
In Vivo Models for Prostate Cancer Research
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
Historical Timeline of PCa Models
2. Patient-Derived Xenografts and Organoids of PCa
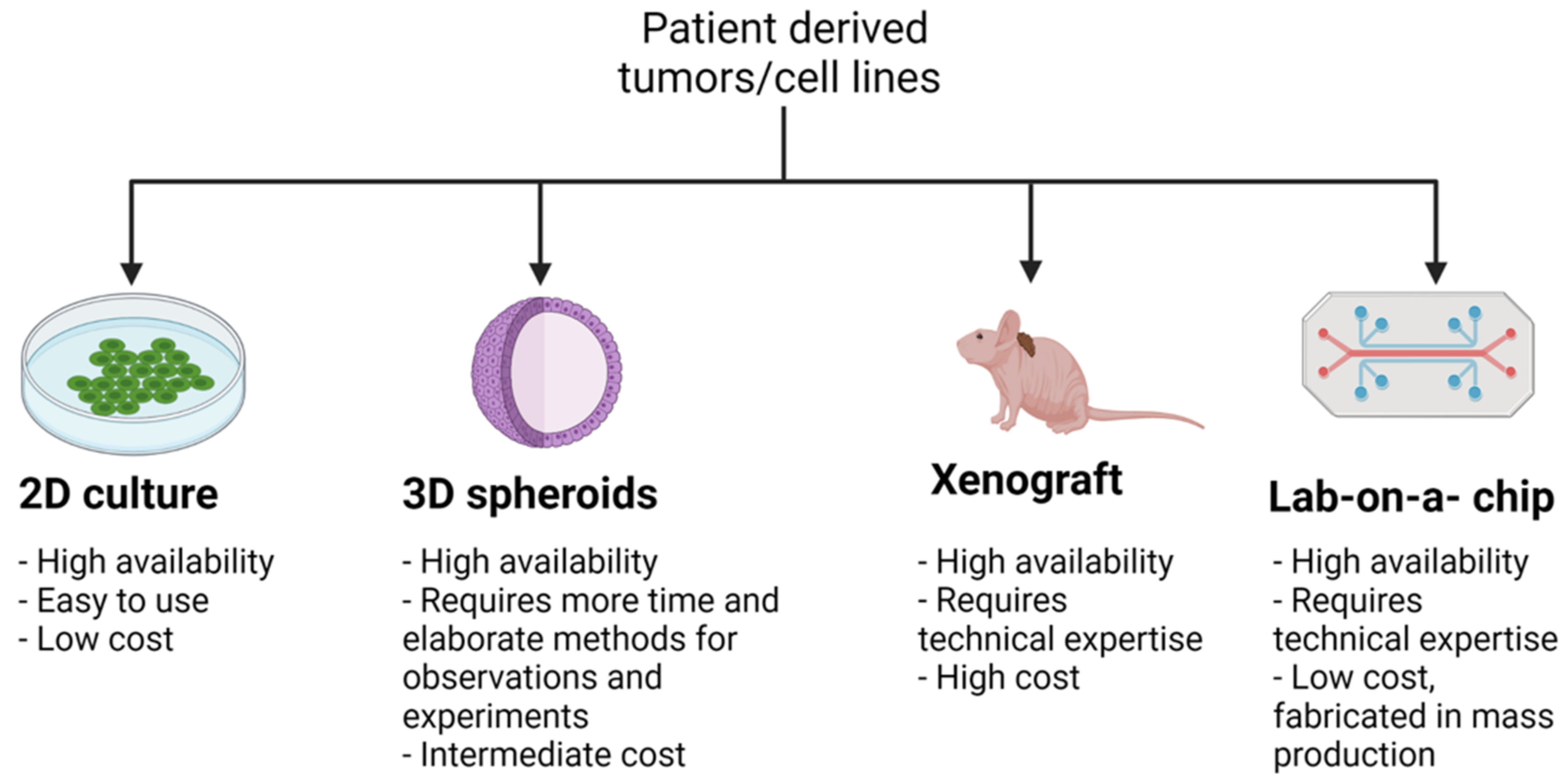
3. Comparing and Contrasting Types of PCa Models
4. Mouse Models Based on Tumor Stage Progression
4.1. Hyperplasia
4.2. Prostatic Intraepithelial Neoplasia
4.3. Microinvasive Carcinoma
4.4. Invasive Carcinoma
4.4.1. Adenocarcinoma
4.4.2. Squamous Cell Carcinoma
4.4.3. Neuroendocrine Carcinoma

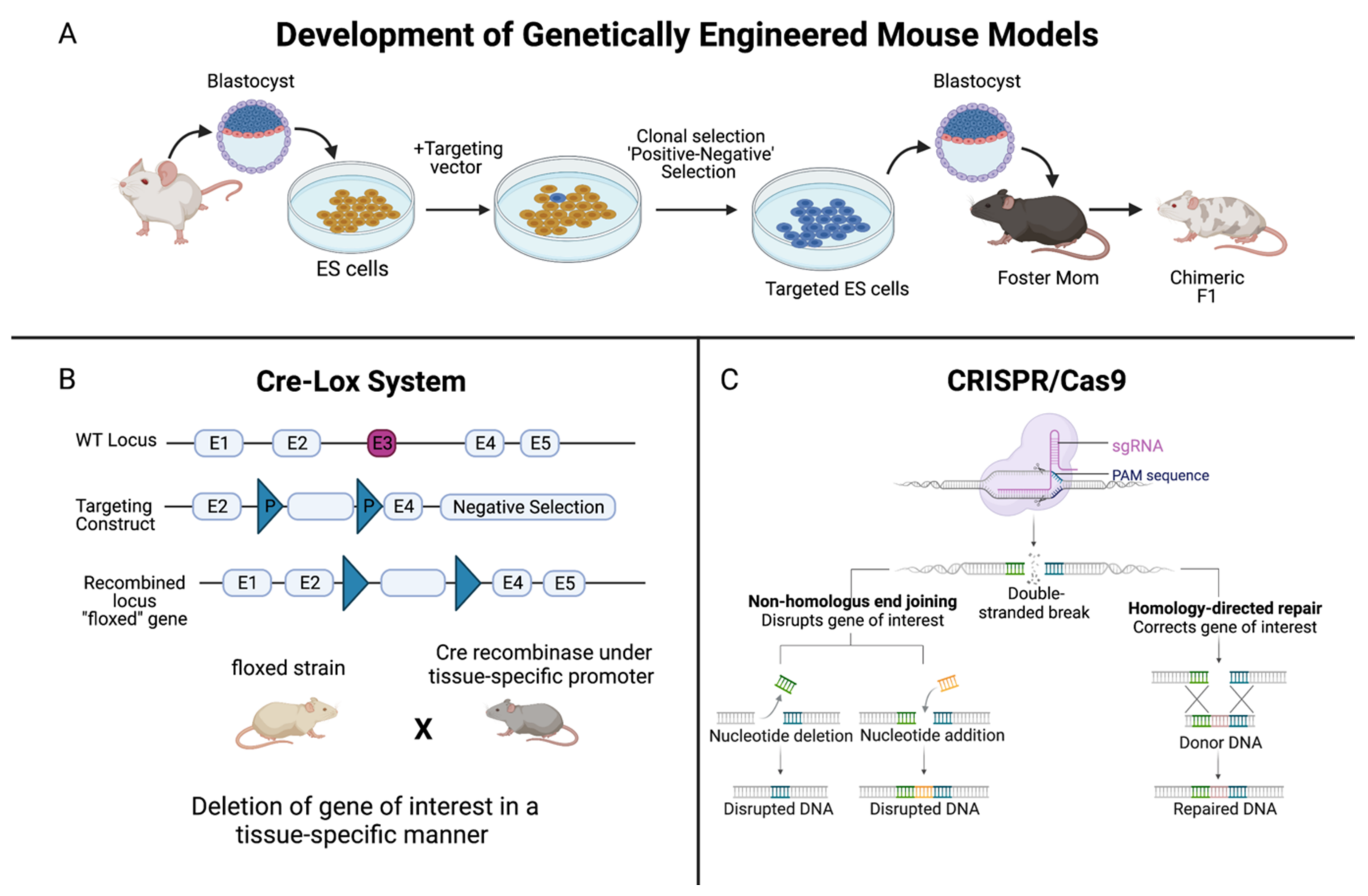{kind=link}
{kind=link}
{kind=link}
{kind=link}
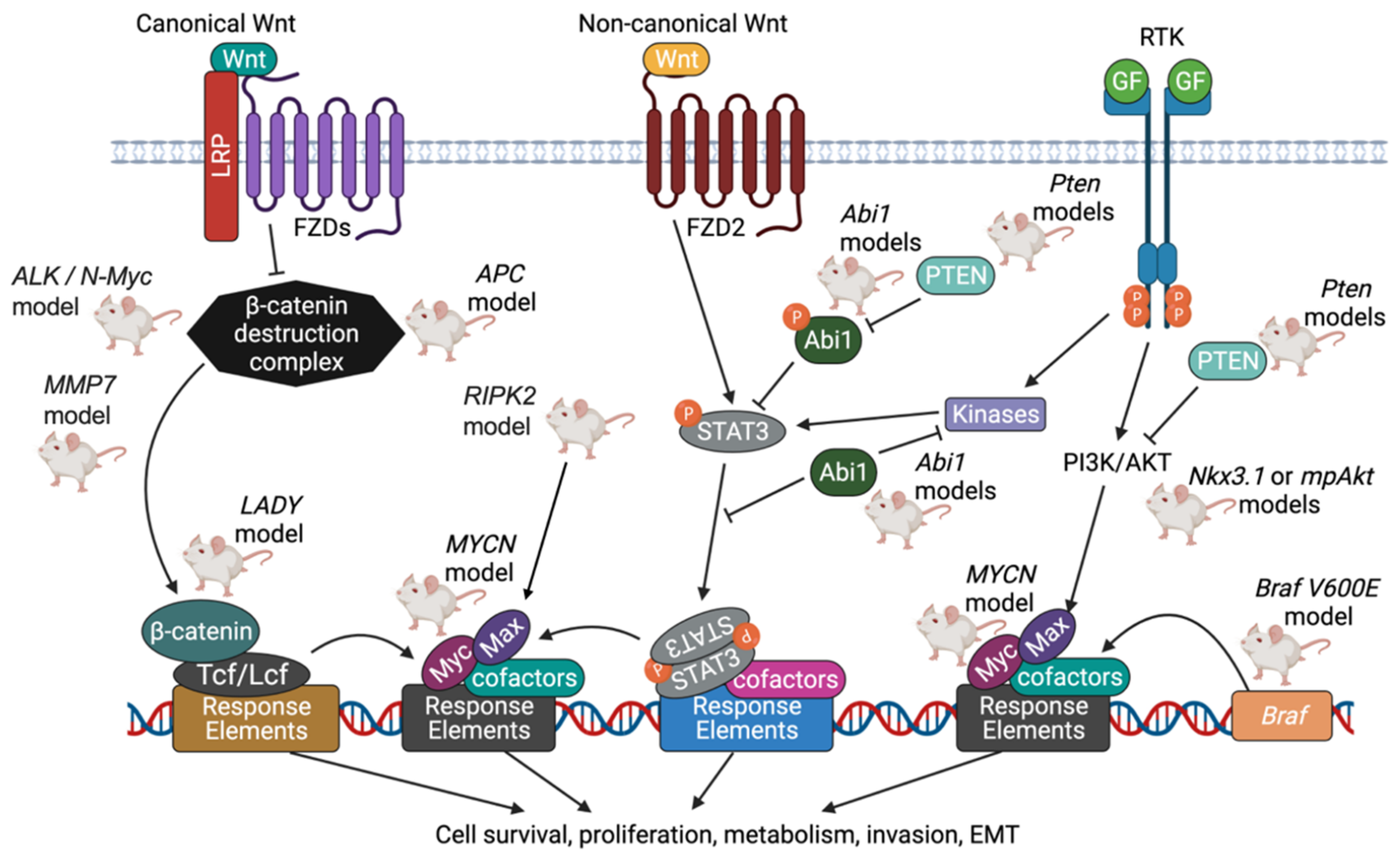{kind=link}
| Model | Alteration | Driver and/or Add. Genetic Alteration | Phenotype | Reference |
|---|---|---|---|---|
| Ptenflox/flox | Loss of expression | Nkx3.1-CreERT2 driver | Microinvasive AD with areas of poorly differentiated AD; squamous metaplasia | [79,80] |
| RARγ | Loss of expression | C57BL/6 F1 background strain | Squamous metaplasia in prostate and seminal vesicles | [81] |
| MYCN | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with neuroendocrine PCa (NEPC) | [82] |
