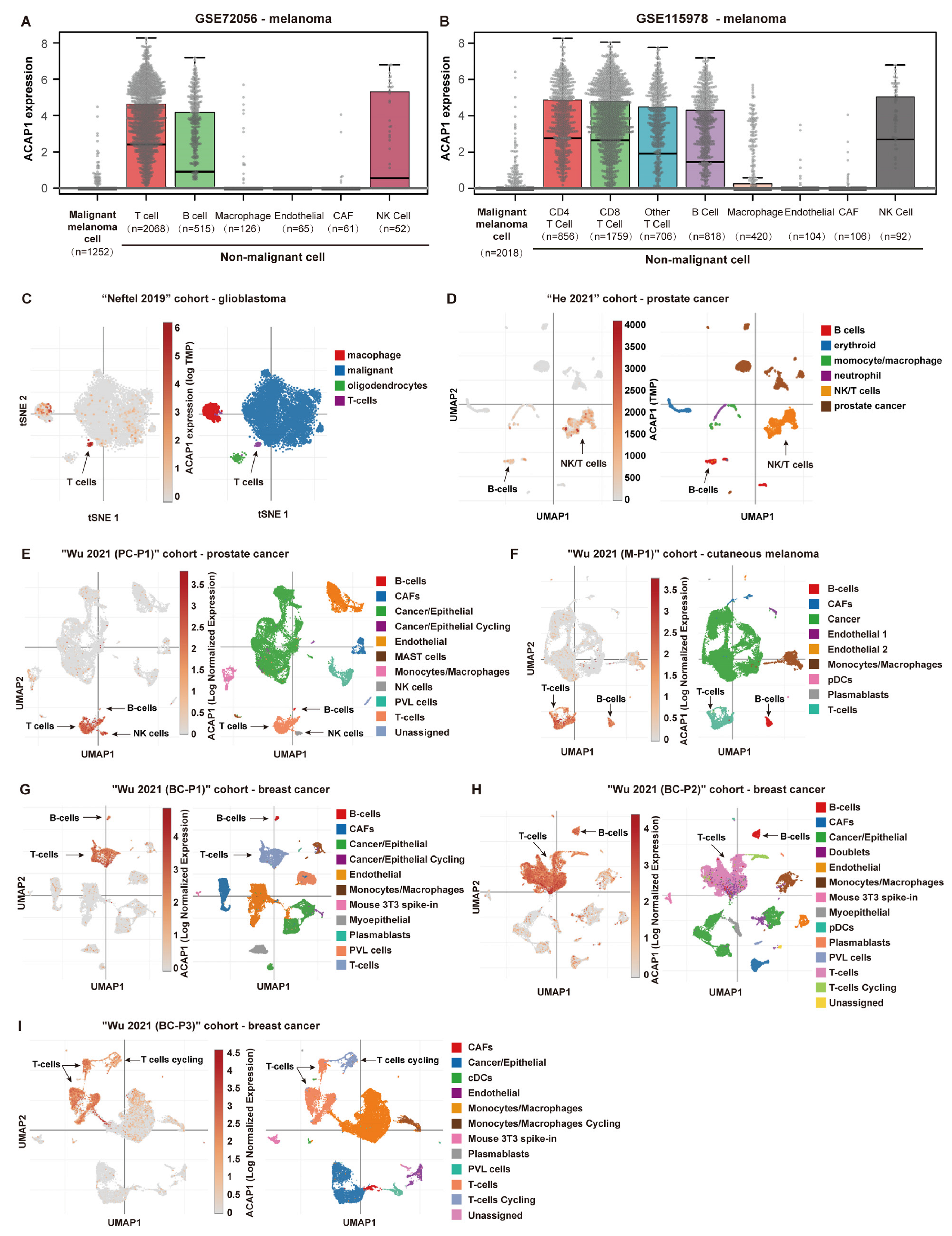
3.1. ACAP1 Is a Marker Gene for Lymphocytes
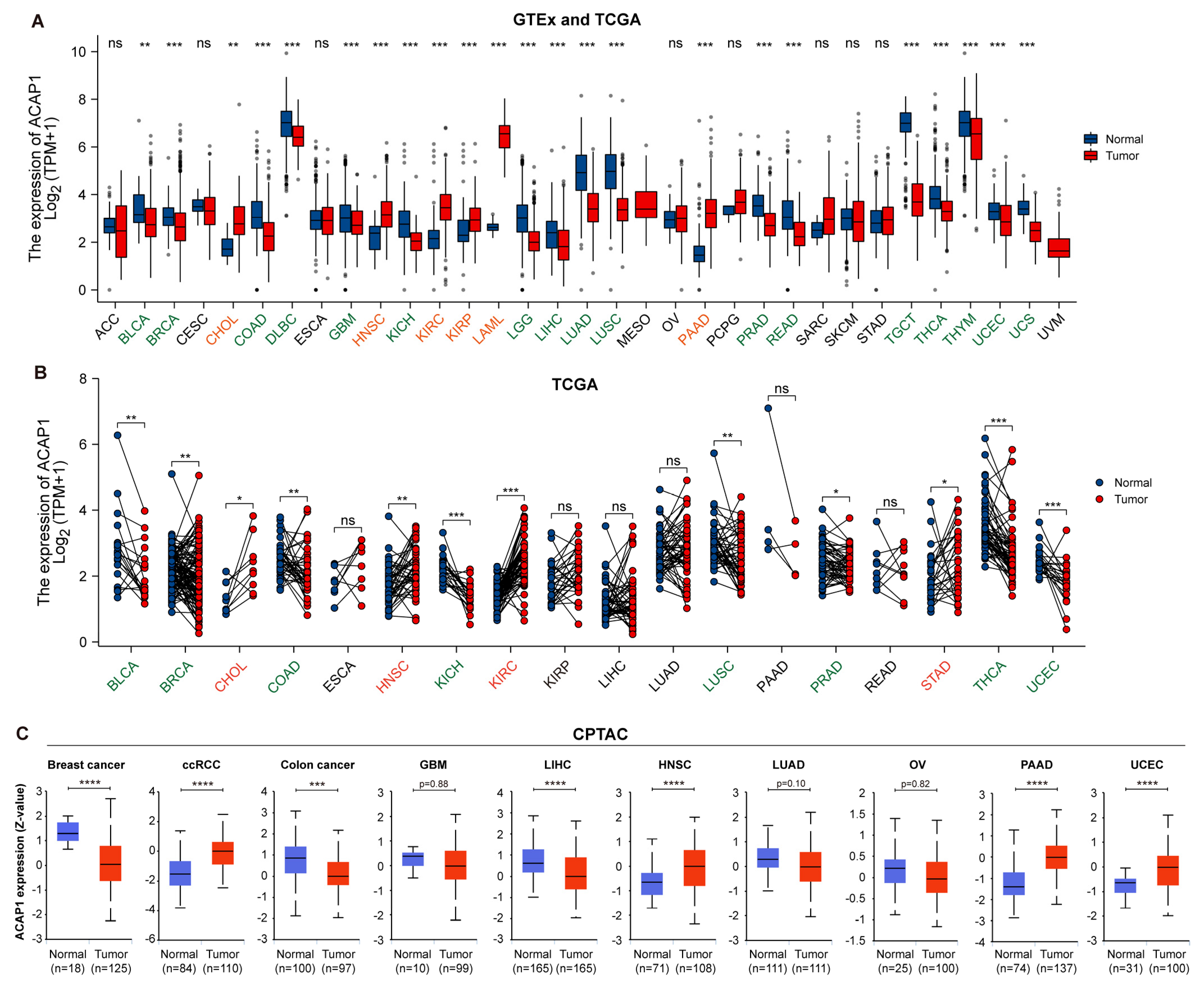
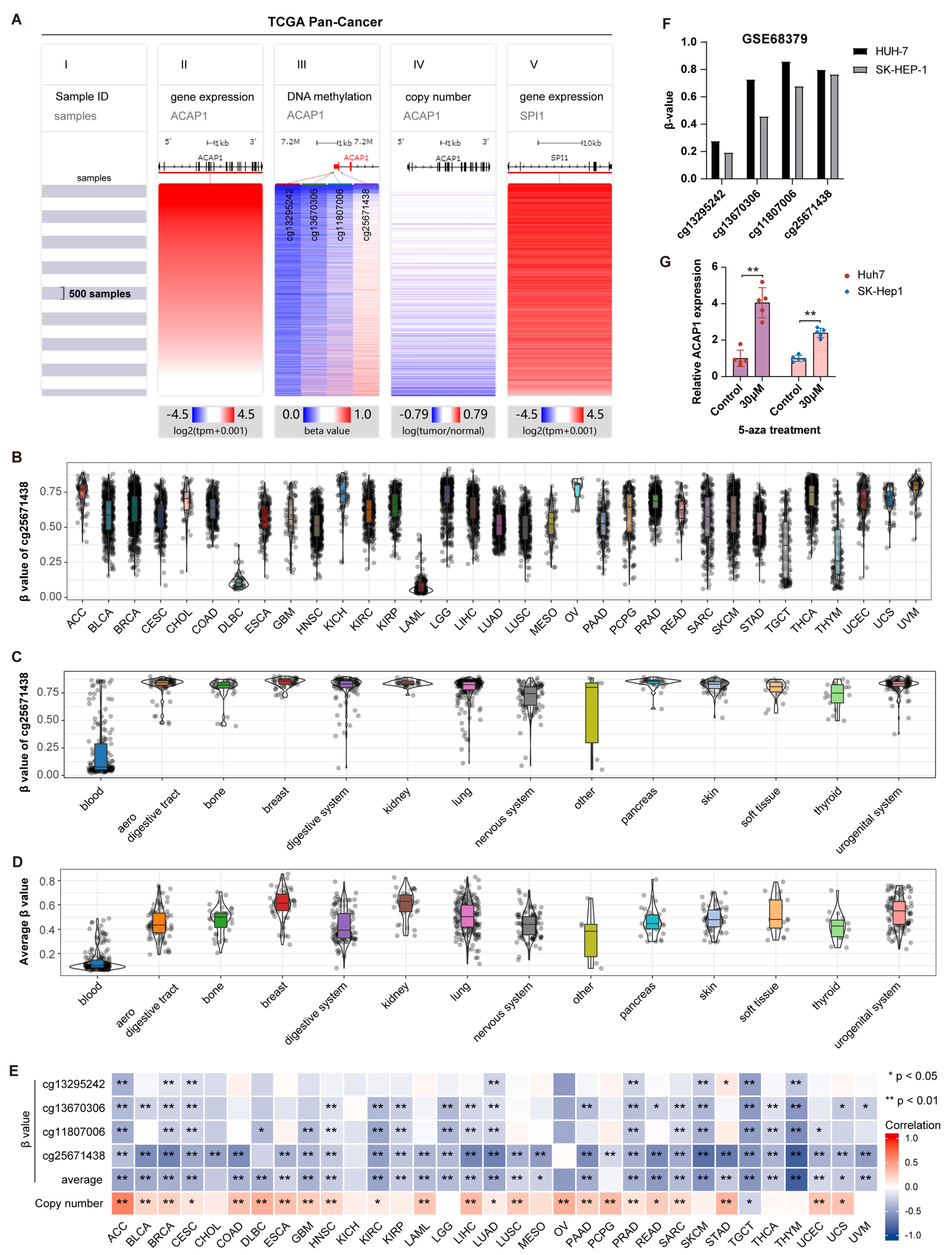
3.2. Pan-Cancer Expression Analysis of ACAP1
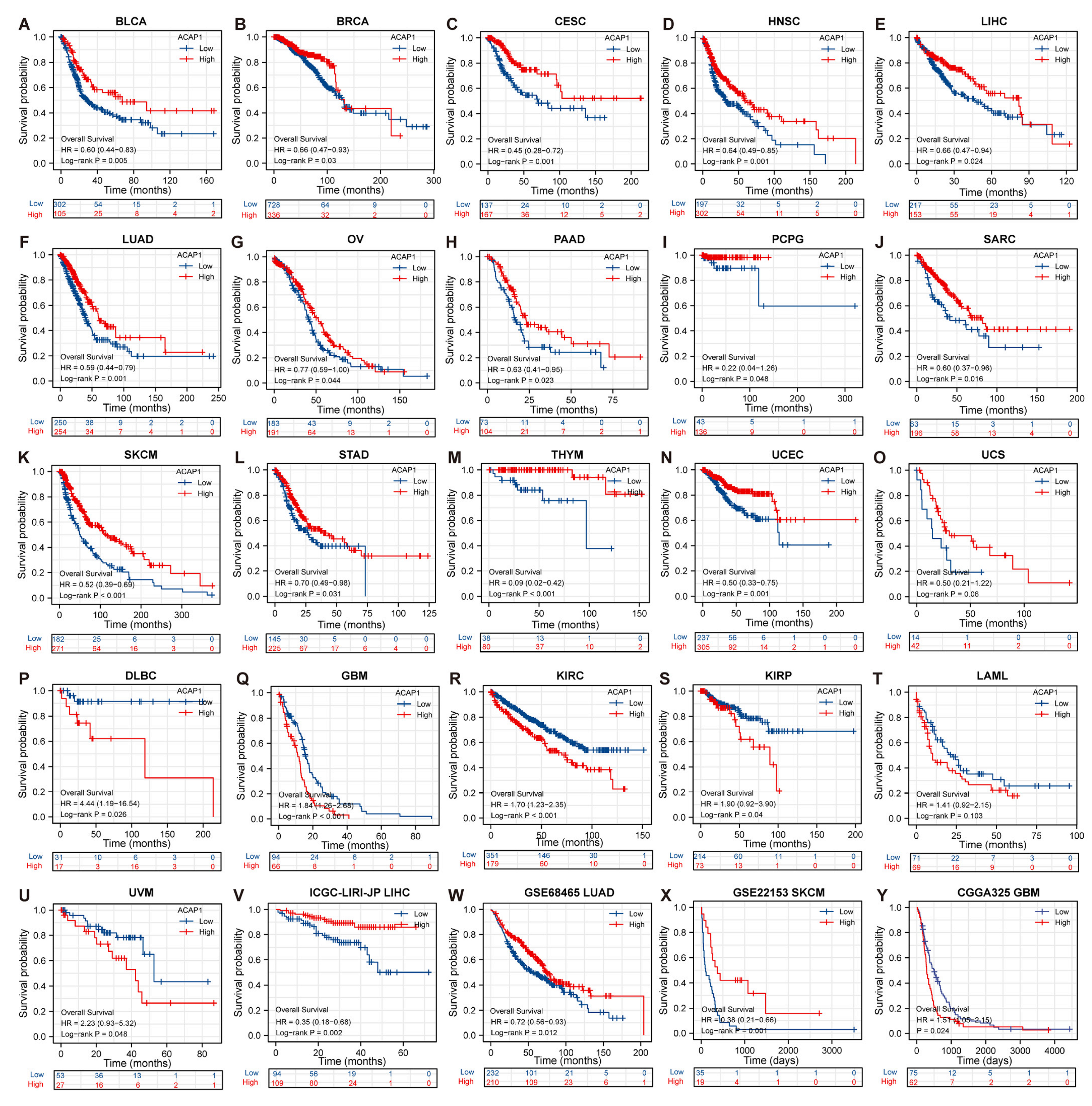
3.3. Prognostic Value of ACAP1 Expression
3.4. Transcriptional Regulation of ACAP1
3.5. ACAP1 Expression Is Positively Associated with TILs and Is Essential for the Cytotoxicity of Lymphocytes
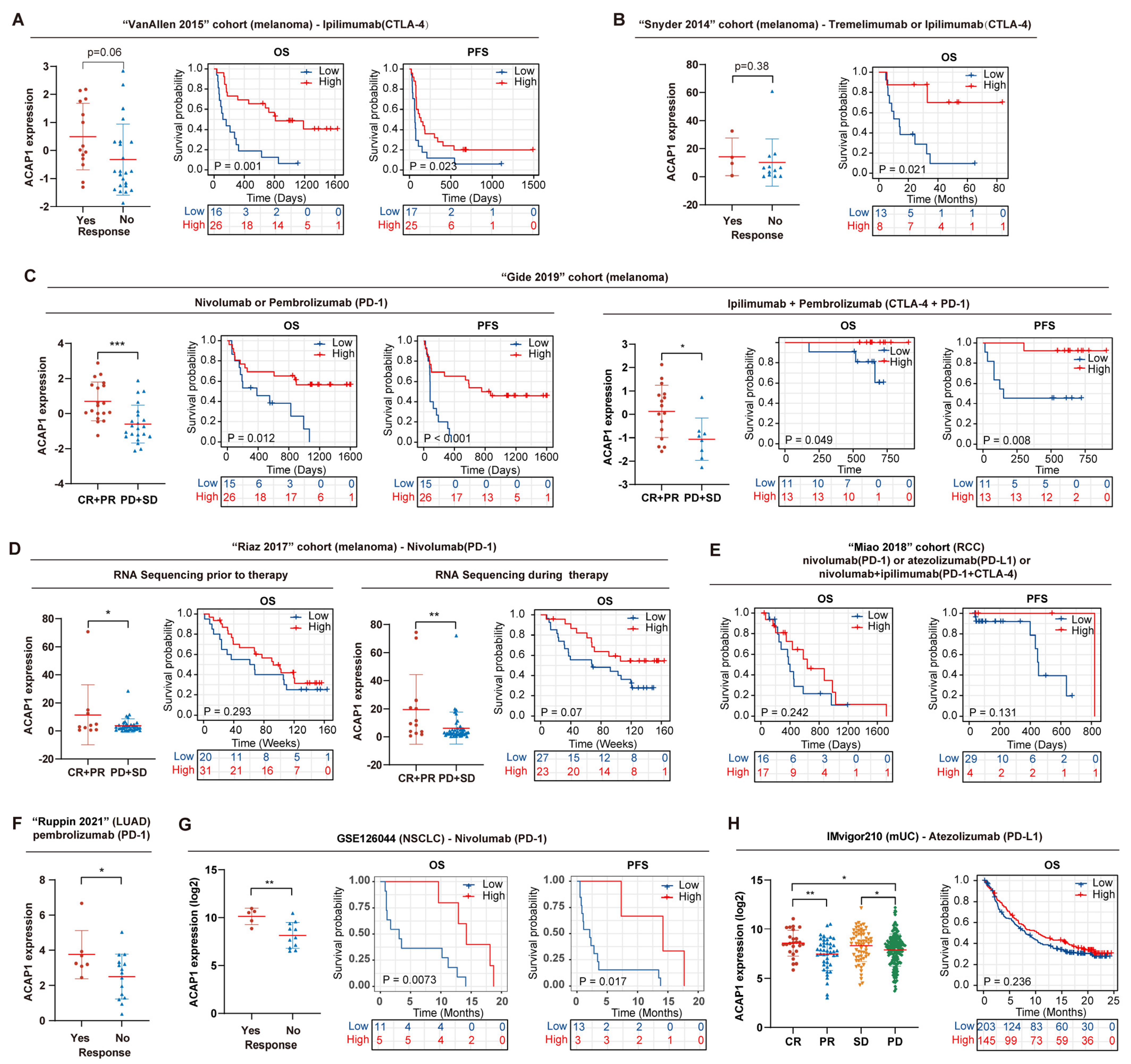
3.6. ACAP1 Level Correlates with Immunotherapy Efficacy and Predicts Prognosis in Cancer Patients Treated with ICT
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, L.B.; Salama, A.K.S. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Depetris, I.; Biason, P.; Intini, R.; Prete, A.A.; Leone, F.; Lombardi, P.; Filippi, R.; Spallanzani, A.; Cascinu, S.; et al. Prediction of Benefit from Checkpoint Inhibitors in Mismatch Repair Deficient Metastatic Colorectal Cancer: Role of Tumor Infiltrating Lymphocytes. Oncologist 2020, 25, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Pages, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Dai, J.; Li, J.; Bos, E.; Porcionatto, M.; Premont, R.T.; Bourgoin, S.; Peters, P.J.; Hsu, V.W. ACAP1 promotes endocytic recycling by recognizing recycling sorting signals. Dev. Cell 2004, 7, 771–776. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ballif, B.A.; Powelka, A.M.; Dai, J.; Gygi, S.P.; Hsu, V.W. Phosphorylation of ACAP1 by Akt regulates the stimulation-dependent recycling of integrin beta1 to control cell migration. Dev. Cell 2005, 9, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Peters, P.J.; Bai, M.; Dai, J.; Bos, E.; Kirchhausen, T.; Kandror, K.V.; Hsu, V.W. An ACAP1-containing clathrin coat complex for endocytic recycling. J. Cell Biol. 2007, 178, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.W.; Bai, M.; Li, K.; Yang, J.S.; Chu, N.; Cole, P.A.; Eck, M.J.; Li, J.; Hsu, V.W. The protein kinase Akt acts as a coat adaptor in endocytic recycling. Nat. Cell Biol. 2020, 22, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto-Furusho, J.K.; Barnich, N.; Xavier, R.; Hisamatsu, T.; Podolsky, D.K. Centaurin beta1 down-regulates nucleotide-binding oligomerization domains 