
Role of PARP Inhibitors in Cancer Immunotherapy: Potential Friends to Immune Activating Molecules and Foes to Immune Checkpoints

Abstract
:Simple Summary
Abstract
1. Introduction
2. Influence of PARP Inhibition on Immune Response to Tumors
2.1. Well Established Effects of PARP Inhibition on the Different Features of TME
2.2. PARP-1 Inhibition Favors NKG2D Activity
3. What the Future Holds: Identification of Additional Targets Modulated by PARPi Supporting Their Combination with ICIs
3.1. PARPi Positively Impact CD155 Expression: A Delicate Balance between T Cell Co-Stimulation and Co-Inhibition
3.2. PARPi Hamper the Consumption of NAD+: Potential Role in the Anti-Tumor T Cell Function
3.3. Effect of PARP Inhibition on Two-Faced Janus Nuclear Factor of Activated T-Cells (NFAT)
3.4. PARPi Take Advantage of LKB1 Deficiency
3.5. PARP Inhibition Potentially Influences the Permanence and Function of Intra-Tumor Resident T Cells
3.6. PARP Inhibition Activates AKT Kinase: Not Only a Detriment
3.7. PARP Inhibition Mimics a GSK-3 Defect
4. Clinical Trials Combining PARPi with ICIs
4.1. Completed Clinical Studies
4.2. Ongoing Phase 3 Clinical Studies
5. Considerations about the Use of PARPi for Hematological Malignancies
6. Delivery of PARPi at the Tumor Site: Nanoparticle-Based Formulation
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T.; Ahel, I.; Chang, P. Family-wide analysis of poly (ADP-ribose) polymerase activity. Nat. Commun. 2014, 5, 4426. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.E.; Hochegger, H.; Takeda, S.; Caldecott, K.W. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydro-lase. Mol. Cell. Biol. 2007, 27, 5597–5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alemasova, E.E.; Larvik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, K.; Takebayashi, S.; Taguchi, H.; Takeda, S.; Okumura, K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008, 183, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Couto, C.A.; Wang, H.Y.; Green, J.C.; Kiely, R.; Siddaway, R.; Borer, C.; Pears, C.J.; Lakin, N.D. PARP regulates nonhomologous end joining through retention of Ku at double-strand breaks. J. Cell Biol. 2011, 194, 367–375. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, S.B.; Patel, A.G.; Hurley, R.M.; Kaufmann, S.H. The Elephant and the Blind Men: Making Sense of PARP Inhibitors in Homologous Recombination Deficient Tumor Cells. Front. Oncol. 2013, 3, 228. [Google Scholar] [CrossRef] [Green Version]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell. 2018, 71, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Luo, W.; Wang, Y. PARP-1 and its associated nucleases in DNA damage response. DNA Rep. 2019, 81, 102651. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell. 2010, 39, 8–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, A.N.; Yang, E.S. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front. Oncol. 2013, 3, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langelier, M.F.; Riccio, A.A.; Pascal, J.M. PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014, 42, 7762–7775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. J. Clin. Oncol. 2018, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Bryant, H.E.; Schultz, N. Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle 2005, 4, 1176–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Petit, S.A.; Ficarro, S.B.; Toomire, K.J.; Xie, A.; Lim, E.; Cao, S.A.; Park, E.; Eck, M.J.; Scully, R.; et al. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov. 2014, 4, 1430–1447. [Google Scholar] [CrossRef] [Green Version]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Marks, J.H.; Mandell, J.B. New York Breast Cancer Study Group. Breast and ovarian cancer risk due to in-herited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- Cavanagh, H.; Rogers, K.M. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Her. Cancer Clin. Pract. 2015, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizukami, K.; Iwasaki, Y.; Kawakami, E.; Hirata, M.; Kamatani, Y.; Matsuda, K.; Endo, M.; Sugano, K.; Yoshida, T.; Murakami, Y.; et al. Genetic characterization of pancreatic cancer patients and prediction of carrier status of germline pathogenic variants in cancer-predisposing genes. EBioMed 2020, 60, 103033. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabiliza-tion: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19, e46263. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly(ADP-ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, M.; Liu, H.; Zhou, H.; Zhao, Y.; Geng, Y.; Jiang, B.; Zhang, K.; Zhang, B.; Han, Z.; et al. Comparison of the Efficacy and Safety of PARP Inhibitors as a Monotherapy for Platinum-Sensitive Recurrent Ovarian Cancer: A Network Meta-Analysis. Front. Oncol. 2021, 11, 785102. [Google Scholar] [CrossRef]
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 44657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussios, S.; Abson, C.; Moschetta, M.; Rassy, E.; Karathanasi, A.; Bhat, T.; Ghumman, F.; Sheriff, M.; Pavlidis, N. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs R D 2020, 20, 55–73. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Mullard, A. European regulators approve first PARP inhibitor. Nat. Rev. Drug Discov. 2014, 13, 877. [Google Scholar] [CrossRef]
- Taylor, A.M.; Chan, D.; Tio, M.; Patil, S.M.; Traina, T.A.; Robson, M.E.; Khasraw, M. PARP (Poly ADP-Ribose Polymer-ase) inhibitors for locally advanced or metastatic breast cancer. Cochrane Database Syst. Rev. 2021, 4, CD011395. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell. Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Tew, W.P.; Lacchetti, C.; Ellis, A.; Maxian, K.; Banerjee, S.; Bookman, M.; Jones, M.B.; Lee, J.M.; Lheureux, S.; Liu, J.F.; et al. PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3468–3493. [Google Scholar] [CrossRef]
- Tattersall, A.; Ryan, N.; Wiggans, A.J.; Rogozińska, E.; Morrison, J. Poly(ADP-ribose) polymerase (PARP) inhibitors for the treatment of ovarian cancer. Cochrane Database Syst. Rev. 2022, 2, CD007929. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; et al. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1721–1731. [Google Scholar] [CrossRef]
- Zam, W.; Ali, L. Immune Checkpoint Inhibitors in the Treatment of Cancer. Curr. Rev. Clin. Exp. Pharmacol. 2022, 17, 103–113. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell. 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Vokes, N.I.; Liu, D.; Taylor-Weiner, A.; Wankowicz, S.M.; Adeegbe, D.; Keliher, D.; Schilling, B.; Tracy, A.; et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 2018, 50, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Qin, S.; Zhao, W.; Yu, S.; Chu, Q.; Wu, K. The role of neoantigen in immune checkpoint blockade therapy. Exp. Hematol. Oncol. 2018, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front. Immunol. 2021, 12, 672356. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.S.; Gao, Y.; Welte, T.; Wang, H.; Liu, J.; Janghorban, M.; Sheng, K.; Niu, Y.; Goldstein, A.; Zhao, N.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell. Biol. 2019, 21, 1113–1126. [Google Scholar] [CrossRef]
- Martinez, A.; Delord, J.P.; Ayyoub, M.; Devaud, C. Preclinical and Clinical Immunotherapeutic Strategies in Epithelial Ovarian Cancer. Cancers 2020, 12, 1761. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, A.; Morra, F.; Celetti, A. Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: The rationale for their inclusion in the clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179. [Google Scholar] [CrossRef] [Green Version]