| TRAMP | Gain of expression | PB promoter driving expression of SV40 early region | Androgen independent tumors are 100% synaptophysin positive, and metastases are 67% positive | [83] |
| LADY | Gain of expression | Large PB (LPB) promoter driving expression of SV40 large T-antigen (Tag) | Visceral metastasis; NEPC | [84] |
| LADY | Gain of expression | LPB driver, 12T-7s line; crossed with PB-Hepsin | Adenocarcinoma with neuroendocrine differentiation (NED); NE metastasis to liver, lung, and bone | [85] |
| LADY | Gain of expression | LPB driver, 12T-7s line; crossed with β-catenin | Adenocarcinoma with focal NED, but without apparent NEPC | [86] |
| Kras G12D | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma, sarcomatoid differentiation, with extensive metastasis | [87,88] |
| Pten and p53 | Loss of expression | PB-Cre mediated deletion of Pten and Trp53; activation of ROSA-LSL luciferase reporter | Fast-growing, lethal sarcomatoid tumors; local invasive PCa | [89] |
| ALK and N-myc | Gain of expression | FVB/NJ and NSG background strains | Neuroblastoma development; metastasis with NED | [90] |
4.4.4. Sarcomatoid Carcinoma
4.5. Metastasis
5. Mouse Models Based on Signaling Pathways
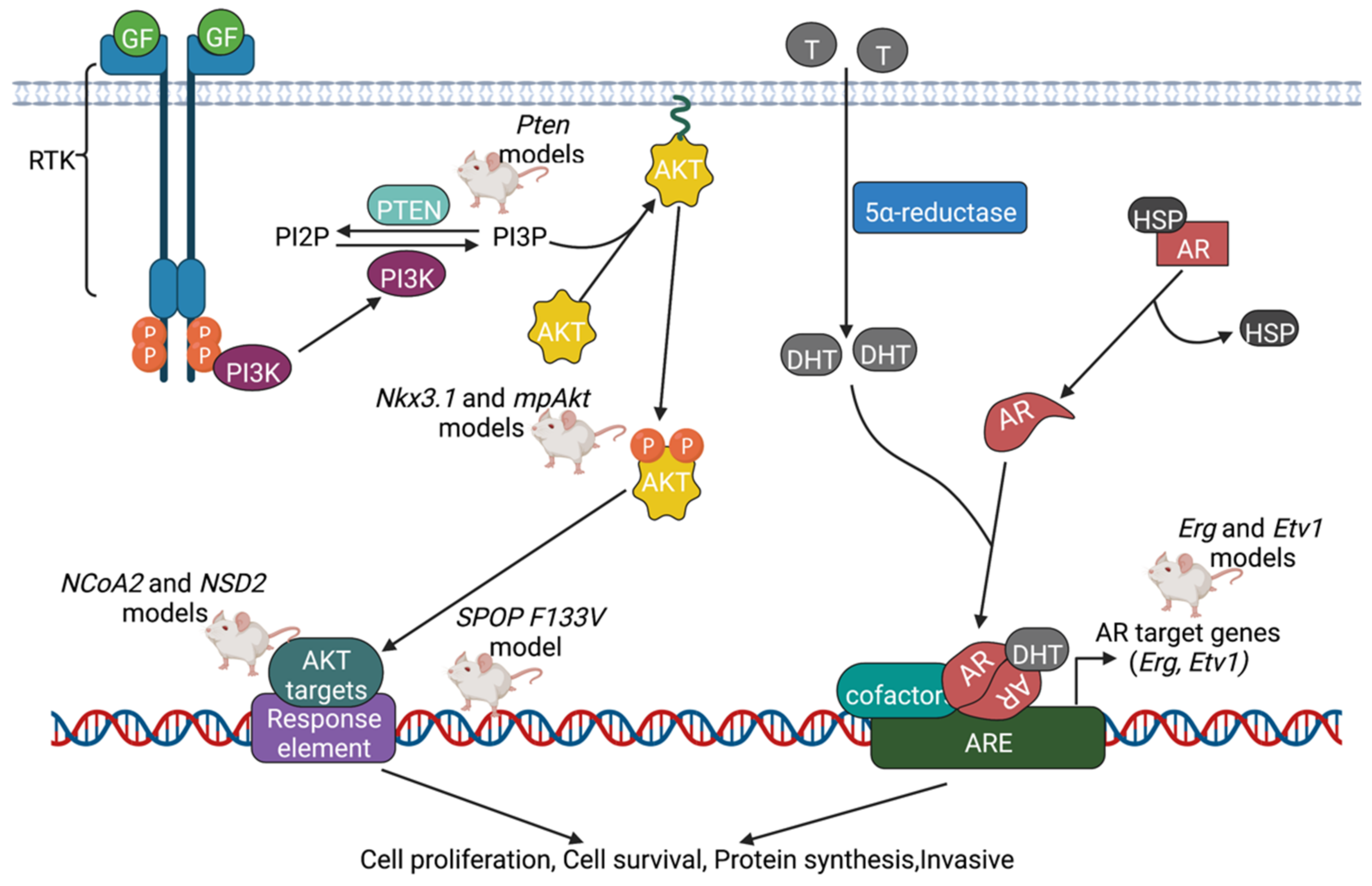
5.1. AR Pathway
5.2. PI3K Pathway