1- and 2-dependent NF-kappaB activation. J. Biol. Chem. 2006, 281, 36060–36070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto-Furusho, J.K.; Furuzawa-Carballeda, J.; Fonseca-Camarillo, G. Gene and protein expression of centaurin beta 1 (CENTB1) are up-regulated in patients with ulcerative colitis. J. Crohns Colitis 2013, 7, e238–e239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q.; Zhang, J.; Wang, Q. Expression of ACAP1 Is Associated with Tumor Immune Infiltration and Clinical Outcome of Ovarian Cancer. DNA Cell Biol. 2020, 39, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Noorbakhsh, J.; Vazquez, F.; McFarland, J.M. Bridging the gap between cancer cell line models and tumours using gene expression data. Br. J. Cancer 2021, 125, 311–312. [Google Scholar] [CrossRef]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattin, J.E.; Schroder, K.; Su, A.I.; Walker, J.R.; Zhang, J.; Wiltshire, T.; Saijo, K.; Glass, C.K.; Hume, D.A.; Kellie, S.; et al. Expression analysis of G Protein-Coupled Receptors in mouse macrophages. Immunome Res. 2008, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repecka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849 e821. [Google Scholar] [CrossRef]
- He, M.X.; Cuoco, M.S.; Crowdis, J.; Bosma-Moody, A.; Zhang, Z.; Bi, K.; Kanodia, A.; Su, M.J.; Ku, S.Y.; Garcia, M.M.; et al. Transcriptional mediators of treatment resistance in lethal prostate cancer. Nat. Med. 2021, 27, 426–433. [Google Scholar] [CrossRef]
- Wu, S.Z.; Roden, D.L.; Al-Eryani, G.; Bartonicek, N.; Harvey, K.; Cazet, A.S.; Chan, C.L.; Junankar, S.; Hui, M.N.; Millar, E.A.; et al. Cryopreservation of human cancers conserves tumour heterogeneity for single-cell multi-omics analysis. Genome Med. 2021, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e924. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255 e236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martin-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949 e916. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Nair, N.U.; Dinstag, G.; Chapman, L.; Chung, Y.; Wang, K.; Sinha, S.; Cha, H.; Kim, D.; Schperberg, A.V.; et al. Synthetic lethality-mediated precision oncology via the tumor transcriptome. Cell 2021, 184, 2487–2502 e2413. [Google Scholar] [CrossRef]