- Keung, M.; Wu, Y.; Vadgama, J.V. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germani, A.; Petrucci, S.; De Marchis, L.; Libi, F.; Savio, C.; Amanti, C.; Bonifacino, A.; Campanella, B.; Capalbo, C.; Lombardi, A.; et al. Beyond BRCA1 and BRCA2: Deleterious Variants in DNA Repair Pathway Genes in Italian Families with Breast/Ovarian and Pancreatic Cancers. J. Clin. Med. 2020, 9, 3003. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, B.; Puccini, A.; Catalano, F.; Borea, R.; Iaia, M.L.; Bruno, W.; Fornarini, G.; Sciallero, S.; Rebuzzi, S.E.; Ghiorzo, P. Beyond BRCA: The Emerging Significance of DNA Damage Response and Personalized Treatment in Pancreatic and Prostate Cancer Patients. Int. J. Mol. Sci. 2022, 23, 4709. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P. Environmental exposures and mutational patterns of cancer genomes. Genome Med. 2010, 2, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, Y.R.; Weigelt, B.; Levine, D.A.; Schoolmeester, J.K.; Dao, L.N.; Balzer, B.L.; Liles, G.; Karlan, B.; Köbel, M.; Lee, C.H.; et al. Clinicopathological analysis of endometrial carcinomas harboring somatic POLE exonuclease domain mutations. Modern Pathol. 2015, 28, 505–514. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Freeman, G.J. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015, 5, 16–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstein, R.M.; Riaz, N. The DNA damage response in immunotherapy and radiation. Adv. Rad. Oncol. 2018, 3, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidotto, T.; Nersesian, S.; Graham, C.; Siemens, D.R.; Koti, M. DNA damage repair gene mutations and their association with tumor immune regulatory gene expression in muscle invasive bladder cancer subtypes. J. Immunother. Cancer 2019, 7, 148. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.L.; Li, X.B.; Ke, H.; Zhang, G.Y.; Tang, Q.; Yuan, J.; Zhou, C.J.; Zhang, J.L.; Zhang, R.; Chen, W.Y. Prognostic Value of Neoantigen Load in Immune Checkpoint Inhibitor Therapy for Cancer. Front. Immunol. 2021, 12, 689076. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage response pathway: Biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm. Sin. B 2021, 11, 2983–2994. [Google Scholar] [CrossRef]
- Tang, M.L.; Khan, M.K.; Croxford, J.L.; Tan, K.W.; Angeli, V.; Gasser, S. The DNA damage response induces antigen presenting cell-like functions in fibroblasts. Eur. J. Immunol. 2014, 44, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Seyedin, S.N.; Hasibuzzaman, M.M.; Pham, V.; Petronek, M.S.; Callaghan, C.; Kalen, A.L.; Mapuskar, K.A.; Mott, S.L.; Spitz, D.R.; Allen, B.G.; et al. Combination Therapy with Radiation and PARP Inhibition Enhances Responsiveness to Anti-PD-1 Therapy in Colorectal Tumor Models. Int. Ernational J. Rad. Oncol. Biol. Phys. 2020, 108, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Chen, Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 2012, 5, ra20. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.S.; Zhang, W.Y.; Tan, N.Y.; Khatoo, M.; Suter, M.A.; Tripathi, S.; Cheung, F.S.; Lim, W.K.; Tan, P.H.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Barber, G. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, G. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrales, L.; McWhirter, S.M.; Dubensky, T.W., Jr.; Gajewski, T.F. The host STING pathway at the interface of cancer and immunity. J. Clin. Investig. 2016, 126, 2404–2411. [Google Scholar] [CrossRef] [Green Version]
- Jing, W.; McAllister, D.; Vonderhaar, E.P.; Palen, K.; Riese, M.J.; Gershan, J.; Johnson, B.D.; Dwinell, M.B. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J. Immunother. Cancer 2019, 7, 115. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9, 1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufeldt, C.J.; Cerikan, B.; Cortese, M.; Frankish, J.; Lee, J.Y.; Plociennikowska, A.; Heigwer, F.; Prasad, V.; Joecks, S.; Burkart, S.S.; et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-κB. Commun. Biol. 2022, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Sign. Trans. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [Green Version]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-phase-Specific DNA Damage in Breast Cancer. J. Nat. Cancer Inst. 2016, 109, djw199. [Google Scholar] [CrossRef] [Green Version]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8+ T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.K.; Konstantinopoulos, P.A. PARP inhibition and immune modulation: Scientific rationale and perspectives for the treatment of gynecologic cancers. Ther. Adv. Med. Oncol. 2020, 12, 1758835920944116. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Yélamos, J.; Moreno-Lama, L.; Jimeno, J.; Ali, S.O. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers 2020, 12, 392. [Google Scholar] [CrossRef]
- Tang, T.; Cheng, X.; Truong, B.; Sun, L.; Yang, X.; Wang, H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol. Ther. 2021, 219, 107709. [Google Scholar] [CrossRef]
- Zhao, Y.; Lee, C.K.; Lin, C.H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.F.; Bui, J.D.; et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 2019, 51, 1059–1073. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Azuma, K.; Kawahara, A.; Yamada, K.; Imamura, Y.; Tokito, T.; Kinoshita, T.; Kage, M.; Hoshino, T. Significance of programmed cell death-ligand 1 expression and its association with survival in patients with small cell lung cancer. J. Thor. Oncol. 2015, 10, 426–430. [Google Scholar] [CrossRef] [Green Version]
- Paglialunga, L.; Salih, Z.; Ricciuti, B.; Califano, R. Immune checkpoint blockade in small cell lung cancer: Is there a light at the end of the tunnel? ESMO 2016, 1, e000022. [Google Scholar] [CrossRef]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(-/-) murine model of ovarian cancer. Biochem. Biophys. Res. Comm. 2015, 463, 551–556. [Google Scholar] [CrossRef]
- Staniszewska, A.D.; Armenia, J.; King, M.; Michaloglou, C.; Reddy, A.; Singh, M.; San Martin, M.; Prickett, L.; Wilson, Z.; Proia, T.; et al. PARP inhibition is a modulator of anti-tumor immune response in BRCA-deficient tumors. Oncoimmunology 2022, 11, 2083755. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Iurescia, S.; Fioretti, D.; Rinaldi, M. Targeting Cytosolic Nucleic Acid-Sensing Pathways for Cancer Immunotherapies. Front. Immunol. 2018, 9, 711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüler, T.; Qin, Z.; Ibe, S.; Noben-Trauth, N.; Blumenstein, T. T helper cell type 1-associated and cytotoxic T lymphocyte-mediated tumor immunity is impaired in interleukin 4-deficient mice. J. Exp. Med. 1999, 189, 803–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Xiao, Z.; Zhao, Q.; Li, M.; Wu, X.; Zhang, L.; Hu, W.; Cho, C.H. Anti-cancer therapy with TNFα and IFNγ: A comprehensive review. Cell Prolif. 2018, 51, e12441. [Google Scholar] [CrossRef] [Green Version]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saenz, L.; Lozano, J.J.; Valdor, R.; Baroja-Mazo, A.; Ramirez, P.; Parrilla, P.; Aparicio, P.; Sumoy, L.; Yélamos, J. Transcriptional regulation by poly(ADP-ribose) polymerase-1 during T cell activation. BMC Genom. 2008, 9, 171. [Google Scholar] [CrossRef] [Green Version]
- Datta, R.; Naura, A.S.; Zerfaoui, M.; Errami, Y.; Oumouna, M.; Kim, H.; Ju, J.; Ronchi, V.P.; Haas, A.L.; Boulares, A.H. PARP-1 deficiency blocks IL-5 expression through calpain-dependent degradation of STAT-6 in a murine asthma model. Allergy 2011, 66, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Ghonim, M.A.; Pyakurel, K.; Ibba, S.V.; Al-Khami, A.A.; Wang, J.; Rodriguez, P.; Rady, H.F.; El-Bahrawy, A.H.; Lammi, M.R.; Mansy, M.S.; et al. PARP inhibition by olaparib or gene knockout blocks asthma-like manifestation in mice by modulating CD4(+) T cell function. J. Transl. Med. 2015, 13, 225. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yamane, H.; Cote-Sierra, J.; Guo, L.; Paul, W.E. GATA-3 promotes Th2 responses through three different mechanisms: Induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 2005, 16, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.; Gong, Y.; Sun, W.; Li, T.; Wang, Z.Q. PARP1: Liaison of Chromatin Remodeling and Transcription. Cancers 2022, 14, 4162. [Google Scholar] [CrossRef]
- Ansel, K.M.; Djuretic, I.; Tanasa, B.; Rao, A. Regulation of Th2 differentiation and Il4 locus accessibility. Ann. Rev. Immunol. 2006, 24, 607–656. [Google Scholar] [CrossRef]
- Zhang, P.; Maruyama, T.; Konkel, J.E.; Abbatiello, B.; Zamarron, B.; Wang, Z.Q.; Chen, W. PARP-1 controls immunosuppressive function of regulatory T cells by destabilizing Foxp3. PLoS ONE 2013, 8, e71590. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Nie, J.; Wang, S.; Chen, Z.; Chen, W.; Li, D.; Hu, H.; Li, B. Poly(ADP-ribosyl)ation of FOXP3 Protein Mediated by PARP-1 Protein Regulates the Function of Regulatory T Cells. J. Biol. Chem. 2015, 290, 28675–28682. [Google Scholar] [CrossRef] [Green Version]
- Nasta, F.; Laudisi, F.; Sambucci, M.; Rosado, M.M.; Pioli, C. Increased Foxp3+ regulatory T cells in poly(ADP-Ribose) polymerase-1 deficiency. J. Immunol. 2010, 184, 3470–3477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tong, Z.; Zhang, W.; Zhang, W.; Buzdin, A.; Mu, X.; Yan, Q.; Zhao, X.; Chang, H.H.; Duhon, M.; et al. FDA-Approved and Emerging Next Generation Predictive Biomarkers for Immune Checkpoint Inhibitors in Cancer Patients. Front. Oncol. 2021, 11, 683419. [Google Scholar] [CrossRef] [PubMed]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expres-sion and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. 2001, 2, 769–776. [Google Scholar] [CrossRef]
