
Genetic Inactivation of Notch1 Synergizes with Loss of Trp53 to Induce Tumor Formation in the Adult Mouse Forebrain

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Tamoxifen Administration
2.3. Tissue Preparation and Immunohistochemistry
3. Results
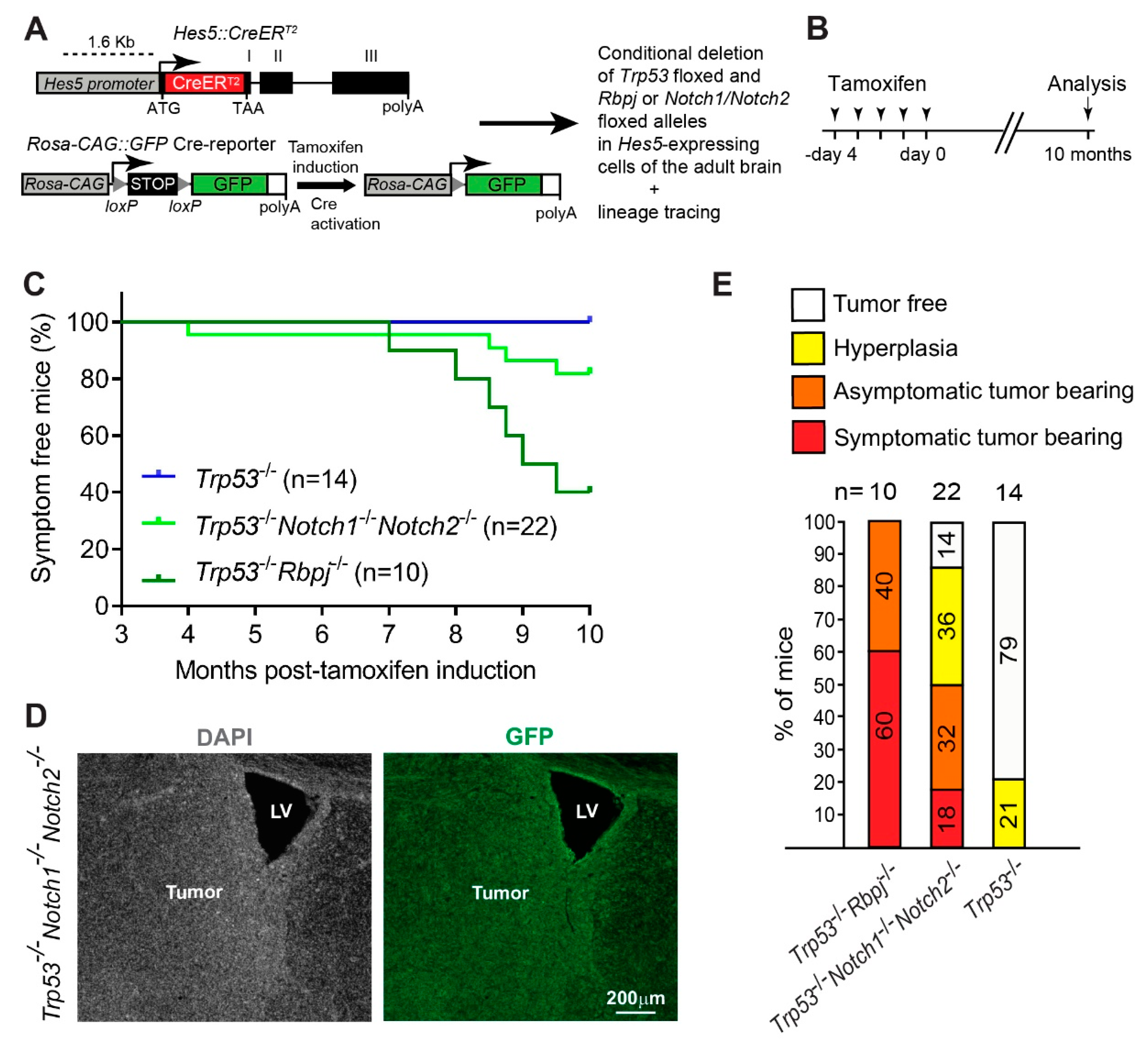
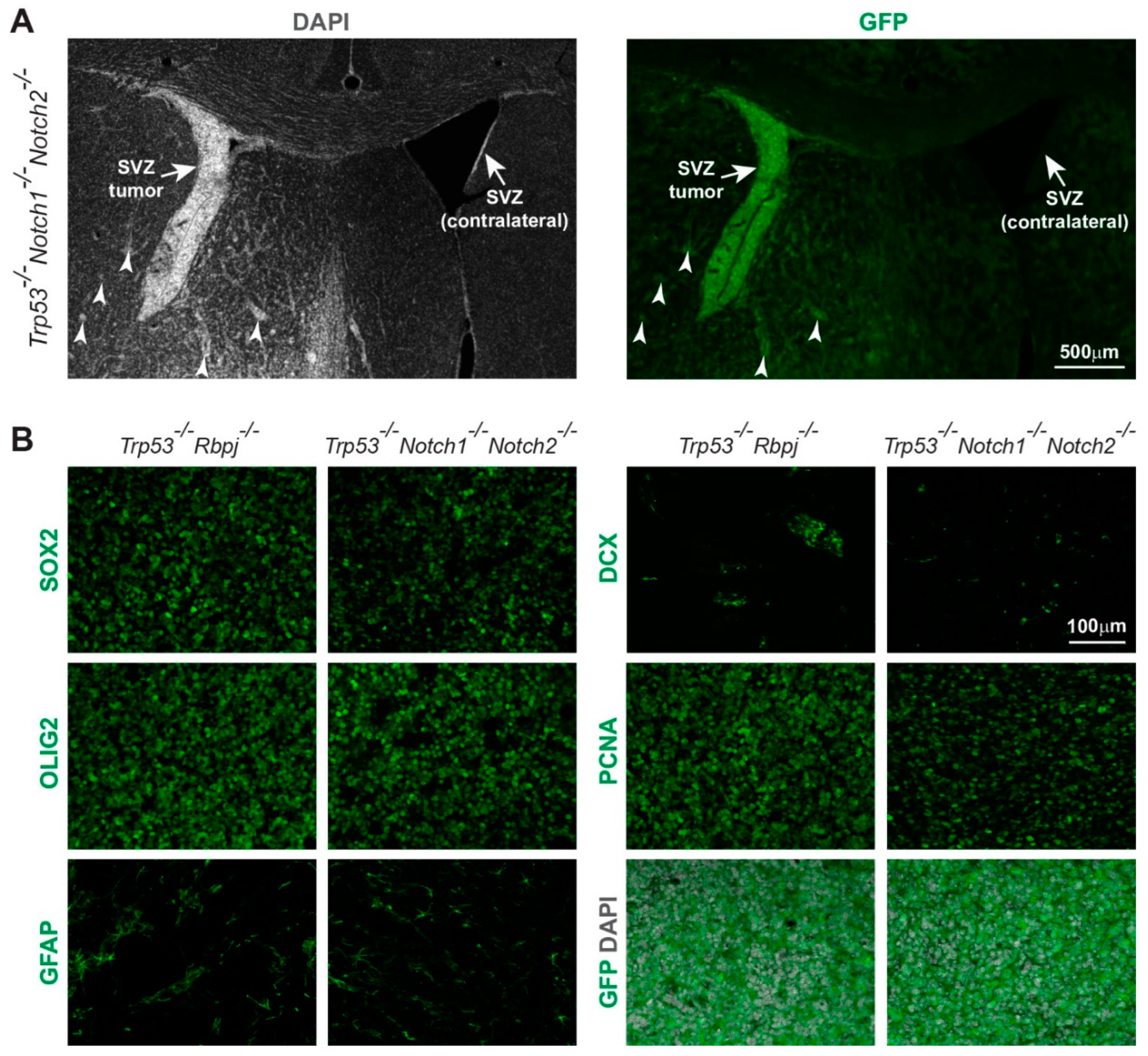
3.1. Combined Deletion of Notch1/Notch2 and Trp53 in Hes5+ Cells Leads to the Formation of Forebrain Tumors in Adult Mice
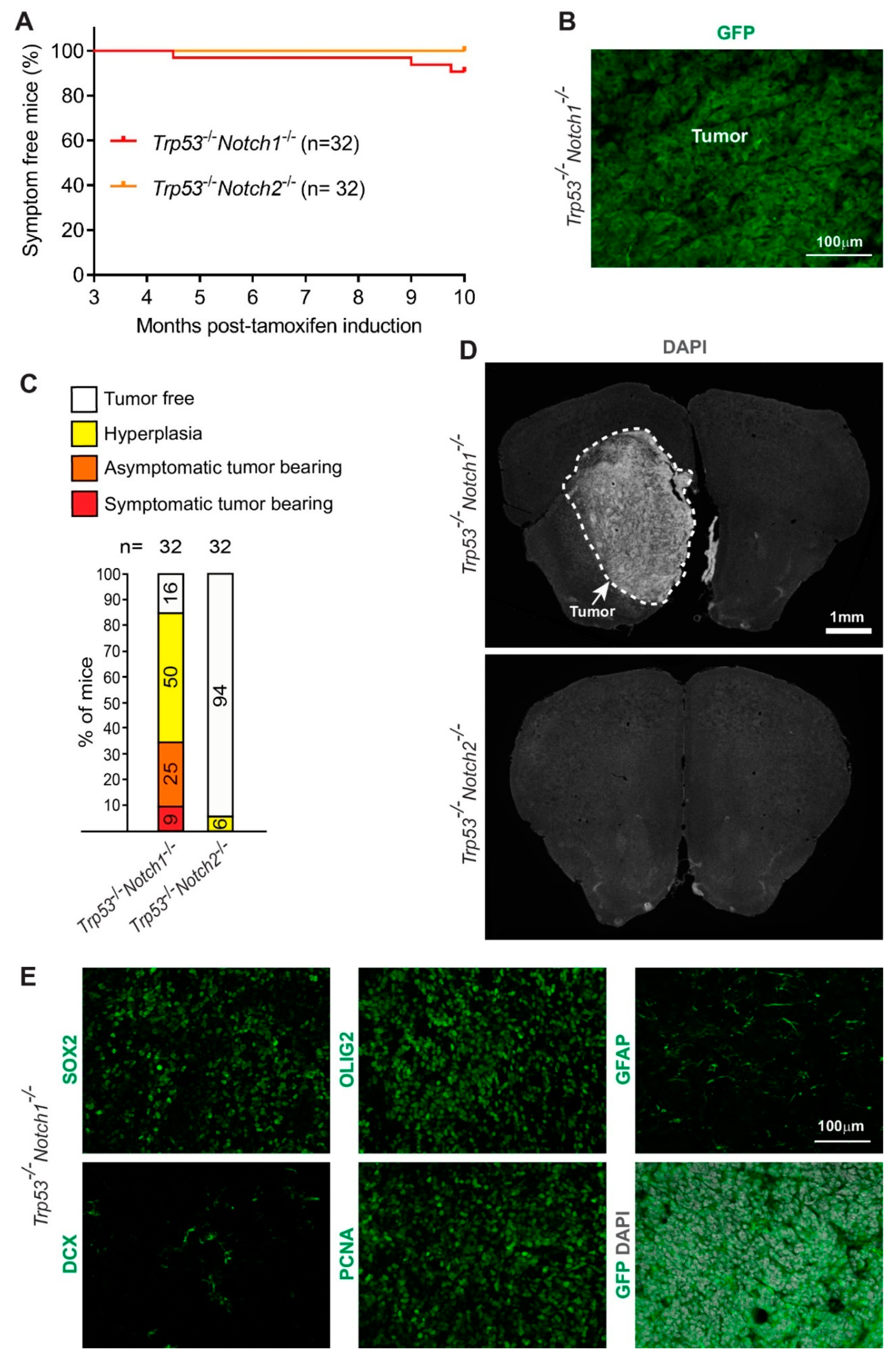
3.2. Deletion of Notch1 and Trp53 in Hes5+ Cells Leads to the Formation of Forebrain Tumors in Adult Mice
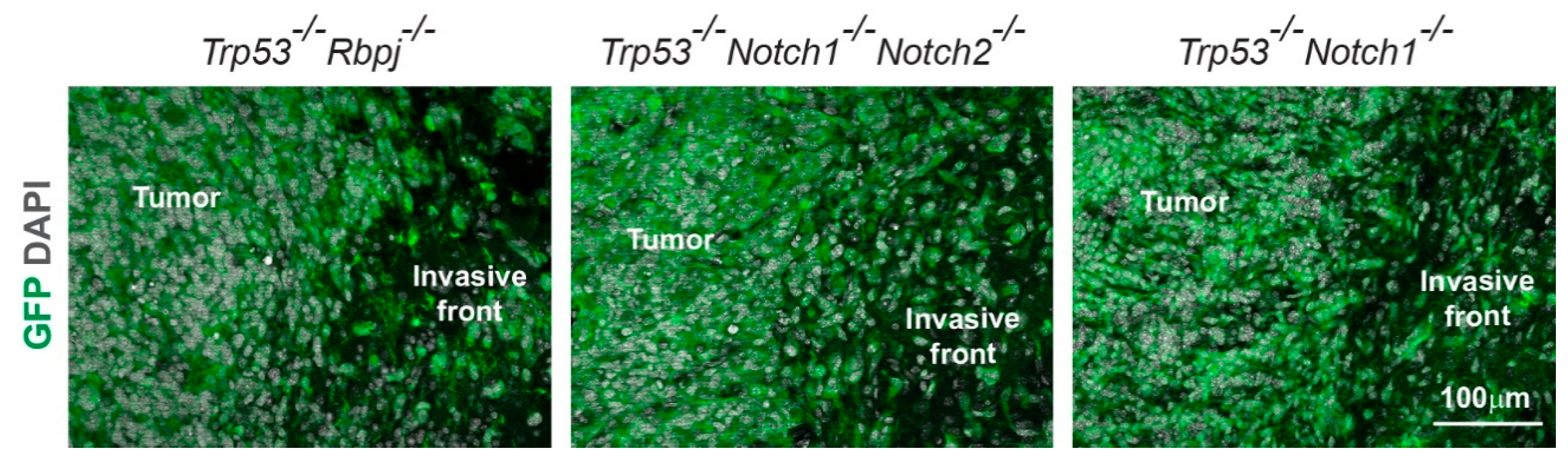
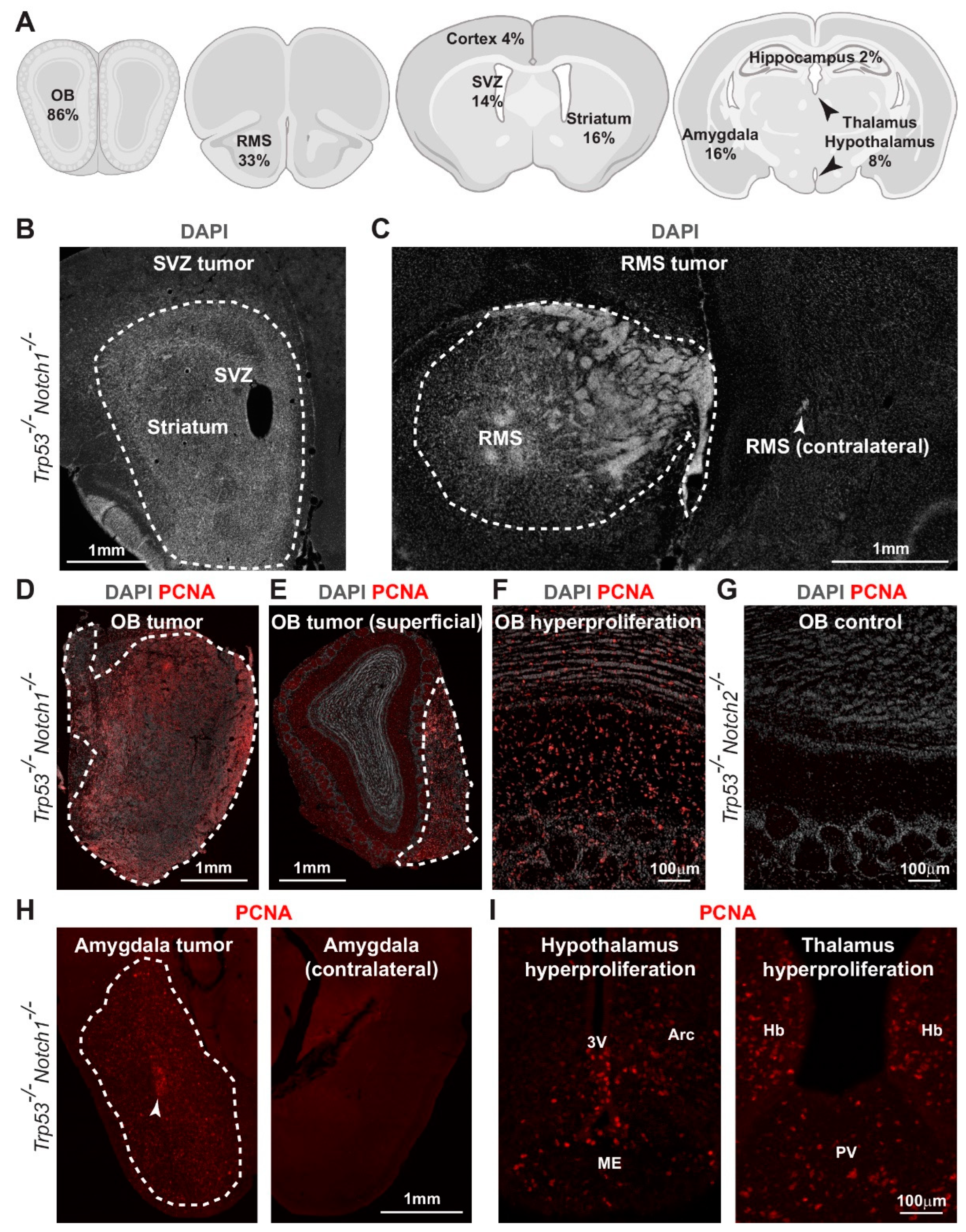
3.3. Hyperplasia and Tumors Are Preferentially Located in Ventral Forebrain Regions and OBs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ntziachristos, P.; Lim, J.S.; Sage, J.; Aifantis, I. From fly wings to targeted cancer therapies: A centennial for notch signaling. Cancer Cell 2014, 25, 318–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, C.S.; Radtke, F. Notch as a tumour suppressor. Nat. Rev. Cancer 2017, 17, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Ibaseta, A.; Fischer, M.M.; Cancilla, B.; O’Young, G.; Cristea, S.; Luca, V.C.; Yang, D.; Jahchan, N.S.; Hamard, C.; et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017, 545, 360–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmigiani, E.; Taylor, V.; Giachino, C. Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma. Cells 2020, 9, e2304. [Google Scholar] [CrossRef]
- Klinakis, A.; Lobry, C.; Abdel-Wahab, O.; Oh, P.; Haeno, H.; Buonamici, S.; van De Walle, I.; Cathelin, S.; Trimarchi, T.; Araldi, E.; et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature 2011, 473, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Jung, E.; Osswald, M.; Ratliff, M.; Dogan, H.; Xie, R.; Weil, S.; Hoffmann, D.C.; Kurz, F.T.; Kessler, T.; Heiland, S.; et al. Tumor cell plasticity, heterogeneity, and resistance in crucial microenvironmental niches in glioma. Nat. Commun. 2021, 12, 1014. [Google Scholar] [CrossRef]