- Cho, J.W.; Hong, M.H.; Ha, S.J.; Kim, Y.J.; Cho, B.C.; Lee, I.; Kim, H.R. Genome-wide identification of differentially methylated promoters and enhancers associated with response to anti-PD-1 therapy in non-small cell lung cancer. Exp. Mol. Med. 2020, 52, 1550–1563. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bosse, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, T.; Ahuja, A.; Rubinsteyn, A.; Aksoy, B.A.; Hellmann, M.D.; Miao, D.; Van Allen, E.; Merghoub, T.; Wolchok, J.D.; Snyder, A.; et al. Somatic Mutations and Neoepitope Homology in Melanomas Treated with CTLA-4 Blockade. Cancer Immunol. Res. 2017, 5, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauss, M.; Donia, M.; Harbst, K.; Andersen, R.; Mitra, S.; Rosengren, F.; Salim, M.; Vallon-Christersson, J.; Torngren, T.; Kvist, A.; et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 2017, 8, 1738. [Google Scholar] [CrossRef] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Sant’ Angelo, M.; Forman, J.; Ross-Macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Uppaluri, R.; Campbell, K.M.; Egloff, A.M.; Zolkind, P.; Skidmore, Z.L.; Nussenbaum, B.; Paniello, R.C.; Rich, J.T.; Jackson, R.; Pipkorn, P.; et al. Neoadjuvant and Adjuvant Pembrolizumab in Resectable Locally Advanced, Human Papillomavirus-Unrelated Head and Neck Cancer: A Multicenter, Phase II Trial. Clin. Cancer Res. 2020, 26, 5140–5152. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Director’s Challenge Consortium for the Molecular Classification of Lung, A.; Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, G.; Busch, C.; Knappskog, S.; Geisler, J.; Miletic, H.; Ringner, M.; Lillehaug, J.R.; Borg, A.; Lonning, P.E. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin. Cancer Res. 2010, 16, 3356–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Zhang, K.N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genom. Proteom. Bioinform. 2021, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.V.; Krebs, W.; Ulas, T.; Xue, J.; Bassler, K.; Gunther, P.; Hardt, A.L.; Schultze, H.; Sander, J.; Klee, K.; et al. The transcriptional regulator network of human inflammatory macrophages is defined by open chromatin. Cell Res. 2016, 26, 151–170. [Google Scholar] [CrossRef] [Green Version]