- Yélamos, J.; Monreal, Y.; Saenz, L.; Aguado, E.; Schreiber, V.; Mota, R.; Fuente, T.; Minguela, A.; Parrilla, P.; de Murcia, G.; et al. PARP-2 deficiency affects the survival of CD4+CD8+ double-positive thymocytes. EMBO J. 2006, 25, 4350–4360. [Google Scholar] [CrossRef]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP inhibition elicits STING-dependent antitumor immunity in brca1-deficient ovarian cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M.; Krishna, C.; Ma, X.; Pei, X.; Lee, K.W.; Makarov, V.; Kuo, F.; Chung, J.; Srivastava, R.M.; Purohit, T.A.; et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat. Cancer 2021, 1, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Chen, L.; Wei, X. Inflammatory Cytokines in Cancer: Comprehensive Understanding and Clinical Progress in Gene Therapy. Cells 2021, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediat. Inflamm. 2014, 2014, 292376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalid, A.; Wolfram, J.; Ferrari, I.; Mu, C.; Mai, J.; Yang, Z.; Zhao, Y.; Ferrari, M.; Ma, X.; Shen, H. Recent Advances in Discovering the Role of CCL5 in Metastatic Breast Cancer. Mini Rev. Med. Chem. 2015, 15, 1063–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, T.; Xu, S.; Jin, P.; Zhou, D.; Wang, Z.; Li, H.; Yang, Z.; Chen, G.; Zheng, X.; et al. PARP inhibitors promote stromal fibroblast activation by enhancing CCL5 autocrine signaling in ovarian cancer. NPJ Precis. Oncol. 2021, 5, 49. [Google Scholar] [CrossRef]
- Huang, C.Y.; Fong, Y.C.; Lee, C.Y.; Chen, M.Y.; Tsai, H.C.; Hsu, H.C.; Tang, C.H. CCL5 increases lung cancer migration via PI3K, Akt and NF-kappaB pathways. Biochem. Pharmacol. 2009, 77, 794–803. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, H.S.; Shin, Y.J.; Lee, C.S.; Won, C.; Lee, S.A.; Lee, J.W.; Kim, Y.; Kang, J.S.; Ye, S.K.; et al. LYR71, a derivative of trimeric resveratrol, inhibits tumorigenesis by blocking STAT3-mediated matrix metalloproteinase 9 expression. Exp. Mol. Med. 2008, 40, 514–522. [Google Scholar] [CrossRef]
- Kato, T.; Fujita, Y.; Nakane, K.; Mizutani, K.; Terazawa, R.; Ehara, H.; Kanimoto, Y.; Kojima, T.; Nozawa, Y.; Deguchi, T.; et al. CCR1/CCL5 interaction promotes invasion of taxane-resistant PC3 prostate cancer cells by increasing secretion of MMPs 2/9 and by activating ERK and Rac signaling. Cytokine 2013, 64, 251–257. [Google Scholar] [CrossRef]
- Li, F.; Wu, X.; Fu, X.; Liu, J.; Song, W.; Xiao, G.G.; Lu, A.; Zhang, G. Poly (ADP-ribose) polymerase 1 (PARP1) inhibition promotes pulmonary metastasis of osteosarcoma by boosting ezrin phosphorylation. Int. J. Biol. Sci. 2022, 18, 1238–1253. [Google Scholar] [CrossRef] [PubMed]
- Clucas, J.; Valderrama, F. ERM proteins in cancer progression. J. Cell Sci. 2014, 127, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moilanen, J.; Lassus, H.; Leminen, A.; Vaheri, A.; Bützow, R.; Carpén, O. Ezrin immunoreactivity in relation to survival in serous ovari-an carcinoma patients. Gynecol. Oncol. 2003, 90, 273–281. [Google Scholar] [CrossRef]
- Horwitz, V.; Davidson, B.; Stern, D.; Tropé, C.G.; Tavor Re’em, T.; Reich, R. Ezrin Is Associated with Disease Progression in Ovarian Carcinoma. PLoS ONE 2016, 11, e0162502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houchins, J.P.; Yabe, T.; McSherry, C.; Bach, F.H. DNA sequence analysis of NKG2, a family of related cDNA clones en-coding type II integral membrane proteins on human natural killer cells. J. Exp. Med. 1991, 173, 1017–1020. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The role of the NKG2D immuno-receptor in immune cell activation and natural killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Fuertes, M.B.; Domaica, C.I.; Zwirner, N.W. Leveraging NKG2D Ligands in Immuno-Oncology. Front. Immunol. 2021, 12, 713158. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, A.; Jamieson, A.M.; Liu, S.D.; Shastri, N.; Raulet, D.H. Ligands for the murine NKG2D receptor: Expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 2000, 1, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Maasho, K.; Opoku-Anane, J.; Marusina, A.I.; Coligan, J.E.; Borrego, F. NKG2D is a costimulatory receptor for human naive CD8+ T cells. J. Immunol. 2005, 174, 4480–4484. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, M.A.; Carayannopoulos, L.N.; Naidenko, O.V.; Matsui, K.; Burack, W.R.; Wise, E.L.; Fremont, D.H.; Allen, P.M.; Yokoyama, W.M.; Colonna, M.; et al. Costimulation through NKG2D enhances murine CD8+ CTL function: Similarities and differences between NKG2D and CD28 costimulation. J. Immunol. 2005, 175, 2825–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prajapati, K.; Perez, C.; Rojas, L.; Burke, B.; Guevara-Patino, J.A. Functions of NKG2D in CD8+ T cells: An opportunity for immunotherapy. Cell. Mol. Immunol. 2018, 15, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Rajasekaran, K.; Xiong, V.; Fong, L.; Gorski, J.; Malarkannan, S. Functional dichotomy between NKG2D and CD28-mediated co-stimulation in human CD8+ T cells. PLoS ONE 2010, 5, e12635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.P.; Taylor, C.; Petryniak, B.; Turka, L.A.; June, C.H.; Thompson, C.B. The genomic organization of the CD28 gene. Implications for the regulation of CD28 mRNA expression and heterogeneity. J. Immunol. 1990, 145, 344–352. [Google Scholar] [PubMed]
- Plunkett, F.J.; Franzese, O.; Finney, H.M.; Fletcher, J.M.; Belaramani, L.L.; Salmon, M.; Dokal, I.; Webster, D.; Lawson, A.D.; Akbar, A. The loss of telomerase activity in highly differentiated CD8+CD28-CD27- T cells is associated with decreased Akt (Ser473) phosphorylation. J. Immunol. 2007, 178, 7710–7719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Cerwenka, A.; Bakker, A.B.; McClanahan, T.; Wagner, J.; Wu, J.; Phillips, J.H.; Lanier, L.L. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity 2000, 12, 721–727. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, C.A.; Cerwenka, A.; Willcox, B.E.; Lanier, L.L.; Bjorkman, P.J. Molecular competition for NKG2D: H60 and RAE1 compete unequally for NKG2D with dominance of H60. Immunity 2001, 15, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Champsaur, M.; Lanier, L.L. Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 2010, 235, 267–285. [Google Scholar] [CrossRef] [Green Version]
- Kuylenstierna, C.; Björkström, N.K.; Andersson, S.K.; Sahlström, P.; Bosnjak, L.; Paquin-Proulx, D.; Malmberg, K.J.; Ljunggren, H.G.; Moll, M.; Sandberg, J.K. NKG2D performs two functions in invariant NKT cells: Direct TCR-independent activation of NK-like cytolysis and co-stimulation of activation by CD1d. Eur. J. Immunol. 2011, 41, 1913–1923. [Google Scholar] [CrossRef]
- Lanier, L.L. NKG2D receptor and its ligands in host defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowbakht, P.; Ionescu, M.C.; Rohner, A.; Kalberer, C.P.; Rossy, E.; Mori, L.; Cosman, D.; De Libero, G.; Wodnar-Filipowicz, A. Ligands for natural killer cell-activating receptors are expressed upon the maturation of normal myelo-monocytic cells but at low levels in acute myeloid leukemias. Blood 2005, 105, 3615–3622. [Google Scholar] [CrossRef]
- Hilpert, J.; Grosse-Hovest, L.; Grünebach, F.; Buechele, C.; Nuebling, T.; Raum, T.; Steinle, A.; Salih, H.R. Comprehensive analysis of NKG2D ligand expression and release in leukemia: Implications for NKG2D-mediated NK cell responses. J. Immunol. 2012, 189, 1360–1371. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Kitamura, H.; Hijikata, A.; Tomizawa-Murasawa, M.; Tanaka, S.; Takagi, S.; Uchida, N.; Suzuki, N.; Sone, A.; Najima, Y.; et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci. Transl. Med. 2010, 2, 17ra9. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimov, N.; Arruda, A.; Popescu, A.; Gupta, V.; Shimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Fauriat, C.; Just-Landi, S.; Mallet, F.; Arnoulet, C.; Sainty, D.; Olive, D.; Costello, R.T. Deficient expression of NCR in NK cells from acute myeloid leukemia: Evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood 2007, 109, 323–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Vairy, S.; Garcia, J.L.; Teira, P.; Bittencourt, H. CTL019 (tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia. Drug Des.Dev. Ther. 2018, 12, 3885–3898. [Google Scholar] [CrossRef] [Green Version]
- Nikiforow, S.; Murad, J.; Daley, H.; Negre, H.; Reder, J.; Sentman, C.L.; Lehmann, F.; Snykers, S.; Allen, R.; Galinsky, I.; et al. A first-in-human phase I trial of NKG2D chimeric antigen receptor-T cells in AML/MDS and multiple myeloma. J. Clin. Oncol. 2016, 34 (Suppl. 15), TPS3102. [Google Scholar] [CrossRef]