- Koch, U.; Lehal, R.; Radtke, F. Stem cells living with a Notch. Development 2013, 140, 689–704. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Kageyama, R. Lysosomes and signaling pathways for maintenance of quiescence in adult neural stem cells. FEBS J. 2021, 288, 3082–3093. [Google Scholar] [CrossRef]
- Basak, O.; Giachino, C.; Fiorini, E.; Macdonald, H.R.; Taylor, V. Neurogenic subventricular zone stem/progenitor cells are Notch1-dependent in their active but not quiescent state. J. Neurosci. 2012, 32, 5654–5666. [Google Scholar] [CrossRef] [Green Version]
- Basak, O.; Taylor, V. Identification of self-replicating multipotent progenitors in the embryonic nervous system by high Notch activity and Hes5 expression. Eur. J. Neurosci. 2007, 25, 1006–1022. [Google Scholar] [CrossRef]
- Ehm, O.; Goritz, C.; Covic, M.; Schaffner, I.; Schwarz, T.J.; Karaca, E.; Kempkes, B.; Kremmer, E.; Pfrieger, F.W.; Espinosa, L.; et al. RBPJkappa-dependent signaling is essential for long-term maintenance of neural stem cells in the adult hippocampus. J. Neurosci. 2010, 30, 13794–13807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, A.; Rolando, C.; Giachino, C.; Saotome, I.; Erni, A.; Brien, C.; Zhang, R.; Zimber-Strobl, U.; Radtke, F.; Artavanis-Tsakonas, S.; et al. Notch2 Signaling Maintains NSC Quiescence in the Murine Ventricular-Subventricular Zone. Cell Rep. 2018, 22, 992–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giachino, C.; Basak, O.; Lugert, S.; Knuckles, P.; Obernier, K.; Fiorelli, R.; Frank, S.; Raineteau, O.; Alvarez-Buylla, A.; Taylor, V. Molecular diversity subdivides the adult forebrain neural stem cell population. Stem Cells 2014, 32, 70–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imayoshi, I.; Sakamoto, M.; Yamaguchi, M.; Mori, K.; Kageyama, R. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J. Neurosci. 2010, 30, 3489–3498. [Google Scholar] [CrossRef] [Green Version]
- Lugert, S.; Basak, O.; Knuckles, P.; Haussler, U.; Fabel, K.; Gotz, M.; Haas, C.A.; Kempermann, G.; Taylor, V.; Giachino, C. Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell 2010, 6, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Lugert, S.; Vogt, M.; Tchorz, J.S.; Muller, M.; Giachino, C.; Taylor, V. Homeostatic neurogenesis in the adult hippocampus does not involve amplification of Ascl1(high) intermediate progenitors. Nat. Commun. 2012, 3, 670. [Google Scholar] [CrossRef] [Green Version]
- Sueda, R.; Imayoshi, I.; Harima, Y.; Kageyama, R. High Hes1 expression and resultant Ascl1 suppression regulate quiescent vs. active neural stem cells in the adult mouse brain. Genes Dev. 2019, 33, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Boareto, M.; Engler, A.; Louvi, A.; Giachino, C.; Iber, D.; Taylor, V. Id4 Downstream of Notch2 Maintains Neural Stem Cell Quiescence in the Adult Hippocampus. Cell Rep. 2019, 28, 1485–1498.e6. [Google Scholar] [CrossRef]
- Imayoshi, I.; Isomura, A.; Harima, Y.; Kawaguchi, K.; Kori, H.; Miyachi, H.; Fujiwara, T.; Ishidate, F.; Kageyama, R. Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science 2013, 342, 1203–1208. [Google Scholar] [CrossRef] [Green Version]
- Parmigiani, E.; Ivanek, R.; Rolando, C.; Hafen, K.; Turchinovich, G.; Lehmann, F.M.; Gerber, A.; Brkic, S.; Frank, S.; Meyer, S.C.; et al. Interferon-gamma resistance and immune evasion in glioma develop via Notch-regulated co-evolution of malignant and immune cells. Dev. Cell 2022, 57, 1847–1865. [Google Scholar] [CrossRef]
- Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.W.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Nakamura, H.; Suzuki, H.; Matsuo, K.; Kataoka, K.; Shimamura, T.; Motomura, K.; Ohka, F.; Shiina, S.; Yamamoto, T.; et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro. Oncol. 2018, 20, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Bai, H.; Harmanci, A.S.; Erson-Omay, E.Z.; Li, J.; Coskun, S.; Simon, M.; Krischek, B.; Ozduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Giachino, C.; Boulay, J.L.; Ivanek, R.; Alvarado, A.; Tostado, C.; Lugert, S.; Tchorz, J.; Coban, M.; Mariani, L.; Bettler, B.; et al. A Tumor Suppressor Function for Notch Signaling in Forebrain Tumor Subtypes. Cancer Cell 2015, 28, 730–742. [Google Scholar] [CrossRef] [Green Version]
- Halani, S.H.; Yousefi, S.; Velazquez Vega, J.; Rossi, M.R.; Zhao, Z.; Amrollahi, F.; Holder, C.A.; Baxter-Stoltzfus, A.; Eschbacher, J.; Griffith, B.; et al. Multi-faceted computational assessment of risk and progression in oligodendroglioma implicates NOTCH and PI3K pathways. NPJ Precis. Oncol. 2018, 2, 24. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Somasundaram, K.; Reddy, S.P.; Vinnakota, K.; Britto, R.; Subbarayan, M.; Nambiar, S.; Hebbar, A.; Samuel, C.; Shetty, M.; Sreepathi, H.K.; et al. Upregulation of ASCL1 and inhibition of Notch signaling pathway characterize progressive astrocytoma. Oncogene 2005, 24, 7073–7083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettegowda, C.; Agrawal, N.; Jiao, Y.; Sausen, M.; Wood, L.D.; Hruban, R.H.; Rodriguez, F.J.; Cahill, D.P.; McLendon, R.; Riggins, G.; et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 2011, 333, 