- Ryan, R.J.; Drier, Y.; Whitton, H.; Cotton, M.J.; Kaur, J.; Issner, R.; Gillespie, S.; Epstein, C.B.; Nardi, V.; Sohani, A.R.; et al. Detection of Enhancer-Associated Rearrangements Reveals Mechanisms of Oncogene Dysregulation in B-cell Lymphoma. Cancer Discov. 2015, 5, 1058–1071. [Google Scholar] [CrossRef] [Green Version]
- Sotoca, A.M.; Prange, K.H.; Reijnders, B.; Mandoli, A.; Nguyen, L.N.; Stunnenberg, H.G.; Martens, J.H. The oncofusion protein FUS-ERG targets key hematopoietic regulators and modulates the all-trans retinoic acid signaling pathway in t(16;21) acute myeloid leukemia. Oncogene 2016, 35, 1965–1976. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Wang, Q.; Dose, M.; Pruett, N.; Kieffer-Kwon, K.R.; Resch, W.; Liang, G.; Tang, Z.; Mathe, E.; Benner, C.; et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 2014, 159, 1524–1537. [Google Scholar] [CrossRef] [Green Version]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaferia, G.R.; Balestrieri, C.; Prosperini, E.; Nicoli, P.; Spaggiari, P.; Zerbi, A.; Natoli, G. Dissection of transcriptional and cis-regulatory control of differentiation in human pancreatic cancer. EMBO J. 2016, 35, 595–617. [Google Scholar] [CrossRef] [Green Version]
- Lay, F.D.; Liu, Y.; Kelly, T.K.; Witt, H.; Farnham, P.J.; Jones, P.A.; Berman, B.P. The role of DNA methylation in directing the functional organization of the cancer epigenome. Genome Res. 2015, 25, 467–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Li, Y.; Lv, J.; Zheng, X.; Wen, H.; Shen, H.; Zhu, G.; Chen, T.Y.; Dhar, S.S.; Kan, P.Y.; et al. ZMYND8 Reads the Dual Histone Mark H3K4me1-H3K14ac to Antagonize the Expression of Metastasis-Linked Genes. Mol. Cell 2016, 63, 470–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Collings, C.K.; Zhao, Z.; Cozzolino, K.A.; Ma, Q.; Liang, K.; Marshall, S.A.; Sze, C.C.; Hashizume, R.; Savas, J.N.; et al. A cytoplasmic COMPASS is necessary for cell survival and triple-negative breast cancer pathogenesis by regulating metabolism. Genes Dev. 2017, 31, 2056–2066. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. elife 2017, 6, e26476. [Google Scholar] [CrossRef]