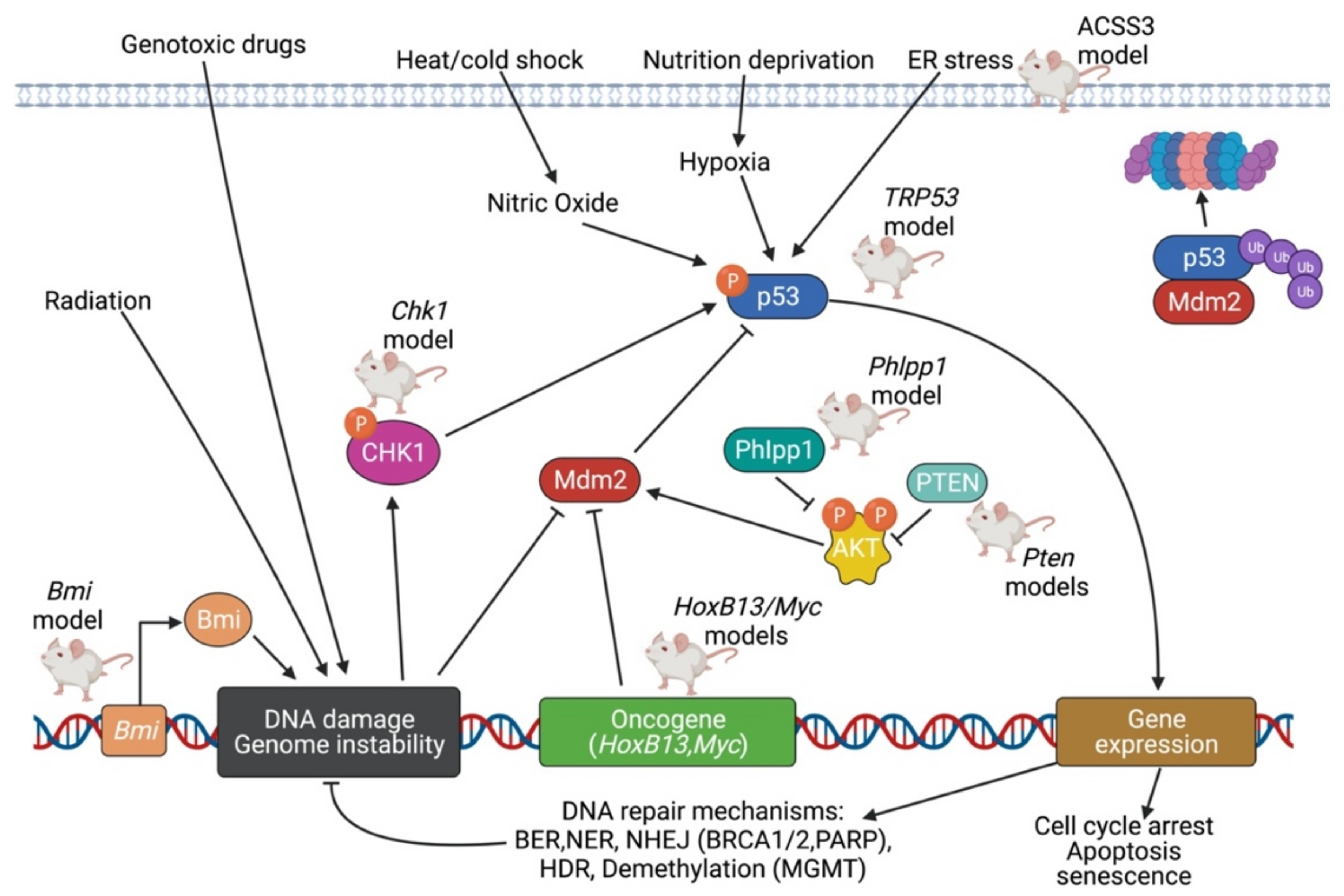
5.3. TP53 Pathway
5.4. DNA Repair Pathway
5.5. MYC Pathway
5.6. Wnt Pathway
6. Future Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Sanford, K.K.; Hobbs, G.L.; Earle, W.R. The tumor-producing capacity of strain L mouse cells after 10 years in vitro. Cancer Res. 1956, 16, 162–166. [Google Scholar] [PubMed]
- Scherer, W.F.; Syverton, J.T.; Gey, G.O. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J. Exp. Med. 1953, 97, 695–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedrzejczak-Silicka, M. History of Cell Culture. In New Insights into Cell Culture Technology; Gowder, S.J.T., Ed.; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef] [Green Version]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar] [PubMed]
- Stone, K.R.; Mickey, D.D.; Wunderli, H.; Mickey, G.H.; Paulson, D.F. Isolation of a human prostate carcinoma cell line (DU 145). Int. J. Cancer. 1978, 21, 274–281. [Google Scholar] [CrossRef]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investig. Urol. 1979, 17, 16–23. [Google Scholar]
- Press Release. Nobel Prize Outreach AB. 2022. Available online: https://www.nobelprize.org/prizes/medicine/2007/press-release/ (accessed on 25 October 2022).