- Curio, S.; Jonsson, G.; Marinović, S. A summary of current NKG2D-based CAR clinical trials. Immunother. Adv. 2021, 1, ltab018. [Google Scholar] [CrossRef] [PubMed]
- Lynn, R.C.; Powell, D.J., Jr. Strain-dependent Lethal Toxicity in NKG2D Ligand-targeted CAR T-cell Therapy. Mol. Ther. 2015, 23, 1559–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemjabbar-Alaoui, H.; Peto, C.J.; Yang, Y.W.; Jablons, D.M. AMXI-5001, a novel dual parp1/2 and microtubule polymerization inhibitor for the treatment of human cancers. Am. J. Cancer Res. 2020, 10, 2649–2676. [Google Scholar] [PubMed]
- Molfetta, R.; Zitti, B.; Lecce, M.; Milito, N.D.; Stabile, H.; Fionda, C.; Cippitelli, M.; Gismondi, A.; Santoni, A.; Paolini, R. CD155: A Multi-Functional Molecule in Tumor Progression. Int. J. Mol. Sci. 2020, 21, 922. [Google Scholar] [CrossRef] [Green Version]
- Castriconi, R.; Dondero, A.; Corrias, M.V.; Lanino, E.; Pende, D.; Moretta, L.; Bottino, C.; Moretta, A. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: Critical role of DNAX accessory molecule-1-poliovirus receptor interaction. Cancer Res. 2004, 64, 9180–9184. [Google Scholar] [CrossRef] [Green Version]
- Vo, A.V.; Takenaka, E.; Shibuya, A.; Shibuya, K. Expression of DNAM-1 (CD226) on inflammatory monocytes. Mol. Immunol. 2016, 69, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.G.; Miller, J.S.; Zheng, S.G. CD226: An Emerging Role in Immunologic Diseases. Front. Cell Dev. Biol. 2020, 8, 564. [Google Scholar] [CrossRef]
- Verhoeven, D.H.; de Hooge, A.S.; Mooiman, E.C.; Santos, S.J.; ten Dam, M.M.; Gelderblom, H.; Melief, C.J.; Hogendoorn, P.C.; Egeler, R.M.; van Tol, M.J.; et al. NK cells recognize and lyse Ewing sarcoma cells through NKG2D and DNAM-1 receptor dependent pathways. Mol. Immunol. 2008, 45, 3917–3925. [Google Scholar] [CrossRef]
- El-Sherbiny, Y.M.; Meade, J.L.; Holmes, T.D.; McGonagle, D.; Mackie, S.L.; Morgan, A.W.; Cook, G.; Feyler, S.; Richards, S.J.; Davies, F.E.; et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007, 67, 8444–8449. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Shin, B.R.; Lee, H.K.; Lee, J.H.; Kim, K.H.; Choi, J.E.; Ji, A.Y.; Hong, J.T.; Kim, Y.; Han, S.B. Cd226-/- natural killer cells fail to establish stable contacts with cancer cells and show impaired control of tumor metastasis in vivo. Oncoimmunology 2017, 6, e1338994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepletier, A.; Lutzky, V.P.; Mittal, D.; Stannard, K.; Watkins, T.S.; Ratnatunga, C.N. The Immune Checkpoint CD96 Defines a Distinct Lymphocyte Phenotype and is Highly Expressed on Tumor-Infiltrating T Cells. Immunol. Cell. Biol. 2019, 97, 152–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, E.; Joller, N.; Cao, Y.; Kuchroo, V.K.; Hafler, D.A. The CD226/CD155 interaction regulates the proinflammatory (Th1/Th17)/anti-inflammatory (Th2) balance in humans. J. Immunol. 2013, 191, 3673–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [Green Version]
- Enqvist, M.; Ask, E.H.; Forslund, E.; Carlsten, M.; Abrahamsen, G.; Béziat, V.; Andersson, S.; Schaffer, M.; Spurkland, A.; Bryceson, Y.; et al. Coordinated expression of DNAM-1 and LFA-1 in educated NK cells. J. Immunol. 2015, 194, 4518–4527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A.K.; Kadri, N.; Snäll, J.; Brodin, P.; Gilfillan, S.; Colonna, M.; Bernhardt, G.; Höglund, P.; Kärre, K.; Chambers, B.J. Expression of CD226 is associated to but not required for NK cell education. Nat. Commun. 2017, 8, 15627. [Google Scholar] [CrossRef] [Green Version]
- Martinet, L.; Andrade, L.F.D.; Guillerey, C.; Lee, J.S.; Liu, J.; Souza-Fonseca-Guimaraes, F.; Hutchinson, D.S.; Klesnik, T.B.; Nicholson, S.E.; Huntington, N.D.; et al. DNAM-1 expression marks an alternative program of NK cell maturation. Cell Rep. 2015, 11, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, K.; Shirakawa, J.; Kameyama, T.; Honda, S.; Tahara-Hanaoka, S.; Miyamoto, A.; Onodera, M.; Sumida, T.; Nakauchi, H.; Miyoshi, H.; et al. CD226 (DNAM-1) is involved in lymphocyte function-associated antigen 1 costimulatory signal for naive T cell differentiation and proliferation. J. Exp. Med. 2015, 198, 1829–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, K.; Lanier, L.L.; Phillips, J.H.; Ochs, H.D.; Shimizu, K.; Nakayama, E.; Nakauchi, H.; Shibuya, A. Physical and functional association of LFA-1 with DNAM-1 adhesion molecule. Immunity 1999, 11, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Iguchi-Manaka, A.; Kai, H.; Yamashita, Y.; Shibata, K.; Tahara-Hanaoka, S.; Honda, S.; Yasui, T.; Kikutani, H.; Shibuya, K.; Shibuya, A. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J. Exp. Med. 2008, 205, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, N.; Lu, Y.; Davidson, D.; Colonna, M.; Veillette, A. DNAM-1 controls NK cell activation via an ITT-like motif. J. Exp. Med. 2015, 212, 2165–2182. [Google Scholar] [CrossRef]
- Yeo, J.; Ko, M.; Lee, D.H.; Park, Y.; Jin, H.S. TIGIT/CD226 Axis Regulates Anti-Tumor Immunity. Pharmaceuticals 2021, 14, 200. [Google Scholar] [CrossRef]
- Niu, C.; Jin, H.; Li, M.; Zhu, S.; Zhou, L.; Jin, F. Low-Dose Bortezomib Increases the Expression of NKG2D and DNAM-1 Ligands and Enhances Induced NK and Gammadelta T Cell-Mediated Lysis in Multiple Myeloma. Oncotarget 2017, 8, 5954–5964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed-Pastor, W.A.; Lambert, L.J.; Ely, Z.A.; Pattada, N.B.; Bhutkar, A.; Eng, G.; Jacks, T. The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell. 2021, 39, 1342–1360.e14. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Inozume, T.; Kawazu, M.; Ueno, T.; Nagasaki, J.; Tanji, E.; Honobe, A.; Ohnuma, T.; Kawamura, T.; Umeda, Y.; et al. TIGIT/CD155 axis mediates resistance to immunotherapy in patients with melanoma with the inflamed tumor microenvironment. J. Immunother. Cancer 2021, 9, e003134. [Google Scholar] [CrossRef]
- Mao, L.; Xiao, Y.; Yang, Q.C.; Yang, S.C.; Yang, L.L.; Sun, Z.J. TIGIT/CD155 blockade enhances anti-PD-L1 therapy in head and neck squamous cell carcinoma by targeting myeloid-derived suppressor cells. Oral Oncol. 2021, 121, 105472. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Inozume, T.; Yaguchi, T.; Furuta, J.; Harada, K.; Kawakami, Y.; Shimada, S. Melanoma Cells Control Antimelanoma CTL Responses via Interaction between TIGIT and CD155 in the Effector Phase. J. Investig. Dermatol. 2016, 136, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Tundo, G.R.; Sbardella, D.; Lacal, P.M.; Graziani, G.; Marini, S. On the Horizon: Targeting Next-Generation Immune Checkpoints for Cancer Treatment. Chemotherapy 2019, 64, 62–80. [Google Scholar] [CrossRef]
- Mahnke, K.; Enk, A.H. TIGIT-CD155 Interactions in Melanoma: A Novel Co-Inhibitory Pathway with Potential for Clinical Intervention. J. Investig. Dermatol. 2016, 136, 9–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Zhao, W.; Li, H.; Chen, Y.; Tian, H.; Li, L.; Zhang, L.; Gao, C.; Zheng, J. Immunoreceptor TIGIT inhibits the cytotoxicity of human cytokine-induced killer cells by interacting with CD155. Cancer Immunol. Immunother. 2016, 65, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, E.Y.; Mellman, I. TIGIT-CD226-PVR axis: Advancing immune checkpoint blockade for cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004711. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.S.; Ko, M.; Choi, D.S.; Kim, J.H.; Lee, D.H.; Kang, S.H.; Kim, I.; Lee, H.J.; Choi, E.K.; Kim, K.P.; et al. CD226hiCD8+ T Cells Are a Prerequisite for Anti-TIGIT Immunotherapy. Cancer Immunol. Res. 2020, 8, 912–925. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; You, X.; Han, S.; Sun, Y.; Zhang, J.; Zhang, Y. CD155/TIGIT, a novel immune checkpoint in human cancers. Oncol. Rep. 2021, 45, 835–845. [Google Scholar] [CrossRef]
- Sato, K.; Yamashita-Kanemaru, Y.; Abe, F.; Murata, R.; Nakamura-Shinya, Y.; Kanemaru, K.; Muratani, M.; Veillette, A.; Goto, M.; Ito, M.; et al. DNAM-1 regulates Foxp3 expression in regulatory T cells by interfering with TIGIT under inflammatory conditions. Proc. Natl. Acad. Sci. USA 2021, 118, e2021309118. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Diff. 2013, 20, 456–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Lee, J.B.; Ha, S.J.; Kim, H.R. Clinical Insights into Novel Immune Checkpoint Inhibitors. Front. Pharmacol. 2021, 12, 681320. [Google Scholar] [CrossRef] [PubMed]
- Sandu, I.; Cerletti, D.; Claassen, M.; Oxenius, A. Exhausted CD8+ T cells exhibit low and strongly inhibited TCR signaling during chronic LCMV infection. Nat. Commun. 2020, 11, 4454. [Google Scholar] [CrossRef]