- Fu, J.; Li, K.; Zhang, W.; Wan, C.; Zhang, J.; Jiang, P.; Liu, X.S. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med. 2020, 12, 21. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Hong, F.; Wang, Z.; Hu, J.; Chen, N.; Lv, L.; Yi, Q. HSD17B6 downregulation predicts poor prognosis and drives tumor progression via activating Akt signaling pathway in lung adenocarcinoma. Cell Death Discov. 2021, 7, 341. [Google Scholar] [CrossRef]
- Sahin, I.; Zhang, S.; Navaraj, A.; Zhou, L.; Dizon, D.; Safran, H.; El-Deiry, W.S. AMG-232 sensitizes high MDM2-expressing tumor cells to T-cell-mediated killing. Cell Death Discov. 2020, 6, 57. [Google Scholar] [CrossRef]
- Kruse, C.A.; Visonneau, S.; Kleinschmidt-DeMasters, B.K.; Gup, C.J.; Gomez, G.G.; Paul, D.B.; Santoli, D. The human leukemic T-cell line, TALL-104, is cytotoxic to human malignant brain tumors and traffics through brain tissue: Implications for local adoptive immunotherapy. Cancer Res. 2000, 60, 5731–5739. [Google Scholar]
- Mandal, R.; Senbabaoglu, Y.; Desrichard, A.; Havel, J.J.; Dalin, M.G.; Riaz, N.; Lee, K.W.; Ganly, I.; Hakimi, A.A.; Chan, T.A.; et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016, 1, e89829. [Google Scholar] [CrossRef] [Green Version]
- Farlow, J.L.; Brenner, J.C.; Lei, Y.L.; Chinn, S.B. Immune deserts in head and neck squamous cell carcinoma: A review of challenges and opportunities for modulating the tumor immune microenvironment. Oral Oncol. 2021, 120, 105420. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.; Chain, B.M.; Peggs, K.S.; Quezada, S.A. The “Achilles’ Heel” of Cancer and Its Implications for the Development of Novel Immunotherapeutic Strategies. Cold Spring Harb. Perspect. Med. 2018, 8, a027086. [Google Scholar] [CrossRef] [Green Version]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gogenur, I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Ball, E.D.; Davis, R.B.; Griffin, J.D.; Mayer, R.J.; Davey, F.R.; Arthur, D.C.; Wurster-Hill, D.; Noll, W.; Elghetany, M.T.; Allen, S.L.; et al. Prognostic value of lymphocyte surface markers in acute myeloid leukemia. Blood 1991, 77, 2242–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Jeune, C.; Bertoli, S.; Elhamri, M.; Vergez, F.; Borel, C.; Huguet, F.; Michallet, M.; Dumontet, C.; Recher, C.; Thomas, X. Initial