- Nagy, A. Cre recombinase: The universal reagent for genome tailoring. Genesis 2000, 26, 99–109. [Google Scholar] [CrossRef]
- Dow, L.E.; Fisher, J.; O’Rourke, K.P.; Muley, A.; Kastenhuber, E.R.; Livshits, G.; Tschaharganeh, D.F.; Socci, N.D.; Lowe, S.W. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [Green Version]
- Abate-Shen, C. A new generation of mouse models of cancer for translational research. Clin. Cancer Res. 2006, 12, 5274–5276. [Google Scholar] [CrossRef]
- Shappell, S.B.; Thomas, G.V.; Roberts, R.L.; Herbert, R.; Ittmann, M.M.; Rubin, M.A.; Humphrey, P.A.; Sundberg, J.P.; Rozengurt, N.; Barrios, R.; et al. Prostate pathology of genetically engineered mice: Definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004, 64, 2270–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpless, N.E.; Depinho, R.A. The mighty mouse: Genetically engineered mouse models in cancer drug development. Nat. Rev. Drug. Discov. 2006, 5, 741–754. [Google Scholar] [CrossRef]
- Singh, M.; Johnson, L. Using genetically engineered mouse models of cancer to aid drug development: An industry perspective. Clin. Cancer Res. 2006, 12, 5312–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, B.; Henderson, N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. USA 1988, 85, 5166–5170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, N.; Hamilton, D. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J. Mol. Biol. 1981, 150, 467–486. [Google Scholar] [CrossRef]
- Trotman, L.C.; Niki, M.; Dotan, Z.A.; Koutcher, J.A.; Di Cristofano, A.; Xiao, A.; Khoo, A.S.; Roy-Burman, P.; Greenberg, N.M.; Van Dyke, T.; et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003, 1, E59. [Google Scholar] [CrossRef]
- Degenhardt, K.; White, E. A mouse model system to genetically dissect the molecular mechanisms regulating tumorigenesis. Clin. Cancer Res. 2006, 12, 5298–5304. [Google Scholar] [CrossRef] [Green Version]
- Carver, B.S.; Pandolfi, P.P. Mouse modeling in oncologic preclinical and translational research. Clin. Cancer Res. 2006, 12, 5305–5311. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [Green Version]
- Woo, X.Y.; Giordano, J.; Srivastava, A.; Zhao, Z.M.; Lloyd, M.W.; de Bruijn, R.; Suh, Y.S.; Patidar, R.; Chen, L.; Scherer, S.; et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat. Genet. 2021, 53, 86–99. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.M.; et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease and Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, J.; Ci, X.; Xue, H.; Wu, R.; Dong, X.; Choi, S.Y.C.; He, H.; Wang, Y.; Zhang, F.; Qu, S.; et al. Patient-derived Hormone-naive Prostate Cancer Xenograft Models Reveal Growth Factor Receptor Bound Protein 10 as an Androgen Receptor-repressed Gene Driving the Development of Castration-resistant Prostate Cancer. Eur. Urol. 2018, 73, 949–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.; Xue, H.; Wang, Y.; Wu, R.; Watahiki, A.; Dong, X.; Cheng, H.; Wyatt, A.W.; Collins, C.C.; Gout, P.W.; et al. Next generation patient-derived prostate cancer xenograft models. Asian J. Androl. 2014, 16, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Corro, C.; Novellasdemunt, L.; Li, V.S.W. A brief history of organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165. [Google Scholar] [CrossRef]
- Beshiri, M.L.; Tice, C.M.; Tran, C.; Nguyen, H.M.; Sowalsky, A.G.; Agarwal, S.; Jansson, K.H.; Yang, Q.; McGowen, K.M.; Yin, J.; et al. A PDX/Organoid Biobank of Advanced Prostate Cancers Captures Genomic and Phenotypic Heterogeneity for Disease Modeling and Therapeutic Screening. Clin. Cancer Res. 2018, 24, 4332–4345. [Google Scholar] [CrossRef] [Green Version]
- Karthaus, W.R.; Iaquinta, P.J.; Drost, J.; Gracanin, A.; van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthel, H.; Sachs, N.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014, 159, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Puca, L.; Bareja, R.; Prandi, D.; Shaw, R.; Benelli, M.; Karthaus, W.R.; Hess, J.; Sigouros, M.; Donoghue, A.; Kossai, M.; et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 2018, 9, 2404. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Kapalczynska, M.; Kolenda, T.; Przybyla, W.; Zajaczkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Blizniak, R.; Luczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Ryu, N.E.; Lee, S.H.; Park, H. Spheroid Culture System Methods and Applications for Mesenchymal Stem Cells. Cells 2019, 8, 1620. [Google Scholar] [CrossRef]
- Volpatti, L.R.; Yetisen, A.K. Commercialization of microfluidic devices. Trends Biotechnol. 2014, 32, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Pawell, R.S.; Inglis, D.W.; Barber, T.J.; Taylor, R.A. Manufacturing and wetting low-cost microfluidic cell separation devices. Biomicrofluidics 2013, 7, 56501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.; Peng, Y.; Li, H.; Chen, W. Organ-on-a-Chip: A New Paradigm for Drug Development. Trends Pharmacol. Sci. 2021, 42, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Ishida, N.; Shirane, M.; Inomata, A.; Inoue, T.; Shishido, N.; Horii, I.; Loh, D.Y.; Nakayama, K. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 1996, 85, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Bhatia-Gaur, R.; Donjacour, A.A.; Sciavolino, P.J.; Kim, M.; Desai, N.; Young, P.; Norton, C.R.; Gridley, T.; Cardiff, R.D.; Cunha, G.R.; et al. Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 1999, 13, 966–977. [Google Scholar] [CrossRef] [Green Version]
- Maddison, L.A.; Sutherland, B.W.; Barrios, R.J.; Greenberg, N.M. Conditional deletion of Rb causes early stage prostate cancer. Cancer Res. 2004, 64, 6018–6025. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, B.W.; Knoblaugh, S.E.; Kaplan-Lefko, P.J.; Wang, F.; Holzenberger, M.; Greenberg, N.M. Conditional deletion of insulin-like growth factor-I receptor in prostate epithelium. Cancer Res. 2008, 68, 3495–3504. [Google Scholar] [CrossRef] [Green Version]
- DeGraff, D.J.; Grabowska, M.M.; Case, T.C.; Yu, X.; Herrick, M.K.; Hayward, W.J.; Strand, D.W.; Cates, J.M.; Hayward, S.W.; Gao, N.; et al. FOXA1 deletion in luminal epithelium causes prostatic hyperplasia and alteration of differentiated phenotype. Lab. Investig. 2014, 94, 726–739. [Google Scholar] [CrossRef] [Green Version]
- Grabowska, M.M.; DeGraff, D.J.; Yu, X.; Jin, R.J.; Chen, Z.; Borowsky, A.D.; Matusik, R.J. Mouse models of prostate cancer: Picking the best model for the question. Cancer Metastasis Rev. 2014, 33, 377–397. [Google Scholar] [CrossRef]