- Deaglio, S.; Robson, S.C. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv. Pharmacol. 2011, 61, 301–332. [Google Scholar] [CrossRef] [Green Version]
- Bono, M.R.; Fernandez, D.; Flores-Santibanez, F.; Rosemblatt, M.; Sauma, D. CD73 and CD39 ectonucleotidases in T cell differentiation: Beyond immunosuppression. FEBS Lett. 2015, 589, 3454–3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Daenthanasanmak, A.; Chakraborty, P.; Wyatt, M.W.; Dhar, P.; Selvam, S.P.; Fu, J.; Zhang, J.; Nguyen, H.; Kang, I.; et al. CD38-NAD (+) Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell. Metab. 2018, 27, 85–100.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Yang, J.; Zhou, J.; Gao, W.; Zhang, Y.; Lin, Y.; Wang, H.; Ruan, Z.; Ni, B. Prognostic Values of CD38+CD101+PD1+CD8+ T Cells in Pancreatic Cancer. Immunol. Investig. 2019, 48, 466–479. [Google Scholar] [CrossRef]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, A.; Mehrotra, S.; Chatterjee, S. CD38: T Cell Immuno-Metabolic Modulator. Cells 2020, 9, 1716. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; Escande, C.; White, T.A.; Thompson, M.; Soares, S.; Benech, J.C.; Chini, E.N. Regulation of SIRT 1 mediated NAD dependent deacetylation: A novel role for the multifunctional enzyme CD38. Biochem. Biophys. Res. Commun. 2006, 349, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.H.; Castellano, G.; Bonet, J.; Le Dily, F.; Font-Mateu, J.; Ballaré, C.; Nacht, A.S.; Soronellas, D.; Oliva, B.; Beato, M. CDK2-dependent activation of PARP-1 is required for hormonal gene regulation in breast cancer cells. Genes Develop. 2012, 26, 1972–1983. [Google Scholar] [CrossRef] [Green Version]
- Fouquerel, E.; Goellner, E.M.; Yu, Z.; Gagné, J.P.; Barbi de Moura, M.; Feinstein, T.; Wheeler, D.; Redpath, P.; Li, J.; Romero, G.; et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014, 8, 1819–1831. [Google Scholar] [CrossRef] [Green Version]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell. 2019, 30, 2584–2597. [Google Scholar] [CrossRef]
- Geltink, R.K.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 26, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Horenstein, A.L.; Malavasi, F. The Key Role of NAD+ in Anti-Tumor Immune Response: An Update. Front. Immunol. 2021, 12, 658263. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Liu, J.; Guo, M.; Gao, X.; Wu, X.; Bai, N.; Guo, W.; Li, N.; Yi, F.; Cheng, R.; et al. The NAD-dependent deacetylase SIRT2 regulates T cell differentiation involved in tumor immune response. Int. J. Biol Sci. 2020, 16, 3075–3084. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Sun, L.; Shen, M.; Zhang, X.; Xiao, Z.; Wang, J.; Zhang, L. Functional assessment of the cell-autonomous role of NADase CD38 in regulating CD8+ T cell exhaustion. iScience 2022, 25, 104347. [Google Scholar] [CrossRef]
- Kwong, A.K.; Chen, Z.; Zhang, H.; Leung, F.P.; Lam, C.M.; Ting, K.Y.; Zhang, L.; Hao, Q.; Zhang, L.H.; Lee, H.C. Catalysis-based inhibitors of the calcium signaling function of CD38. Biochemistry 2012, 51, 555–564. [Google Scholar] [CrossRef]
- Lin, W.K.; Bolton, E.L.; Cortopassi, W.A.; Wang, Y.; O’Brien, F.; Maciejewska, M.; Jacobson, M.P.; Garnham, C.; Ruas, M.; Parrington, J.; et al. Synthesis of the Ca(2+)-mobilizing messengers NAADP and cADPR by intracellular CD38 enzyme in the mouse heart: Role in beta-adrenoceptor signaling. J. Biol. Chem. 2017, 292, 13243–13257. [Google Scholar] [CrossRef] [PubMed]
- States, D.J.; Walseth, T.F.; Lee, H.C. Similarities in amino acid sequences of Aplysia ADP-ribosyl cyclase and human lymphocyte antigen CD38. Trends Biochem. Sci. 1992, 17, 495. [Google Scholar] [CrossRef]
- Sato, K.; Yamashita-Kanemaru, Y.; Abe, F.; Murata, R.; Nakamura-Shinya, Y.; Kanemaru, K.; Muratani, M.; Veillette, A.; Goto, M.; Ito, M.; et al. CD38 identifies pre-activated CD8+ T cells which can be reinvigorated by anti-PD-1 blockade in human lung cancer. Cancer Immunol. Immunother. 2021, 70, 3603–3616. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, F.; Wang, L.; Qiu, S.; Yao, Y.; Yan, C.; Xiong, X.; Chen, X.; Ji, Q.; Cao, J.; et al. NAD+ supplement potentiates tumor-killing function by rescuing defective TUB-mediated NAMPT transcription in tumor-infiltrated T cells. Cell Rep. 2021, 36, 109516. [Google Scholar] [CrossRef]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, G.R.; Olson, E.N. NFAT signaling: Choreographing the social lives of cells. Cell 2002, 109, S67–S79. [Google Scholar] [CrossRef] [Green Version]
- Macian, F.; Lopez-Rodriguez, C.; Rao, A. Partners in transcription: NFAT and AP-1. Oncogene 2001, 20, 2476–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaffrey, P.G.; Luo, C.; Kerppola, T.K.; Jain, J.; Badalian, T.M.; Ho, A.M.; Burgeon, E.; Lane, W.S.; Lambert, J.N.; Curran, T. Isolation of the cyclosporin-sensitive T cell transcription factor NFATp. Science 1993, 262, 750–754. [Google Scholar] [CrossRef]
- García-Rodríguez, C.; Rao, A. Nuclear factor of activated T cells (NFAT)-dependent transactivation regulated by the coactivators p300/CREB-binding protein (CBP). J. Exp. Med. 1998, 187, 2031–2036. [Google Scholar] [CrossRef] [Green Version]
- Baksh, S.; Widlund, H.R.; Frazer-Abel, A.A.; Du, J.; Fosmire, S.; Fisher, D.E.; DeCaprio, J.A.; Modiano, J.F.; Breakoff, S.J. NFATc2-mediated repression of cyclin-dependent kinase 4 expression. Mol. Cell. 2002, 10, 1071–1081. [Google Scholar] [CrossRef]
- Dai, Y.S.; Xu, J.; Molkentin, J.D. The DnaJ-related factor Mrj interacts with nuclear factor of activated T cells c3 and mediates transcriptional repression through class II histone deacetylase recruitment. Mol. Cell. Biol. 2005, 25, 9936–9948. [Google Scholar] [CrossRef]
- Valdor, R.; Schreiber, V.; Saenz, L.; Martinez, T.; Munoz-Suano, A.; Dominguez-Villar, M.; Ramirez, P.; Parrilla, P.; Aguado, E.; Garcia-Cozar, F.; et al. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Mol. Immunol. 2008, 45, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Olabisi, O.A.; Soto-Nieves, N.; Nieves, E.; Yang, T.T.; Yang, X.; Yu, R.Y.; Suk, H.Y.; Macian, F.; Chow, C.W. Regulation of transcription factor NFAT by ADP-ribosylation. Mol. Cell. Biol. 2008, 28, 2860–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.R.; Rao, A. NFAT, immunity and cancer: A transcription factor comes of age. Nat. Rev. Immunol. 2010, 10, 645–656. [Google Scholar] [CrossRef]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laderian, B.; Mundi, P.; Fojo, T.E.; Bates, S. Emerging Therapeutic Implications of STK11 Mutation: Case Series. Oncologist 2020, 25, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Chen, J.; Cui, F.; Wang, H.; Wang, S.; Hang, W.; Zeng, Q.; Quan, C.S.; Zhai, Y.X.; Wang, J.W.; et al. LKB1 is a DNA damage response protein that regulates cellular sensitivity to PARP inhibitors. Oncotarget 2016, 7, 73389–73401. [Google Scholar] [CrossRef] [Green Version]
- Della Corte, C.M.; Byers, L.A. Evading the STING: LKB1 Loss Leads to STING Silencing and Immune Escape in KRAS-Mutant Lung Cancers. Cancer Discov. 2019, 9, 16–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, L.; Long, Z.; Guo, Y.; Zhang, Z.; Dong, O.A. PARP1 Inhibitors Enhanced IFNγ-Induced PD-L1 Expression in LKB1-Mutant Lung Cancer. J. Thor. Oncol. 2021, 16, S117–S118. [Google Scholar] [CrossRef]
- Zhang, P.; Nakatsukasa, H.; Tu, E.; Kasagi, S.; Cui, K.; Ishikawa, M.; Konkel, J.E.; Maruyama, T.; Wei, G.; Abbatiello, B.; et al. PARP-1 regulates expression of TGF-β receptors in T cells. Blood 2013, 122, 2224–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.A.; Li, M.O. TGF-β: Guardian of T cell function. J. Immunol. 2013, 191, 3973–3979. [Google Scholar] [CrossRef] [PubMed]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.C.; et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat. Immunol. 2008, 9, 1347–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutet, M.; Gauthier, L.; Leclerc, M.; Gros, G.; de Montpreville, V.; Théret, N.; Donnadieu, E.; Mami-Chouaib, F. TGFβ Signaling Intersects with CD103 Integrin Signaling to Promote T-Lymphocyte Accumulation and Antitumor Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 1757–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Duhen, T.; Duhen, R.; Montler, J.; Moses, T.; Moudgil, N.F.; de Miranda, C.P.; Goodall, T.C.; Blair, B.A.; Fox, J.E.; McDermott, J.E.; et al. Co-expression of CD39 and CD103 identifies tumor reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 2724. [Google Scholar] [CrossRef] [Green Version]
- McPhee, T.R.; McDonald, P.C.; Oloumi, A.; Dedhar, S. Integrin-linked kinase regulates E-cadherin expression through PARP-1. Dev. Dyn. 2008, 237, 2737–2747. [Google Scholar] [CrossRef]