1453–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besseyrias, V.; Fiorini, E.; Strobl, L.J.; Zimber-Strobl, U.; Dumortier, A.; Koch, U.; Arcangeli, M.L.; Ezine, S.; Macdonald, H.R.; Radtke, F. Hierarchy of Notch-Delta interactions promoting T cell lineage commitment and maturation. J. Exp. Med. 2007, 204, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Tanigaki, K.; Yamamoto, N.; Kuroda, K.; Yoshimoto, M.; Nakahata, T.; Ikuta, K.; Honjo, T. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int. Immunol. 2002, 14, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Peterse, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001, 29, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Radtke, F.; Wilson, A.; Stark, G.; Bauer, M.; van Meerwijk, J.; MacDonald, H.R.; Aguet, M. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 1999, 10, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Tchorz, J.S.; Suply, T.; Ksiazek, I.; Giachino, C.; Cloetta, D.; Danzer, C.P.; Doll, T.; Isken, A.; Lemaistre, M.; Taylor, V.; et al. A modified RMCE-compatible Rosa26 locus for the expression of transgenes from exogenous promoters. PLoS ONE 2012, 7, e30011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessandrini, F.; Ceresa, D.; Appolloni, I.; Pagani, F.; Poliani, P.L.; Marubbi, D.; Malatesta, P. Glioblastoma models driven by different mutations converge to the proneural subtype. Cancer Lett. 2020, 469, 447–455. [Google Scholar] [CrossRef]
- Braune, E.B.; Tsoi, Y.L.; Phoon, Y.P.; Landor, S.; Silva Cascales, H.; Ramskold, D.; Deng, Q.; Lindqvist, A.; Lian, X.; Sahlgren, C.; et al. Loss of CSL Unlocks a Hypoxic Response and Enhanced Tumor Growth Potential in Breast Cancer Cells. Stem Cell Rep. 2016, 6, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Kulic, I.; Robertson, G.; Chang, L.; Baker, J.H.; Lockwood, W.W.; Mok, W.; Fuller, M.; Fournier, M.; Wong, N.; Chou, V.; et al. Loss of the Notch effector RBPJ promotes tumorigenesis. J. Exp. Med. 2015, 212, 37–52. [Google Scholar] [CrossRef]