- Li, G.; Kanagasabai, T.; Lu, W.; Zou, M.R.; Zhang, S.M.; Celada, S.I.; Izban, M.G.; Liu, Q.; Lu, T.; Ballard, B.R.; et al. KDM5B Is Essential for the Hyperactivation of PI3K/AKT Signaling in Prostate Tumorigenesis. Cancer Res. 2020, 80, 4633–4643. [Google Scholar] [CrossRef]
- Thomsen, M.K.; Ambroisine, L.; Wynn, S.; Cheah, K.S.; Foster, C.S.; Fisher, G.; Berney, D.M.; Moller, H.; Reuter, V.E.; Scardino, P.; et al. SOX9 elevation in the prostate promotes proliferation and cooperates with PTEN loss to drive tumor formation. Cancer Res. 2010, 70, 979–987. [Google Scholar] [CrossRef] [Green Version]
- Choi, N.; Zhang, B.; Zhang, L.; Ittmann, M.; Xin, L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell 2012, 21, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cristofano, A.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef]
- Couto, S.S.; Cao, M.; Duarte, P.C.; Banach-Petrosky, W.; Wang, S.; Romanienko, P.; Wu, H.; Cardiff, R.D.; Abate-Shen, C.; Cunha, G.R. Simultaneous haploinsufficiency of Pten and Trp53 tumor suppressor genes accelerates tumorigenesis in a mouse model of prostate cancer. Differentiation 2009, 77, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Chorzalska, A.; Dubielecka, P.M.; White, J.R.; Vedvyas, Y.; Hedvat, C.V.; Haimovitz-Friedman, A.; Koutcher, J.A.; Reimand, J.; Bader, G.D.; et al. Disruption of Abi1/Hssh3bp1 expression induces prostatic intraepithelial neoplasia in the conditional Abi1/Hssh3bp1 KO mice. Oncogenesis 2012, 1, e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, L.E.; Rigatti, L.H.; Ai, J.; Zhang, A.; Zhou, J.; Nelson, J.B.; Wang, Z. EAF2 loss induces prostatic intraepithelial neoplasia from luminal epithelial cells in mice. Am. J. Clin. Exp. Urol. 2020, 8, 18–27. [Google Scholar] [PubMed]
- Zhou, L.; Song, Z.; Hu, J.; Liu, L.; Hou, Y.; Zhang, X.; Yang, X.; Chen, K. ACSS3 represses prostate cancer progression through downregulating lipid droplet-associated protein PLIN3. Theranostics 2021, 11, 841–860. [Google Scholar] [CrossRef]
- Kwon, O.J.; Zhang, B.; Jia, D.; Zhang, L.; Wei, X.; Zhou, Z.; Liu, D.; Huynh, K.T.; Zhang, K.; Zhang, Y.; et al. Elevated expression of the colony-stimulating factor 1 (CSF1) induces prostatic intraepithelial neoplasia dependent of epithelial-Gp130. Oncogene 2022, 41, 1309–1323. [Google Scholar] [CrossRef]
- Adissu, H.A.; McKerlie, C.; Di Grappa, M.; Waterhouse, P.; Xu, Q.; Fang, H.; Khokha, R.; Wood, G.A. Timp3 loss accelerates tumour invasion and increases prostate inflammation in a mouse model of prostate cancer. Prostate 2015, 75, 1831–1843. [Google Scholar] [CrossRef]
- Clegg, N.J.; Couto, S.S.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Taylor, B.S.; Ellwood-Yen, K.; Gerald, W.L.; Sander, C.; Sawyers, C.L. MYC cooperates with AKT in prostate tumorigenesis and alters sensitivity to mTOR inhibitors. PLoS ONE 2011, 6, e17449. [Google Scholar] [CrossRef]
- Anderson, P.D.; McKissic, S.A.; Logan, M.; Roh, M.; Franco, O.E.; Wang, J.; Doubinskaia, I.; van der Meer, R.; Hayward, S.W.; Eischen, C.M.; et al. Nkx3.1 and Myc crossregulate shared target genes in mouse and human prostate tumorigenesis. J. Clin. Investig. 2012, 122, 1907–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Eltoum, I.E.; Roh, M.; Wang, J.; Abdulkadir, S.A. Interactions between cells with distinct mutations in c-MYC and Pten in prostate cancer. PLoS Genet. 2009, 5, e1000542. [Google Scholar] [CrossRef] [PubMed]
- Abate-Shen, C.; Banach-Petrosky, W.A.; Sun, X.; Economides, K.D.; Desai, N.; Gregg, J.P.; Borowsky, A.D.; Cardiff, R.D.; Shen, M.M. Nkx3.1; Pten mutant mice develop invasive prostate adenocarcinoma and lymph node metastases. Cancer Res. 2003, 63, 3886–3890. [Google Scholar] [PubMed]
- Kim, M.J.; Bhatia-Gaur, R.; Banach-Petrosky, W.A.; Desai, N.; Wang, Y.; Hayward, S.W.; Cunha, G.R.; Cardiff, R.D.; Shen, M.M.; Abate-Shen, C. Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer Res. 2002, 62, 2999–3004. [Google Scholar] [PubMed]
- Di Cristofano, A.; De Acetis, M.; Koff, A.; Cordon-Cardo, C.; Pandolfi, P.P. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat. Genet. 2001, 27, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Ouyang, X.; Banach-Petrosky, W.; Borowsky, A.D.; Lin, Y.; Kim, M.; Lee, H.; Shih, W.J.; Cardiff, R.D.; Shen, M.M.; et al. A critical role for p27kip1 gene dosage in a mouse model of prostate carcinogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 17204–17209. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Xu, D.; Ding, H.F.; Kim, J.; Zhang, J.; Hai, T.; Yan, C. Loss of ATF3 promotes Akt activation and prostate cancer development in a Pten knockout mouse model. Oncogene 2015, 34, 4975–4984. [Google Scholar] [CrossRef] [Green Version]
- Bruxvoort, K.J.; Charbonneau, H.M.; Giambernardi, T.A.; Goolsby, J.C.; Qian, C.N.; Zylstra, C.R.; Robinson, D.R.; Roy-Burman, P.; Shaw, A.K.; Buckner-Berghuis, B.D.; et al. Inactivation of Apc in the mouse prostate causes prostate carcinoma. Cancer Res. 2007, 67, 2490–2496. [Google Scholar] [CrossRef] [Green Version]
- Nacerddine, K.; Beaudry, J.B.; Ginjala, V.; Westerman, B.; Mattiroli, F.; Song, J.Y.; van der Poel, H.; Ponz, O.B.; Pritchard, C.; Cornelissen-Steijger, P.; et al. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. J Clin. Investig. 2012, 122, 1920–1932. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Teruya-Feldstein, J.; Behrendt, N.; Chen, Z.; Noda, T.; Hino, O.; Cordon-Cardo, C.; Pandolfi, P.P. Genetic analysis of Pten and Tsc2 functional interactions in the mouse reveals asymmetrical haploinsufficiency in tumor suppression. Genes Dev. 2005, 19, 1779–1786. [Google Scholar] [CrossRef]
- Chen, M.; Pratt, C.P.; Zeeman, M.E.; Schultz, N.; Taylor, B.S.; O’Neill, A.; Castillo-Martin, M.; Nowak, D.G.; Naguib, A.; Grace, D.M.; et al. Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell 2011, 20, 173–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunardi, A.; Varmeh, S.; Chen, M.; Taulli, R.; Guarnerio, J.; Ala, U.; Seitzer, N.; Ishikawa, T.; Carver, B.S.; Hobbs, R.M.; et al. Suppression of CHK1 by ETS Family Members Promotes DNA Damage Response Bypass and Tumorigenesis. Cancer Discov. 2015, 5, 550–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, R.; Blando, J.M.; Perez, C.J.; Abba, M.C.; Benavides, F.; Kazanietz, M.G. Protein Kinase C Epsilon Cooperates with PTEN Loss for Prostate Tumorigenesis through the CXCL13-CXCR5 Pathway. Cell Rep. 2017, 19, 375–388. [Google Scholar] [CrossRef]
- Nguyen, A.H.; Tremblay, M.; Haigh, K.; Koumakpayi, I.H.; Paquet, M.; Pandolfi, P.P.; Mes-Masson, A.M.; Saad, F.; Haigh, J.J.; Bouchard, M. Gata3 antagonizes cancer progression in Pten-deficient prostates. Hum. Mol. Genet. 2013, 22, 2400–2410. [Google Scholar] [CrossRef] [PubMed]