absolute lymphocyte count as a prognostic factor for outcome in acute myeloid leukemia. Leuk. Lymphoma 2013, 55, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.M.; Abdulateef, N.A.B. Absolute Monocyte Count is Superior than Absolute Lymphocyte Count at Day 28 as an Independent Prognostic Factor in Acute Myeloid Leukemia. Indian J. Hematol. Blood Transfus. 2018, 35, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Bar, M.; Othus, M.; Park, H.M.; Sandhu, V.; Chen, X.; Wood, B.L.; Estey, E. Elevated lymphocyte count at time of acute myeloid leukemia diagnosis is associated with shorter remission. Leuk. Lymphoma 2015, 56, 3109–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhigarev, D.; Varshavsky, A.; MacFarlane, A.W.; Jayaguru, P.; Barreyro, L.; Khoreva, M.; Dulaimi, E.; Nejati, R.; Drenberg, C.; Campbell, K.S. Lymphocyte Exhaustion in AML Patients and Impacts of HMA/Venetoclax or Intensive Chemotherapy on Their Biology. Cancers 2022, 14, 3352. [Google Scholar] [CrossRef]
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.; Montiel-Esparza, R.; Mukhopadhyay, R.; Vanura, K.; Blazar, B.R.; Karp, J.E.; et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. J. Clin. Investig. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Philip, M.; Ferrell, P.B. Alterations of T-cell-mediated immunity in acute myeloid leukemia. Oncogene 2020, 39, 3611–3619. [Google Scholar] [CrossRef]
- Swatler, J.; Turos-Korgul, L.; Kozlowska, E.; Piwocka, K. Immunosuppressive Cell Subsets and Factors in Myeloid Leukemias. Cancers 2021, 13, 1203. [Google Scholar] [CrossRef]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Neckers, L.M.; Yenokida, G.; James, S.P. The role of the transferrin receptor in human B lymphocyte activation. J. Immunol. 1984, 133, 2437–2441. [Google Scholar] [PubMed]
- Bayer, A.L.; Baliga, P.; Woodward, J.E. Transferrin receptor in T cell activation and transplantation. J. Leukoc. Biol. 1998, 64, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Nakayamada, S.; Saito, K.; Nakano, K.; Tanaka, Y. Activation signal transduction by beta1 integrin in T cells from patients with systemic lupus erythematosus. Arthritis Rheumtol. 2007, 56, 1559–1568. [Google Scholar] [CrossRef]