- Chow, R.D.; Guzman, C.D.; Wang, G.; Schmidt, F.; Youngblood, M.W.; Ye, L.; Errami, Y.; Dong, M.B.; Martinez, M.A.; Zhang, S.; et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 2017, 20, 1329–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Mizuno, H.; Imayoshi, I.; Furusawa, C.; Shirahige, K.; Kageyama, R. The cyclic gene Hes1 contributes to diverse differentiation responses of embryonic stem cells. Genes Dev. 2009, 23, 1870–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, H.; Kawaguchi, D.; Kuebrich, B.D.; Kitamoto, T.; Yamaguchi, M.; Gotoh, Y.; Furutachi, S. Area-Specific Regulation of Quiescent Neural Stem Cells by Notch3 in the Adult Mouse Subependymal Zone. J. Neurosci. 2017, 37, 11867–11880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alunni, A.; Krecsmarik, M.; Bosco, A.; Galant, S.; Pan, L.; Moens, C.B.; Bally-Cuif, L. Notch3 signaling gates cell cycle entry and limits neural stem cell amplification in the adult pallium. Development 2013, 140, 3335–3347. [Google Scholar] [CrossRef] [Green Version]
- Alcantara Llaguno, S.; Sun, D.; Pedraza, A.M.; Vera, E.; Wang, Z.; Burns, D.K.; Parada, L.F. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat. Neurosci. 2019, 22, 545–555. [Google Scholar] [CrossRef]
- Hu, Q.D.; Ang, B.T.; Karsak, M.; Hu, W.P.; Cui, X.Y.; Duka, T.; Takeda, Y.; Chia, W.; Sankar, N.; Ng, Y.K.; et al. F3/contactin acts as a functional ligand for Notch during oligodendrocyte maturation. Cell 2003, 115, 163–175. [Google Scholar] [CrossRef] [Green Version]
- John, G.R.; Shankar, S.L.; Shafit-Zagardo, B.; Massimi, A.; Lee, S.C.; Raine, C.S.; Brosnan, C.F. Multiple sclerosis: Re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat. Med. 2002, 8, 1115–1121. [Google Scholar] [CrossRef]