- Somasiri, A.; Howarth, A.; Goswami, D.; Dedhar, S.; Roskelley, C.D. Overexpression ofthe integrin-linked kinase mesenchymally transforms mammary epithelial cells. J. Cell. Sci. 2001, 114, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Mami-Chouaib, F.; Blanc, C.; Corgnac, S.; Hans, S.; Malenica, I.; Granier, C.; Tihy, I.; Tartour, E. Resident memory T cells, critical components in tumor immunology. J. Immunother. Cancer 2018, 6, 87. [Google Scholar] [CrossRef] [Green Version]
- Quinn, E.; Hawkins, N.; Yip, Y.L.; Suter, C.; Ward, R. CD103+ intraepithelial lymphocytes-a unique population in microsatellite unstable sporadic colorectal cancer. Eur. J. Cancer 2003, 2003, 469–475. [Google Scholar] [CrossRef]
- Wang, B.; Wu, S.; Zeng, H.; Liu, Z.; Dong, W.; He, W.; Chen, X.; Dong, X.; Zheng, L.; Lin, T.; et al. CD103+ Tumor Infiltrating Lymphocytes Predict a Favorable Prognosis in Urothelial Cell Carcinoma of the Bladder. J. Urol. 2015, 194, 556–562. [Google Scholar] [CrossRef]
- Corgnac, S.; Boutet, M.; Kfoury, M.; Naltet, C.; Mami-Chouaib, F. The Emerging Role of CD8+ Tissue Resident Memory T (TRM) Cells in Antitumor Immunity: A Unique Functional Contribution of the CD103 Integrin. Front. Immunol. 2018, 9, 1904. [Google Scholar] [CrossRef] [PubMed]
- Banh, C.; Fugère, C.; Brossay, L. Immunoregulatory functions of KLRG1 cadherin interactions are dependent on forward and reverse signaling. Blood 2009, 114, 5299–5306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Kalia, V.; Haining, W.N.; Konieczny, B.T.; Subramaniam, S.; Ahmed, R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J. Exp. Med. 2008, 205, 625–640. [Google Scholar] [CrossRef] [Green Version]
- Xin, A.; Masson, F.; Liao, Y.; Preston, S.; Guan, T.; Gloury, R.; Olshansky, M.; Lin, J.X.; Li, P.; Speed, T.P.; et al. A molecular threshold for effector CD8(+) T cell differentiation controlled by transcription factors Blimp-1 and T-bet. Nat. Immunol. 2016, 17, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Wherry, E.J.; Goldrath, A.W. Molecular regulation of effector and memory T cell differentiation. Nat. Immunol. 2014, 15, 1104–1115. [Google Scholar] [CrossRef]
- Buchholz, V.R.; Schumacher, T.N.; Busch, D.H. T Cell Fate at the Single-Cell Level. Ann. Rev. Immunol. 2016, 34, 65–92. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zheng, Y.; Han, Z.; Zhang, X. Functions and clinical significance of KLRG1 in the development of lung adenocarcinoma and immunotherapy. BMC Cancer 2021, 29, 752. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapodi, A.; Bognar, Z.; Szabo, C.; Gallyas, F.; Sumegi, B.; Hocsak, E. PARP inhibition induces Akt-mediated cytoprotective effects through the formation of a mitochondria-targeted phospho-ATM-NEMO-Akt-mTOR signalosome. Biochem. Pharmacol. 2019, 162, 98–108. [Google Scholar] [CrossRef]
- Ethier, C.; Tardif, M.; Arul, L.; Poirier, G.G. PARP-1 modulation of mTOR signaling in response to a DNA alkylating agent. PLoS ONE 2012, 7, e47978. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Qin, B.; Wu, F.; Qin, S.; Nowsheen, S.; Shan, S.; Zayas, J.; Pei, H.; Lou, Z.; Wang, L. Regulation of Serine-Threonine Kinase Akt Activation by NAD(+)-Dependent Deacetylase SIRT7. Cell Rep. 2017, 18, 1229–1240. [Google Scholar] [CrossRef]
- Ding, M.; Hu, L.; Yang, H.; Gao, C.; Zeng, K.; Yu, M.; Feng, J.; Qiu, J.; Liu, C.; Fu, F.; et al. Reduction of SIRT1 blunts the protective effects of ischemic post-conditioning in diabetic mice by impairing the Akt signaling pathway. Biochem. Biophys. Acta 2019, 1865, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Shu, R.; Yu, C.; Fu, Z.; Li, Z. Mammalian AKT, the Emerging Roles on Mitochondrial Function in Diseases. Aging Dis. 2022, 13, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharm. 2021, 12, 628690. [Google Scholar] [CrossRef]
- Macintyre, A.N.; Finlay, D.; Preston, G.; Sinclair, L.V.; Waugh, C.M.; Tamas, P.; Feijoo, C.; Okkenhaug, K.; Cantrell, D.A. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity 2011, 34, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.H.; Suresh, M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front. Immunol. 2013, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Henson, S.M.; Franzese, O.; Macaulay, R.; Libri, V.; Azevedo, R.I.; Kiani-Alikhan, S.; Plunkett, F.J. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8C T cells. Blood 2009, 113, 6619–6628. [Google Scholar] [CrossRef]
- Franzese, O.; Henson, S.M.; Naro, C.; Bonmassar, E. Defect in HSP90 expression in highly differentiated human CD8(+) T lymphocytes. Cell Death Dis. 2014, 5, e1294. [Google Scholar] [CrossRef] [Green Version]
- Kane, L.P.; Weiss, A. The PI-3 kinase/Akt pathway and T cell activation: Pleiotropic pathways downstream of PIP3. Immunol. Rev. 2003, 192, 7–20. [Google Scholar] [CrossRef]
- Finlay, D.; Cantrell, D.A. Metabolism, migration and memory in cytotoxic T cells. Nat. Rev. Immunol. 2011, 11, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzese, O.; Palermo, B.; Di Donna, C.; Sperduti, I.; Ferraresi, V.; Stabile, H.; Gismondi, A.; Santoni, A.; Nisticò, P. Polyfunctional Melan-A-specific tumor-reactive CD8(+) T cells elicited by dacarbazine treatment before peptide-vaccination depends on AKT activation sustained by ICOS. Oncoimmunology 2016, 5, e1114203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, Y.; Katayama, M.; Mirzoeva, O.K.; Berger, M.S.; Pieper, R.O. Akt activation suppresses Chk2-mediated, methylating agent-induced G2 arrest and protects from temozolomide-induced mitotic catastrophe and cellular senescence. Cancer Res. 2005, 65, 4861–4869. [Google Scholar] [CrossRef] [Green Version]
- Caporali, S.; Levati, L.; Starace, G.; Ragone, G.; Bonmassar, E.; Alvino, E.; D’Atri, S. AKT is activated in an n response to temozolomide and confers protection against drug-induced cell growth inhibition. Mol. Pharm. 2008, 74, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Harker, J.A.; Chanthong, K.; Stevenson, P.G.; Zuniga, E.I.; Rudd, C.E. Glycogen synthase kinase 3 inactivation drives T-bet-mediated downregulation of co- receptor PD-1 to enhance CD8(+) cytolytic T cell responses. Immunity 2016, 44, 274–286. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Rothstein, D.; Rudd, C.E. Small-Molecule Inhibition of PD-1 Transcription Is an Effective Alternative to Antibody Blockade in Cancer Therapy. Cancer Res. 2018, 78, 706–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiorano, B.A.; Lorusso, D.; Maiorano, M.; Ciardiello, D.; Parrella, P.; Petracca, A.; Cormio, G.; Maiello, E. The In-terplay between PARP Inhibitors and Immunotherapy in Ovarian Cancer: The Rationale behind a New Combination Therapy. Int. J. Mol. Sci. 2022, 23, 3871. [Google Scholar] [CrossRef] [PubMed]
- Kazanova, A.; Krueger, J.; Rudd, C.E. CD28 and glycogen synthase kinase-3 (GSK-3) inhibitor combination therapy generates tumor infiltrating T-cells (TILs) with a reversal of markers for T-cell exhaustion. J. Immunol. 2020, 204 (Suppl. 1), 165.35. [Google Scholar]
- Ghonim, M.A.; Ibba, S.V.; Tarhuni, A.F.; Errami, Y.; Luu, H.H.; Dean, M.J.; El-Bahrawy, A.H.; Wyczechowska, D.; Benslimane, I.A.; Del Valle, L.; et al. Targeting PARP-1 with metronomic therapy modulates MDSC suppressive function and enhances anti-PD-1 immunotherapy in colon cancer. J. Immunother. Cancer 2021, 9, e001643. [Google Scholar] [CrossRef]
- Wanderley, C.; Correa, T.S.; Scaranti, M.; Cunha, F.Q.; Barroso-Sousa, R. Targeting PARP1 to Enhance Anticancer Checkpoint Immunotherapy Response: Rationale and Clinical Implications. Front. Immunol. 2022, 13, 816642. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Cimino-Mathews, A.; Peer, C.J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Cao, L.; Harrell, M.I.; Swisher, E.M.; Houston, N.; et al. Safety and Clinical Activity of the Programmed Death-Ligand 1 Inhibitor Durvalumab in Combination with Poly (ADP-Ribose) Polymerase Inhibitor Olaparib or Vascular Endothelial Growth Factor Receptor 1-3 Inhibitor Cediranib in Women’s Cancers: A Dose-Escalation, phase I Study. J. Clin. Oncol. 2017, 35, 2193–2202. [Google Scholar] [CrossRef] [PubMed]
- Drew, Y.; de Jonge, M.; Hong, S.H.; Park, Y.H.; Wolfer, A.; Brown, J.; Ferguson, M.; Gore, M.E.; Alvarez, R.H.; Gresty, C.; et al. An Open- Label, phase II Basket Study of Olaparib and Durvalumab (MEDIOLA): Results in Germline BRCA-Mutated (gBRCAm) Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC). Gynecol. Oncol. 2018, 149, 246–247. [Google Scholar] [CrossRef]