- Morimoto, C.; Kobayashi, H.; Nishijima, R.; Tanaka, H.; Iwata, S. Role of the beta1 integrin molecule in T-cell activation and migration. Mod. Rheumatol. 2000, 10, 8–15. [Google Scholar] [CrossRef]
- Nicolet, B.P.; Guislain, A.; Wolkers, M.C. CD29 Enriches for Cytotoxic Human CD4(+) T Cells. J. Immunol. 2021, 207, 2966–2975. [Google Scholar] [CrossRef]
- Nicolet, B.P.; Guislain, A.; van Alphen, F.P.J.; Gomez-Eerland, R.; Schumacher, T.N.M.; van den Biggelaar, M.; Wolkers, M.C. CD29 identifies IFN-gamma-producing human CD8(+) T cells with an increased cytotoxic potential. Proc. Natl. Acad. Sci. USA 2020, 117, 6686–6696. [Google Scholar] [CrossRef] [PubMed]
- Stotesbury, C.; Alves-Peixoto, P.; Montoya, B.; Ferez, M.; Nair, S.; Snyder, C.M.; Zhang, S.; Knudson, C.J.; Sigal, L.J. alpha2beta1 Integrin Is Required for Optimal NK Cell Proliferation during Viral Infection but Not for Acquisition of Effector Functions or NK Cell-Mediated Virus Control. J. Immunol. 2020, 204, 1582–1591. [Google Scholar] [CrossRef]
- Xiang, R.F.; Li, S.; Ogbomo, H.; Stack, D.; Mody, C.H. beta1 Integrins Are Required To Mediate NK Cell Killing of Cryptococcus neoformans. J. Immunol. 2018, 201, 2369–2376. [Google Scholar] [CrossRef] [Green Version]
- Ritsma, L.; Dey-Guha, I.; Talele, N.; Sole, X.; Chowdhury, J.; Ross, K.N.; Ramaswamy, S. Integrin beta1 activation induces an anti-melanoma host response. PLoS ONE 2017, 12, e0175300. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Cancer Type | Treatment | Number of Pos/Neg Cases | AUC | ||||
|---|---|---|---|---|---|---|---|---|
| ACAP1 | TIDE | MSI Score | TMB | CD274 | ||||
| VanAllen 2015 [25] | Melanoma | CTLA4 | Pos = 19, Neg = 23 | 0.7002 | 0.8032 | 0.7391 | 0.673 | 0.6407 |
| Riaz 2017 [28] | Melanoma | PD1_Prog | Pos = 4, Neg = 22 | 0.8295 | 0.2273 | 0.6932 | 0.4722 | 0.5227 |
| Riaz 2017 [28] | Melanoma | PD_Naive | Pos = 6, Neg = 19 | 0.4474 | 0.5965 | 0.4035 | 0.6204 | 0.2675 |
| Nathanson 2017 [33] | Melanoma | CTLA4_Pre | Pos = 4, Neg = 5 | 0.3 | 0.6 | 0.95 | N/A | 0.65 |
| Nathanson 2017 [33] | Melanoma | CTLA4_Post | Pos = 4, Neg = 11 | 0.75 | 0.25 | 0.5227 | N/A | 0.6591 |
| Liu 2019 [34] | Melanoma | PD1_Prog | Pos = 16, Neg = 31 | 0.6371 | 0.4617 | 0.4456 | N/A | 0.5625 |