- Wang, S.; Sdrulla, A.D.; diSibio, G.; Bush, G.; Nofziger, D.; Hicks, C.; Weinmaster, G.; Barres, B.A. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 1998, 21, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Alcantara Llaguno, S.R.; Wang, Z.; Sun, D.; Chen, J.; Xu, J.; Kim, E.; Hatanpaa, K.J.; Raisanen, J.M.; Burns, D.K.; Johnson, J.E.; et al. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell 2015, 28, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Galvao, R.P.; Kasina, A.; McNeill, R.S.; Harbin, J.E.; Foreman, O.; Verhaak, R.G.; Nishiyama, A.; Miller, C.R.; Zong, H. Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proc. Natl. Acad. Sci. USA 2014, 111, E4214–E4223. [Google Scholar] [CrossRef]
- Sutcliffe, M.D.; Galvao, R.P.; Wang, L.; Kim, J.; Rosenfeld, L.K.; Singh, S.; Zong, H.; Janes, K.A. Premalignant Oligodendrocyte Precursor Cells Stall in a Heterogeneous State of Replication Stress Prior to Gliomagenesis. Cancer Res. 2021, 81, 1868–1882. [Google Scholar] [CrossRef]
- Chen, P.; Wang, W.; Liu, R.; Lyu, J.; Zhang, L.; Li, B.; Qiu, B.; Tian, A.; Jiang, W.; Ying, H.; et al. Olfactory sensory experience regulates gliomagenesis via neuronal IGF1. Nature 2022, 606, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Boda, E.; Lorenzati, M.; Parolisi, R.; Harding, B.; Pallavicini, G.; Bonfanti, L.; Moccia, A.; Bielas, S.; Di Cunto, F.; Buffo, A. Molecular and functional heterogeneity in dorsal and ventral oligodendrocyte progenitor cells of the mouse forebrain in response to DNA damage. Nat. Commun. 2022, 13, 2331. [Google Scholar] [CrossRef] [PubMed]