- Domchek, S.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Gresty, C.; Opincar, L.M.; Herbolsheimer, P.; Kaufman, B. Phase II Study of Olaparib (O) and Durvalumab (D) (MEDIOLA): Updated Results in Patients (Pts) With Germline BRCA-Mutated (gBRCAm), Metastatic Breast Cancer (MBC). Ann. Oncol. 2019, 30, v477. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.E.; Vidal, G.A.; Mita, M.M.; Fleming, G.F.; Holloway, R.W.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. J. Clin. Oncol. 2018, 36, 106. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-Label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, M.; Meniawy, T.; Markman, B.; Mileshkin, L.; Harnett, P.; Millward, M.; Lundy, J.; Freimund, A.; Norris, C.; Mu, S.; et al. Pamiparib in Combination with Tislelizumab in Patients with Advanced Solid Tumours: Results from the Dose-Escalation Stage of a Multicentre, Open-Label, phase 1a/B Trial. Lancet Oncol. 2019, 20, 1306–1315. [Google Scholar] [CrossRef]
- Lampert, E.J.; Zimmer, A.; Padget, M.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: A Proof-of-Concept Phase II Study. Clin. Cancer Res. 2020, 26, 4268–4279. [Google Scholar] [CrossRef]
- Eskander, R.N.; Ledermann, J.A.; Birrer, M.J.; Fujiwara, K.; Gaillard, S.; Richardson, G.E.; Wei, C.; Baig, M.A.; Zohren, F.; Monk, B.J. JAVELIN ovarian PARP 100 study design: Phase III trial of avelumab + chemotherapy followed by avelumab + talazoparib maintenance in previously untreated epithelial ovarian cancer. J. Clin. Oncol. 2019, 37 (Suppl. 8), TPS9. [Google Scholar] [CrossRef]
- Thomas, A.; Vilimas, R.; Trindade, C.; Erwin-Cohen, R.; Roper, N.; Xi, L.; Krishnasamy, V.; Levy, E.; Mammen, A.; Nichols, S.; et al. Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a phase II Study. J. Thor. Oncol. 2019, 14, 1447–1457. [Google Scholar] [CrossRef]
- Hyman, D.M.; Zelnak, A.B.; Bauer, T.M.; Ulahannan, S.V.; Ford, J.M.; Cesari, R.; Hoyle, M.; Chappey, C.; Stewart, R.; Conte, U.; et al. JAVELIN BRCA/ATM: A phase 2 trial of avelumab (anti–PD-L1) plus talazoparib (PARP inhibitor) in patients with advanced solid tumors with a BRCA1/2 or ATM defect. J. Clin. Oncol. 2019, 37, TPS2660. [Google Scholar] [CrossRef]
- Hardy-Bessard, A.C.; Moore, K.N.; Mirza, M.R.; Asselain, B.; Redondo, A.; Pfisterer, J.; Pignata, S.; Provencher, D.M.; Cibula, D.; Reyners, A.K.L.; et al. ENGOT-OV44/FIRST study: A randomized, double-blind, adaptive, phase III study of platinum-based therapy with dostarlimab (TSR-042) + niraparib versus standard-of-care (SOC) platinum-based therapy as first-line treatment of stage 3/4 non-mucinous epithelial ovarian cancer (OC). J. Clin. Oncol. 2019, 37 (Suppl. 15), TPS5600. [Google Scholar] [CrossRef]
- Fujiwara, K.; Vergote, I.; Sehouli, J.; Salutari, V.; Zola, P.; Madry, R.; Wenham, R.M.; Korach, J.; Pautier, P.; Cibula, D.; et al. ENGOT-ov43/KEYLYNK-001: A Phase III trial of pembrolizumab plus chemotherapy with olaparib maintenance for first-line treatment of BRCA’-nonmutated advanced epithelial ovarian cancer. Ann. Oncol. 2019, 30, ix89. [Google Scholar] [CrossRef]
- Monk, B.J.; Coleman, R.L.; Fujiwara, K.; Wilson, M.K.; Oza, A.M.; Oaknin, A.; O’Malley, D.M.; Lorusso, D.; Westin, S.N.; Safra, T.; et al. ATHENA (GOG-3020/ENGOT-ov45): A randomized, Phase III trial to evaluate rucaparib as monotherapy (ATHENA-MONO) and rucaparib in combination with nivolumab (ATHENA-COMBO) as maintenance treatment following frontline platinum-based chemotherapy in ovarian cancer. Int. J. Gynecol. Cancer 2021, 31, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Harter, P.; Budinski, M.; Colombo, N.; Floquet, A.; Rubio Pérez, M.J.; Kim, J.-V.; Lheureux, S.; Marth, C.; Nyvang, G.B.; Okamoto, A.; et al. DUO-O: A randomized Phase III trial of durvalumab (durva) in combination with chemotherapy and bevacizumab (bev), followed by maintenance durva, bev and olaparib (olap), in newly diagnosed advanced ovarian cancer patients. J. Clin. Oncol. 2019, 37 (Suppl. 15), TPS5598. [Google Scholar] [CrossRef]
- Musacchio, L.; Salutari, V.; Pignata, S.; Braicu, E.; Cibula, D.; Colombo, N.; Frenel, J.S.; Zagouri, F.; Carbone, V.; Ghizzoni, V.; et al. Randomized phase III trial on niraparib-TSR-042 (dostarlimab) versus physician’s choice chemotherapy in recurrent ovarian, fallopian tube, or primary peritoneal cancer patients not candidate for platinum retreatment: NItCHE trial (MITO 33). Int. J. Gyn. Cancer 2021, 31, 1369–1373. [Google Scholar] [CrossRef]
- Gonzalez Martin, A.; Sanchez Lorenzo, L.; Colombo, N.; dePont Christensen, R.; Heitz, F.; Meirovitz, M.; Selle, F.; van Gorp, T.; Alvarez, N.; Sanchez, J. A phase III, randomized, double blinded trial of platinum based chemotherapy with or without atezolizumab followed by niraparib maintenance with or without atezolizumab in patients with recurrent ovarian, tubal, or peritoneal cancer and platinum treatment free interval of more than 6 months: ENGOT-Ov41/GEICO 69-O/ANITA Trial. Int. J. Gynecol. Cancer 2021, 31, 617–622. [Google Scholar] [CrossRef]
- Ray-Coquard, I.L.; Leary, A.; Bigot, F.; Montane, L.; Fabbro, M.; Hardy-Bessard, A.-C.; Selle, F.; Chakiba, C.; Lortholary, A.; Berton, D.; et al. ROCSAN trial (GINECO-EN203b/ENGOT-EN8): A multicenter randomized phase 2/3 evaluating dostarlimab in combination with niraparib versus niraparib alone compared to chemotherapy in the treatment of endometrial/ovarian carcinosarcoma after at least one line of platinum-based chemotherapy. J. Clin. Oncol. 2021, 39, 15. [Google Scholar] [CrossRef]
- Neville Westin, S.; Moore, K.N.; Van Nieuwenhuysen, E.; Oza, A.M.; Mileshkin, L.R.; Okamoto, A.; Suzuki, A.; Meyer, K.; Barker, L.; Rhee, J.; et al. DUO-E/GOG-3041/ENGOT-EN10: A randomized Phase III trial of first-line carboplatin (carb) and paclitaxel (pac) in combination with durvalumab (durva), followed by maintenance durva with or without olaparib (ola), in patients (pts) with newly diagnosed (nd) advanced or recurrent endometrial cancer (EC). J. Clin. Oncol. 2020, 38, 15. [Google Scholar] [CrossRef]
- Singh, D.D.; Parveen, A.; Yadav, D.K. Role of PARP in TNBC: Mechanism of Inhibition, Clinical Applications, and Resistance. Biomedicines 2021, 9, 1512. [Google Scholar] [CrossRef]
- Rugo, H.S.; Llombart-Cussac, A.; Andre, F.; Robson, M.E.; Saji, S.; Harbeck, N.; Schmid, P.; Cescon, D.V.; Ahn, J.S.; Nanda, R.; et al. KEYLYNK-009: A Phase II/III, open-label, randomized study of pembrolizumab (pembro) plus olaparib vs pembro plus chemotherapy after induction with first line pembro plus chemotherapy in patients with locally recurrent inoperable or metastatic triple-negative breast cancer (TNBC). J. Clin. Oncol. 2020, 38 (Suppl. 15), TPS596. [Google Scholar]
- Ji, W.; Weng, X.; Xu, D.; Cai, S.; Lou, H.; Ding, L. Non-small cell lung cancer cells with deficiencies in homologous recombination genes are sensitive to PARP inhibitors. Biochem. Biophys. Res. Commun. 2020, 522, 121–126. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; de Castro, G., Jr.; Garassino, M.C.C.; Mazieres, J.; Sanborn, R.E.; Smit, E.F.F.; Spigel, D.R.; Thomas, M.; Velcheti, V.; Zhi, E.; et al. First-line (1L) maintenance therapy with niraparib (nira) + pembrolizumab (pembro) vs placebo + pembro in advanced/ metastatic non-small cell lung cancer (NSCLC): Phase III ZEAL-1L study. Ann. Oncol. 2021, 32, 5. [Google Scholar] [CrossRef]
- Gray, J.E.; Owonikoko, T.K.; Kato, T.; Cardona Zorrilla, A.F.; Wagman, J.; Chen, X.; Zhao, B.; Schulz, C. Randomized Phase III study of first line pembrolizumab plus pemetrexed/platinum followed by pembrolizumab and maintenance olaparib versus pemetrexed in patients with metastatic nonsquamous non-small cell lung cancer (NSCLC): KEYLYNK-006. J. Clin. Oncol. 2020, 38 (Suppl. 15), TPS9632. [Google Scholar] [CrossRef]
- Jabbour, S.K.; Cho, B.C.; Bria, E.; Kato, T.; Bhosle, J.; Gainor, J.F.; Reguart, N.; Wang, L.; Morgensztern, D.; Gurary, E.; et al. Phase 3 study of pembrolizumab with concurrent chemoradiation therapy followed by pembrolizumab with or without olaparib versus concurrent chemoradiation therapy followed by durvalumab in unresectable, locally advanced, stage III non-small cell lung cancer: KEYLYNK-012. J. Clin. Oncol. 2021, 39, 15. [Google Scholar] [CrossRef]
- Gray, J.E.; Owonikoko, T.K.; Kato, T.; Nadal, E.; Greystoke, A.; Cardona, A.F.; Pen-rod, J.; Wei, Z.; Lara-Guerra, H.; Schulz, C. Randomized, placebo-controlled Phase III study of 1L pembrolizumab (Pembro) plus carboplatin/taxane followed by pembro with or without maintenance olaparib in patients (Pts) with metastatic squamous non-small cell lung cancer (sqNSCLC): KEYLYNK-008. Ann. Oncol. 2020, 31, S4. [Google Scholar] [CrossRef]