| Liu 2019 [34] | Melanoma | PD1_Naive | Pos = 33, Neg = 41 | 0.5632 | 0.5506 | 0.5018 | N/A | 0.51 |
| Lauss 2017 [35] | Melanoma | ACT | Pos = 10, Neg = 15 | 0.6867 | 0.54 | 0.4933 | 0.7571 | 0.7333 |
| Hugo 2016 [36] | Melanoma | PD1 | Pos = 14, Neg = 12 | 0.5179 | 0.7024 | 0.6905 | 0.6346 | 0.6012 |
| Gide 2019 [26] | Melanoma | PD1 | Pos = 19, Neg = 22 | 0.8158 | 0.6005 | 0.4306 | N/A | 0.8278 |
| Gide 2019 [26] | Melanoma | PD1 + CTLA4 | Pos = 21, Neg = 11 | 0.6494 | 0.6753 | 0.697 | N/A | 0.7879 |
| Ruppin 2021 [29] | NSCLC | PD1 | Pos = 7, Neg = 15 | 0.8286 | 0.5143 | 0.4571 | N/A | 0.6952 |
| Kim 2018 [37] | Gastric cancer | PD1 | Pos = 12, Neg = 33 | 0.6338 | 0.5985 | 0.75 | N/A | 0.8333 |
| Miao 2018 [31] | ccRcc | PD1 or PD-L1 + CTLA4 | Pos = 20, Neg = 13 | 0.5769 | 0.4808 | 0.2538 | 0.65 | 0.4231 |
| McDermott 2018 [5] | ccRcc | PD-L1 | Pos = 20, Neg = 61 | 0.6057 | 0.5311 | 0.5541 | 0.5357 | 0.6213 |
| Braun 2020 [38] | ccRcc | PD1 | Pos = 201, Neg = 94 | 0.449 | 0.4641 | 0.5289 | 0.5631 | 0.5621 |
| Zhao 2019 [39] | Glioblastoma | PD1_Pre | Pos = 8, Neg = 7 | 0.5 | 0.59 | 0.41 | N/A | 0.68 |
| Zhao 2019 [39] | Glioblastoma | PD1_Post | Pos = 6, Neg = 3 | 0.6667 | 0.6667 | 0.6667 | N/A | 0.6111 |
| Mariathasan 2018 [32] | metastatic urothelial cancer | PD-L1 | Pos = 68, Neg = 230 | 0.4866 | 0.5175 | 0.5551 | 0.7278 | 0.5818 |
| Uppaluri 2020 [40] | HNSC | PD1_Pre | Pos = 8, Neg = 15 | 0.3667 | 0.4833 | 0.6333 | N/A | 0.6917 |
| Uppaluri 2020 [40] | HNSC | PD1_Post | Pos = 9, Neg = 13 | 0.359 | 0.5385 | 0.453 | N/A | 0.7009 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, Q.; Pu, Y.; Chao, F.; Bian, P.; Lv, L. ACAP1 Deficiency Predicts Inferior Immunotherapy Response in Solid Tumors. Cancers 2022, 14, 5951. https://doi.org/10.3390/cancers14235951
Yi Q, Pu Y, Chao F, Bian P, Lv L. ACAP1 Deficiency Predicts Inferior Immunotherapy Response in Solid Tumors. Cancers. 2022; 14(23):5951. https://doi.org/10.3390/cancers14235951
Chicago/Turabian StyleYi, Qiyi, Youguang Pu, Fengmei Chao, Po Bian, and Lei Lv. 2022. "ACAP1 Deficiency Predicts Inferior Immunotherapy Response in Solid Tumors" Cancers 14, no. 23: 5951. https://doi.org/10.3390/cancers14235951