- Kohvakka, A.; Sattari, M.; Shcherban, A.; Annala, M.; Urbanucci, A.; Kesseli, J.; Tammela, T.L.J.; Kivinummi, K.; Latonen, L.; Nykter, M.; et al. AR and ERG drive the expression of prostate cancer specific long noncoding RNAs. Oncogene 2020, 39, 5241–5251. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Tran, J.; Gopalan, A.; Chen, Z.; Shaikh, S.; Carracedo, A.; Alimonti, A.; Nardella, C.; Varmeh, S.; Scardino, P.T.; et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat. Genet. 2009, 41, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Baena, E.; Shao, Z.; Linn, D.E.; Glass, K.; Hamblen, M.J.; Fujiwara, Y.; Kim, J.; Nguyen, M.; Zhang, X.; Godinho, F.J.; et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013, 27, 683–698. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.K.; Bakiri, L.; Hasenfuss, S.C.; Wu, H.; Morente, M.; Wagner, E.F. Loss of JUNB/AP-1 promotes invasive prostate cancer. Cell Death Differ. 2015, 22, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.; Siwko, S.; Zeng, L.; Li, J.; Yi, Z.; Liu, M. Prostate-specific G-protein-coupled receptor collaborates with loss of PTEN to promote prostate cancer progression. Oncogene 2016, 35, 1153–1162. [Google Scholar] [CrossRef]
- Wang, G.; Lunardi, A.; Zhang, J.; Chen, Z.; Ala, U.; Webster, K.A.; Tay, Y.; Gonzalez-Billalabeitia, E.; Egia, A.; Shaffer, D.R.; et al. Zbtb7a suppresses prostate cancer through repression of a Sox9-dependent pathway for cellular senescence bypass and tumor invasion. Nat. Genet. 2013, 45, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandana, S.; Ellwood-Yen, K.; Sawyers, C.; Wills, M.; Weidow, B.; Case, T.; Vasioukhin, V.; Matusik, R. Hepsin cooperates with MYC in the progression of adenocarcinoma in a prostate cancer mouse model. Prostate 2010, 70, 591–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Liu, S.; Parajuli, K.R.; Zhang, W.; Zhang, K.; Mo, Z.; Liu, J.; Chen, Z.; Yang, S.; Wang, A.R.; et al. Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene 2017, 36, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhang, B.; Rowan, B.G.; Jazwinski, S.M.; Abdel-Mageed, A.B.; Steele, C.; Wang, A.R.; Sartor, O.; Niu, T.; Zhang, Q. A Novel Controlled PTEN-Knockout Mouse Model for Prostate Cancer Study. Front. Mol. Biosci. 2021, 8, 696537. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Y.; Zhou, Y.U.; Xu, Z.; Ma, Y.Q. Kindlin-3 in Immune Cells Is Required to Suppress Prostate Cancer Tumor Growth in Mice. Anticancer Res. 2022, 42, 1217–1220. [Google Scholar] [CrossRef] [PubMed]
- Di Sant’Agnese, P.A. Neuroendocrine cells of the prostate and neuroendocrine differentiation in prostatic carcinoma: A review of morphologic aspects. Urology 1998, 51, 121–124. [Google Scholar] [CrossRef]
- Floc’h, N.; Kinkade, C.W.; Kobayashi, T.; Aytes, A.; Lefebvre, C.; Mitrofanova, A.; Cardiff, R.D.; Califano, A.; Shen, M.M.; Abate-Shen, C. Dual targeting of the Akt/mTOR signaling pathway inhibits castration-resistant prostate cancer in a genetically engineered mouse model. Cancer Res. 2012, 72, 4483–4493. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kruithof-de Julio, M.; Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009, 461, 495–500. [Google Scholar] [CrossRef] [Green Version]
- Lohnes, D.; Kastner, P.; Dierich, A.; Mark, M.; LeMeur, M.; Chambon, P. Function of retinoic acid receptor gamma in the mouse. Cell 1993, 73, 643–658. [Google Scholar] [CrossRef]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef]
- Gingrich, J.R.; Barrios, R.J.; Morton, R.A.; Boyce, B.F.; DeMayo, F.J.; Finegold, M.J.; Angelopoulou, R.; Rosen, J.M.; Greenberg, N.M. Metastatic prostate cancer in a transgenic mouse. Cancer Res. 1996, 56, 4096–4102. [Google Scholar] [PubMed]
- Kasper, S.; Sheppard, P.C.; Yan, Y.; Pettigrew, N.; Borowsky, A.D.; Prins, G.S.; Dodd, J.G.; Duckworth, M.L.; Matusik, R.J. Development, progression, and androgen-dependence of prostate tumors in probasin-large T antigen transgenic mice: A model for prostate cancer. Lab. Investig. 1998, 78, 319–333. [Google Scholar] [PubMed]
- Klezovitch, O.; Chevillet, J.; Mirosevich, J.; Roberts, R.L.; Matusik, R.J.; Vasioukhin, V. Hepsin promotes prostate cancer progression and metastasis. Cancer Cell 2004, 6, 185–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Wang, Y.; DeGraff, D.J.; Wills, M.L.; Matusik, R.J. Wnt/beta-catenin activation promotes prostate tumor progression in a mouse model. Oncogene 2011, 30, 1868–1879. [Google Scholar] [CrossRef] [Green Version]
- Aytes, A.; Mitrofanova, A.; Kinkade, C.W.; Lefebvre, C.; Lei, M.; Phelan, V.; LeKaye, H.C.; Koutcher, J.A.; Cardiff, R.D.; Califano, A.; et al. ETV4 promotes metastasis in response to activation of PI3-kinase and Ras signaling in a mouse model of advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E3506–E3515. [Google Scholar] [CrossRef] [Green Version]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [Green Version]
- Yong, C.; Moose, D.L.; Bannick, N.; Gutierrez, W.R.; Vanneste, M.; Svensson, R.; Breheny, P.; Brown, J.A.; Dodd, R.D.; Cohen, M.B.; et al. Locally invasive, castrate-resistant prostate cancer in a Pten/Trp53 double knockout mouse model of prostate cancer monitored with non-invasive bioluminescent imaging. PLoS ONE 2020, 15, e0232807. [Google Scholar] [CrossRef]
- Unno, K.; Chalmers, Z.R.; Pamarthy, S.; Vatapalli, R.; Rodriguez, Y.; Lysy, B.; Mok, H.; Sagar, V.; Han, H.; Yoo, Y.A.; et al. Activated ALK Cooperates with N-Myc via Wnt/beta-Catenin Signaling to Induce Neuroendocrine Prostate Cancer. Cancer Res. 2021, 81, 2157–2170. [Google Scholar] [CrossRef]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Wu, S.P.; Creighton, C.J.; Dai, F.; Xie, X.; Cheng, C.M.; Frolov, A.; Ayala, G.; Lin, X.; Feng, X.H.; et al. COUP-TFII inhibits TGF-beta-induced growth barrier to promote prostate tumorigenesis. Nature 2013, 493, 236–240. [Google Scholar] [CrossRef]
- Qin, J.; Lee, H.J.; Wu, S.P.; Lin, S.C.; Lanz, R.B.; Creighton, C.J.; DeMayo, F.J.; Tsai, S.Y.; Tsai, M.J. Androgen deprivation-induced NCoA2 promotes metastatic and castration-resistant prostate cancer. J. Clin. Investig. 2014, 124, 5013–5026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Xue, W.; Yuan, H.; Dong, B.; Ding, Y.; Liu, Y.; Jiang, M.; Kan, S.; Sun, T.; Ren, J.; et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J. Clin. Investig. 2017, 127, 1284–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Herzka, T.; Zheng, W.; Qi, J.; Wilkinson, J.E.; Bradner, J.E.; Robinson, B.D.; Castillo-Martin, M.; Cordon-Cardo, C.; Trotman, L.C. RapidCaP, a novel GEM model for metastatic prostate cancer analysis and therapy, reveals myc as a driver of Pten-mutant metastasis. Cancer Discov. 2014, 4, 318–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Hubner, A.; Mulholland, D.J.; Standen, C.L.; Karasarides, M.; Cavanagh-Kyros, J.; Barrett, T.; Chi, H.; Greiner, D.L.; Tournier, C.; Sawyers, C.L.; et al. JNK and PTEN cooperatively control the development of invasive adenocarcinoma of the prostate. Proc. Natl. Acad. Sci. USA 2012, 109, 12046–12051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pencik, J.; Schlederer, M.; Gruber, W.; Unger, C.; Walker, S.M.; Chalaris, A.; Marie, I.J.; Hassler, M.R.; Javaheri, T.; Aksoy, O.; et al. STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat. Commun. 2015, 6, 7736. [Google Scholar] [CrossRef] [Green Version]