- Yu, E.Y.; Park, S.H.; Huang, Y.-H.; Bennamoun, M.; Xu, L.; Kim, J.; Antonarakis, E.S. Phase III study of pembrolizumab (pembro) plus olaparib versus enzalutamide (enza) or abiraterone acetate (abi) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) who progressed on chemotherapy: KEYLYNK-010. J. Clin. Oncol. 2020, 38, 6. [Google Scholar] [CrossRef]
- Markham, A. Pamiparib: First Approval. Drugs 2021, 81, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.; Meniawy, T.; Markman, B.; Mileshkin, L.R.; Harnett, P.; Millward, M.; Lundy, J.; Freimund, A.E.; Norris, C.; Wu, J.; et al. A phase 1b study of the anti-PD-1 monoclonal antibody BGB-A317 (A317) in combination with the PARP inhibitor BGB-290 (290) in advanced solid tumors. J. Clin. Oncol. 2018, 36 (Suppl. 48), 48. [Google Scholar] [CrossRef]
- Adams, S.F.; Rixe, O.; Lee, J.H.; McCance, D.J.; Westgate, S.; Eberhardt, S.C.; Rutledge, T.; Muller, C. Phase I Study Combining Olaparib and Tremelimumab for the Treatment of Women With BRCA-Deficient Recurrent Ovarian Cancer. J. Clin. Oncol. 2017, 35, e17052. [Google Scholar] [CrossRef]
- Skelding, K.A.; Lincz, L.F. PARP Inhibitors and Haematological Malignancies-Friend or Foe? Cancers 2021, 13, 5328. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Padella, A.; Ghelli Luserna Di Rorà, A.; Marconi, G.; Ghetti, M.; Martinelli, G.; Simonetti, G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 10. [Google Scholar] [CrossRef]
- Gojo, I.; Beumer, J.H.; Pratz, K.W.; McDevitt, M.A.; Baer, M.R.; Blackford, A.L.; Smith, B.D.; Gore, S.D.; Carraway, H.E.; Showel, M.M.; et al. A Phase 1 Study of the PARP Inhibitor Veliparib in Combination with Temozolomide in Acute Myeloid Leukemia. Clin. Cancer Res. 2017, 23, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Pratz, K.W.; Rudek, M.A.; Gojo, I.; Litzow, M.R.; McDevitt, M.A.; Ji, J.; Karnitz, L.M.; Herman, J.G.; Kinders, R.J.; Smith, B.D.; et al. A Phase I Study of Topotecan, Carboplatin and the PARP Inhibitor Veliparib in Acute Leukemias, Aggressive Myeloproliferative Neoplasms, and Chronic Myelomonocytic Leukemia. Clin. Cancer Res. 2017, 23, 899–907. [Google Scholar] [CrossRef] [Green Version]
- Aurelius, J.; Thorén, F.B.; Akhiani, A.A.; Brune, M.; Palmqvist, L.; Hansson, M.; Hellstrand, K.; Martner, A. Monocytic AML cells inactivate antileukemic lymphocytes: Role of NADPH oxidase/gp91(phox) expression and the PARP-1/PAR pathway of apoptosis. Blood 2012, 119, 5832–5837. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Xu, X.; Chen, W. The Current State of the Art in PARP Inhibitor-Based Delivery Nanosystems. Pharmaceutics 2022, 14, 1647. [Google Scholar] [CrossRef]
- Zhang, D.; Baldwin, P.; Leal, A.S.; Carapellucci, S.; Sridhar, S.; Liby, K.T. A nano-liposome formulation of the PARP inhibitor Talazoparib enhances treatment efficacy and modulates immune cell populations in mammary tumors of BRCA-deficient mice. Theranostics 2019, 9, 6224–6238. [Google Scholar] [CrossRef]
- Sharma, M.R.; Stadler, W.M.; Ratain, M.J. Randomized phase II trials: A long-term investment with promising returns. J. Natl. Cancer Inst. 2011, 103, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.A.; Pilié, P.G.; Yap, T.A. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018, 78, 6717–6725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makvandi, M.; Xu, K.; Lieberman, B.P.; Anderson, R.C.; Effron, S.S.; Winters, H.D.; Zeng, C.; McDonald, E.S.; Pryma, D.A.; Greenberg, R.A.; et al. A Radiotracer Strategy to Quantify PARP-1 Expression In Vivo Provides a Biomarker That Can Enable Patient Selection for PARP Inhibitor Therapy. Cancer Res. 2016, 76, 4516–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, T.A.; Ains-worth, W.B.; Ellis, P.A.; Donawho, C.K.; Di Giammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2019, 17, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Makvandi, M.; Pantel, A.; Schwartz, L.; Schubert, E.; Xu, K.; Hsieh, C.J.; Hou, C.; Kim, H.; Weng, C.C.; Winters, H.; et al. A PET imaging agent for evaluating PARP-1 expression in ovarian cancer. J. Clin. Investig. 2018, 128, 2116–2126. [Google Scholar] [CrossRef]
- McDonald, E.S.; Pantel, A.R.; Shah, P.D.; Farwell, M.D.; Clark, A.S.; Doot, R.K.; Pryma, D.A.; Carlin, S.D. In vivo visualization of PARP inhibitor pharmacodynamics. JCI Insight 2021, 6, e146592. [Google Scholar] [CrossRef] [PubMed]



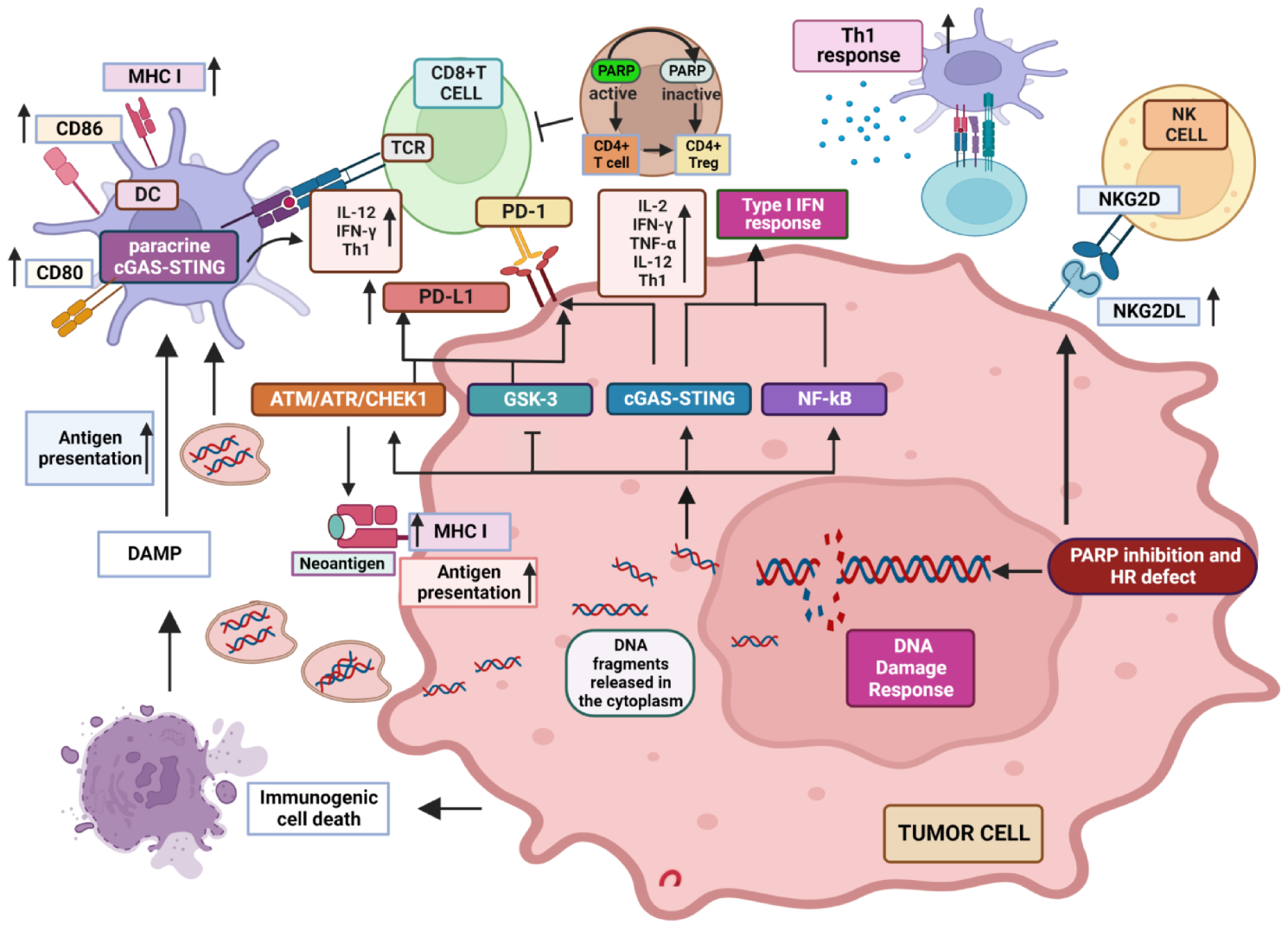{kind=link}
{kind=link}
| PARPi | ICI | Trial | Combinations Including PARPi | Identifier—Reference |
|---|---|---|---|---|
| talazoparib | avelumab (anti PD-L1) | JAVELIN ovarian PARP 100 | Maintenance therapy with talazoparib plus/minus avelumab following avelumab plus chemotherapy in untreated advanced ovarian cancer | NCT03642132 [290] |
| niraparib | dostarlimab (anti PD-1) | ENGOT-0V44/FIRST | First-line treatment with niraparib plus platinum-based therapy and dostarlimab vs. SOC platinum-based therapy of advanced non-mucinous epithelial ovarian cancer | NCT03602859 [293] |
| olaparib | pembrolizumab (anti PD-1) | ENGOT-OV43/ KEYLYNK-001 | Maintenance therapy with olaparib plus/minus pembrolizumab following first line carboplatin/paclitaxel in BRCA-wild-type epithelial ovarian cancer | NCT03740165 [294] |
| rucaparib | nivolumab (anti PD-1) | ATHENA | Maintenance therapy with rucaparib and nivolumab following response to front-line treatment in newly diagnosed ovarian cancer | NCT03522246 [295] |
| olaparib | durvalumab (anti PD-L1) | DUO-O | Maintenance therapy with olaparib plus bevacizumab and durvalumab, preceded by platinum-based chemotherapy plus bevacizumab in newly diagnosed ovarian cancer | NCT03737643 [296] |
| niraparib | dostarlimab (anti PD-1) | NItCHE- MITO33 | Niraparib plus dostarlimab vs. chemotherapy in recurrent ovarian, fallopian tube or primary peritoneal cancer | NCT04679064 [297] |
| niraparib | atezolizumab (anti PD-L1) | ANITA | Maintenance with niraparib plus/minus atezolizumab following platinum-based chemotherapy plus/minus atezolizumabin in recurrent ovarian, tubal or peritoneal cancer | NCT03598270 [298] |
| niraparib | dostarlimab (anti PD-1) | ROCSAN | Niraparib plus dostarlimab and niraparib alone vs. chemotherapy in recurrent ovarian carcinosarcoma | NCT03651206 [299] |
| olaparib | durvalumab (anti PD-L1) | DUO-E | Maintenance therapy with durvalumab plus/minus olaparib after first line treatment in advanced and recurrent endometrial cancer | NCT04269200 [300] |
| niraparib | dostarlimab (anti PD-1) | RUBY part 2 | Niraparib plus dostarlimab after dostarlimab and chemotherapy in recurrent or primary advanced endometrial cancer | NCT03981796 |
| olaparib | pembrolizumab (anti PD-1) | KEYLYNK-009 | Maintenance with olaparib plus pembrolizumab vs. chemotherapy plus pembrolizumab after first-line chemotherapy plus pembrolizumab in TNBC | NCT04191135 [302] |
| niraparib | pembrolizumab (anti PD-1) | ZEAL-1L | Maintenance therapy with pembrolizumab plus/minus niraparib following SOC first-line platinum-based chemotherapy and pembrolizumab in advanced/metastatic NSCLC | NCT04475939 [304] |
| olaparib | pembrolizumab (anti PD-1) | KEYLINK-006 | Pembrolizumab plus maintenance olaparib vs. pembrolizumab plus maintenance pemetrexed in non-squamous NSCLC | NCT03976323 [305] |
| olaparib | pembrolizumab (anti PD-1) | KEYLYNK-012 | Pembrolizumab and CCRT followed by maintenance with pembrolizumab plus/minus olaparib placebo vs. CCRT followed by durvalumab in NSCLC | NCT04380636 [306] |
| olaparib | pembrolizumab (anti PD-1) | KEYLYNK-008 | Maintenance therapy with pembrolizumab plus/minus olaparib in first line squamous NSCLC | NCT03976362 [307] |
| olaparib | pembrolizumab (anti PD-1) | KEYLYNK-010 | Pembrolizumab plus olaparib vs. hormone therapy in mCRPC unselected for HR repair defects resistant to hormone/chemotherapy | NCT03834519 [308] |
| pamiparib | tislelizumab (anti PD-1) | - | Tislelizumab plus or minus pamiparib in advanced malignancies | NCT04164199 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzese, O.; Graziani, G. Role of PARP Inhibitors in Cancer Immunotherapy: Potential Friends to Immune Activating Molecules and Foes to Immune Checkpoints. Cancers 2022, 14, 5633. https://doi.org/10.3390/cancers14225633
Franzese O, Graziani G. Role of PARP Inhibitors in Cancer Immunotherapy: Potential Friends to Immune Activating Molecules and Foes to Immune Checkpoints. Cancers. 2022; 14(22):5633. https://doi.org/10.3390/cancers14225633
Chicago/Turabian StyleFranzese, Ornella, and Grazia Graziani. 2022. "Role of PARP Inhibitors in Cancer Immunotherapy: Potential Friends to Immune Activating Molecules and Foes to Immune Checkpoints" Cancers 14, no. 22: 5633. https://doi.org/10.3390/cancers14225633