- Bigas, A.; Espinosa, L. The multiple usages of Notch signaling in development, cell differentiation and cancer. Curr. Opin. Cell Biol. 2018, 55, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrique, D.; Schweisguth, F. Mechanisms of Notch signaling: A simple logic deployed in time and space. Development 2019, 146, dev172148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcolea, M.P.; Greulich, P.; Wabik, A.; Frede, J.; Simons, B.D.; Jones, P.H. Differentiation imbalance in single oesophageal progenitor cells causes clonal immortalization and field change. Nat. Cell. Biol. 2014, 16, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, K.; Marques-Torrejon, M.A.; Dharmalingham, G.; El-Azhar, Y.; Schneider, M.D.; Pollard, S.M.; Rodriguez, T.A. Glioblastoma stem cells induce quiescence in surrounding neural stem cells via Notch signaling. Genes Dev. 2020, 34, 1599–1604. [Google Scholar] [CrossRef]
- Kessaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trp53-/- | Trp53-/-Rbpj-/- | Trp53-/-Notch1-/-Notch2-/- | Trp53-/-Notch1-/- | Trp53-/-Notch2-/- | |
|---|---|---|---|---|---|
| Hyperplasia | 49.5 ± 10.5 (n = 3) | n/a | 73.6 ± 4.3 (n = 5) | 63.7 ± 9.1 (n = 5) | 55.0 ± 3.8 (n = 2) |
| Tumor | n/a | 89.6 ± 0.6 (n = 4) | 85.4 ± 4.9 (n = 4) | 76.7 ± 5.5 (n = 5) | n/a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parmigiani, E.; Giachino, C. Genetic Inactivation of Notch1 Synergizes with Loss of Trp53 to Induce Tumor Formation in the Adult Mouse Forebrain. Cancers 2022, 14, 5409. https://doi.org/10.3390/cancers14215409
Parmigiani E, Giachino C. Genetic Inactivation of Notch1 Synergizes with Loss of Trp53 to Induce Tumor Formation in the Adult Mouse Forebrain. Cancers. 2022; 14(21):5409. https://doi.org/10.3390/cancers14215409
Chicago/Turabian StyleParmigiani, Elena, and Claudio Giachino. 2022. "Genetic Inactivation of Notch1 Synergizes with Loss of Trp53 to Induce Tumor Formation in the Adult Mouse Forebrain" Cancers 14, no. 21: 5409. https://doi.org/10.3390/cancers14215409