- Kwon, O.J.; Zhang, L.; Wang, J.; Su, Q.; Feng, Q.; Zhang, X.H.; Mani, S.A.; Paulter, R.; Creighton, C.J.; Ittmann, M.M.; et al. Notch promotes tumor metastasis in a prostate-specific Pten-null mouse model. J. Clin. Investig. 2016, 126, 2626–2641. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Wu, C.J.; Chu, G.C.; Xiao, Y.; Ho, D.; Zhang, J.; Perry, S.R.; Labrot, E.S.; Wu, X.; Lis, R.; et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 2011, 470, 269–273. [Google Scholar] [CrossRef]
- Ding, Z.; Wu, C.J.; Jaskelioff, M.; Ivanova, E.; Kost-Alimova, M.; Protopopov, A.; Chu, G.C.; Wang, G.; Lu, X.; Labrot, E.S.; et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell 2012, 148, 896–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, G.K.; Mutton, L.N.; Khalili, M.; McMullin, R.P.; Hicks, J.L.; Bianchi-Frias, D.; Horn, L.A.; Kulac, I.; Moubarek, M.S.; Nelson, P.S.; et al. Combined MYC Activation and Pten Loss Are Sufficient to Create Genomic Instability and Lethal Metastatic Prostate Cancer. Cancer Res. 2016, 76, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Kobayashi, T.; Floc’h, N.; Kinkade, C.W.; Aytes, A.; Dankort, D.; Lefebvre, C.; Mitrofanova, A.; Cardiff, R.D.; McMahon, M.; et al. B-Raf activation cooperates with PTEN loss to drive c-Myc expression in advanced prostate cancer. Cancer Res. 2012, 72, 4765–4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Flesken-Nikitin, A.; Corney, D.C.; Wang, W.; Goodrich, D.W.; Roy-Burman, P.; Nikitin, A.Y. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006, 66, 7889–7898. [Google Scholar] [CrossRef] [Green Version]
- Arriaga, J.M.; Panja, S.; Alshalalfa, M.; Zhao, J.; Zou, M.; Giacobbe, A.; Madubata, C.J.; Kim, J.Y.; Rodriguez, A.; Coleman, I.; et al. A MYC and RAS co-activation signature in localized prostate cancer drives bone metastasis and castration resistance. Nat. Cancer 2020, 1, 1082–1096. [Google Scholar] [CrossRef]
- Han, Q.; Xie, Q.R.; Li, F.; Cheng, Y.; Wu, T.; Zhang, Y.; Lu, X.; Wong, A.S.T.; Sha, J.; Xia, W. Targeted inhibition of SIRT6 via engineered exosomes impairs tumorigenesis and metastasis in prostate cancer. Theranostics 2021, 11, 6526–6541. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, B.; Qian, C.; Vasquez, A.; Kamra, M.; Chatterjee, A.; Lee, Y.J.; Yuan, X.; Ellis, L.; Di Vizio, D.; et al. Receptor-interacting protein kinase 2 (RIPK2) stabilizes c-Myc and is a therapeutic target in prostate cancer metastasis. Nat. Commun. 2022, 13, 669. [Google Scholar] [CrossRef]
- Roy, A.K.; Lavrovsky, Y.; Song, C.S.; Chen, S.; Jung, M.H.; Velu, N.K.; Bi, B.Y.; Chatterjee, B. Regulation of androgen action. Vitam. Horm. 1999, 55, 309–352. [Google Scholar] [CrossRef]
- Heemers, H.V.; Tindall, D.J. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex. Endocr. Rev. 2007, 28, 778–808. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, R.A. The Biology of Cancer, 2nd ed.; Garland Science: New York, NY, USA, 2014. [Google Scholar]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccarese, C.; Massari, F.; Iacovelli, R.; Fiorentino, M.; Montironi, R.; Di Nunno, V.; Giunchi, F.; Brunelli, M.; Tortora, G. Prostate cancer heterogeneity: Discovering novel molecular targets for therapy. Cancer Treat. Rev. 2017, 54, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Cardiff, R.D.; Desai, N.; Banach-Petrosky, W.A.; Parsons, R.; Shen, M.M.; Abate-Shen, C. Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 2884–2889. [Google Scholar] [CrossRef] [Green Version]
- Bowen, C.; Bubendorf, L.; Voeller, H.J.; Slack, R.; Willi, N.; Sauter, G.; Gasser, T.C.; Koivisto, P.; Lack, E.E.; Kononen, J.; et al. Loss of NKX3.1 expression in human prostate cancers correlates with tumor progression. Cancer Res. 2000, 60, 6111–6115. [Google Scholar]
- Hong, H.; Kohli, K.; Garabedian, M.J.; Stallcup, M.R. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol. Cell. Biol. 1997, 17, 2735–2744. [Google Scholar] [CrossRef] [Green Version]
- Toufektchan, E.; Toledo, F. The Guardian of the Genome Revisited: p53 Downregulates Genes Required for Telomere Maintenance, DNA Repair, and Centromere Structure. Cancers 2018, 10, 135. [Google Scholar] [CrossRef] [Green Version]
- Brognard, J.; Newton, A.C. PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol. Metab. 2008, 19, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Soejima, H.; Zhao, W.; Mukai, T. Epigenetic silencing of the MGMT gene in cancer. Biochem. Cell Biol. 2005, 83, 429–437. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeltser, L.; Desplan, C.; Heintz, N. Hoxb-13: A new Hox gene in a distant region of the HOXB cluster maintains colinearity. Development 1996, 122, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Cai, B. G84E mutation in HOXB13 is firmly associated with prostate cancer risk: A meta-analysis. Tumour Biol. 2014, 35, 1177–1182. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Karsy, M.; Zhuge, J.; Zhong, M.; Liu, D. B-Raf and the inhibitors: From bench to bedside. J. Hematol. Oncol. 2013, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrell, E.M.; Morrison, D.K. Ras-Mediated Activation of the Raf Family Kinases. Cold Spring Harb. Perspect. Med. 2019, 9, a033746. [Google Scholar] [CrossRef]
- Nusse, R. Wnt signaling in disease and in development. Cell Res. 2005, 15, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Arriaga, J.M.; Abate-Shen, C. Genetically Engineered Mouse Models of Prostate Cancer in the Postgenomic Era. Cold Spring Harb. Perspect. Med. 2019, 9, a030528. [Google Scholar] [CrossRef]
- Leibold, J.; Ruscetti, M.; Cao, Z.; Ho, Y.J.; Baslan, T.; Zou, M.; Abida, W.; Feucht, J.; Han, T.; Barriga, F.M.; et al. Somatic Tissue Engineering in Mouse Models Reveals an Actionable Role for WNT Pathway Alterations in Prostate Cancer Metastasis. Cancer Discov. 2020, 10, 1038–1057. [Google Scholar] [CrossRef]
- Sciarra, A.; Mariotti, G.; Gentile, V.; Voria, G.; Pastore, A.; Monti, S.; Di Silverio, F. Neuroendocrine differentiation in human prostate tissue: Is it detectable and treatable? BJU Int. 2003, 91, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Nath, D.; Li, X.; Mondragon, C.; Post, D.; Chen, M.; White, J.R.; Hryniewicz-Jankowska, A.; Caza, T.; Kuznetsov, V.A.; Hehnly, H.; et al. Abi1 loss drives prostate tumorigenesis through activation of EMT and non-canonical WNT signaling. Cell Commun. Signal. CCS 2019, 17, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacobbe, A.; Abate-Shen, C. Modeling metastasis in mice: A closer look. Trends Cancer 2021, 7, 916–929. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Le Magnen, C.; Mitrofanova, A.; Ouyang, X.; Califano, A.; Abate-Shen, C. Identification of an NKX3.1-G9a-UTY transcriptional regulatory network that controls prostate differentiation. Science 2016, 352, 1576–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardella, C.; Lunardi, A.; Patnaik, A.; Cantley, L.C.; Pandolfi, P.P. The APL paradigm and the “co-clinical trial” project. Cancer Discov. 2011, 1, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, A.; Panja, S.; Virk, R.K.; Kim, J.Y.; Zott, R.; Cremers, S.; Golombos, D.M.; Liu, D.; Mosquera, J.M.; Mostaghel, E.A.; et al. Co-clinical Analysis of a Genetically Engineered Mouse Model and Human Prostate Cancer Reveals Significance of NKX3.1 Expression for Response to 5alpha-reductase Inhibition. Eur. Urol. 2017, 72, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Clohessy, J.G.; Pandolfi, P.P. The Mouse Hospital and Its Integration in Ultra-Precision Approaches to Cancer Care. Front. Oncol. 2018, 8, 340. [Google Scholar] [CrossRef] [Green Version]
- Moreira, D.; Adamus, T.; Zhao, X.; Su, Y.L.; Zhang, Z.; White, S.V.; Swiderski, P.; Lu, X.; DePinho, R.A.; Pal, S.K.; et al. STAT3 Inhibition Combined with CpG Immunostimulation Activates Antitumor Immunity to Eradicate Genetically Distinct Castration-Resistant Prostate Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 5948–5962. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.K.; Kortylewski, M. Breaking bad habits: Targeting MDSCs to alleviate immunosuppression in prostate cancer. Oncoimmunology 2016, 5, e1078060. [Google Scholar] [CrossRef]





| Model | Availability, Ease of Use, Cost | Major Applications | Advantages | Disadvantages |
|---|---|---|---|---|
| 2D Cell Lines | -High availability -Easy to use -Low cost | -Molecular and mechanistic studies -Drug screening studies -Validation experiments -Epigenetic studies | -Relatively quick results -Data may be made available online -Phenotypic analysis by microscopy studies | -Results may only apply to particular cell line, unless repeated in multiple cell lines -Lacks intra-tumor heterogeneity -Intrinsic effects due to high level passaging -Lacks phenotypic characteristics of parental tumors |
| 3D Organoids, Spheroids | -High availability -Requires more time and elaborate methods for observations and experiments -Intermediate cost | -Molecular and mechanistic studies -Drug screening studies -Imaging and observational studies | -Data can be easily obtained in a relatively short time -Phenotypic analysis by microscopy studies -High content data such as drug screens -More relevant to human cancer -Easily accessible for DNA and mRNA sequence analysis -Presence of ECM | -Longer timescale than 2D cell lines -Requires more extensive analysis -Data obtained is dependent on environment, causing high variability |
| Xenografts, Allografts | -High availability -Technical expertise required to use -High cost | -Drug response studies and novel drug screening studies -Confirmation of molecular and mechanistic studies | -Comparable to in vivo context -Drug approval studies -Easily accessible for DNA and mRNA sequence analysis | -More time-consuming than 2D or 3D models -Drug response associated with genotype -Immunosuppression limits understanding the role of the immune system in tumor response (alleviated for allografts) |
| Patient Derived Xenografts (PDXs) | -Limited availability -Patient consent required to use -Technical expertise required to use -High cost | -Heterotopic injection or transplant -Understanding tumor heterogeneity -Personalized drug testing for effective therapy -Maintenance of tumor architecture -In vivo physiology | -Easily accessible for DNA and mRNA sequence analysis -Understanding of drug resistance response -May lead to specific identification for treatment target | -Subsequent confirmation studies more difficult as each model is unique to each patient -Immunosuppression limits understanding the role of the immune system in tumor response |
| Genetically Engineered (GE) Mice -With transgene expression -Knockouts (KOs) -Constitutive -Conditional | -Limited number of models -Animal colony maintenance required -High cost | -Establishment of in vivo functions of oncogenes and tumor suppressor genes -Genetic interactions -Tumor progression studies -Epithelial-stromal interactions -Immunological studies -Platform for studying metastasis | -Definitive functional studies, including metastatic potential -Establishment of in vivo functions in context of different tissues -Easily accessible for DNA and mRNA sequence analysis | -Long-term studies, with long tumor latency -Time and high cost associated with breeding skills and genotyping -Many common genotypes not represented -Patented strains unavailable -Phenotype may be influenced by strain -Spontaneous, strain-dependent tumorigenesis independent from genetic engineering |
| Model | Alteration | Driver and/or Add. Genetic Alteration | Phenotype | Reference |
|---|---|---|---|---|
| p27Kip1(1) | Loss of expression | Created by gene targeting in embryonic stem cells | Hyperplasia of multiple organs, including prostate, testis, and thymus | [36] |
| Nkx3.1 | Loss of expression | Genomic clones isolated from λFIXII library from 129Sv/J genomic DNA | Prostatic epithelial hyperplasia and dysplasia; decreased bulbourethral gland size | [37] |
| Rbflox | Loss of expression | PBCre4 driver (2) | Focal areas of epithelial hyperplasia; loss of basement membrane and smooth muscle layer integrity | [38] |
| IGF-1flox | Gain of expression | PBCre4 driver (2) | Cell autonomous proliferation; hyperplasia | [39] |
| FOXA1 | Loss of expression | PBCre4 driver (2) | Progressive hyperplasia with extensive cribriform patterning | [40] |
| Model | Alteration | Driver and/or Add. Genetic Alteration | Phenotype | Reference |
|---|---|---|---|---|
| KDM5B | Gain of expression | Loss of Pten function | HGPIN | [42] |
| Sox9 | Gain of expression | Hemizygous loss of Pten (germline heterozygous Pten allele) | HGPIN | [43] |
| Ptenflox/flox | Loss of expression | K14-CreERT2 driver (1) | PIN development | [44] |
| Pten | Loss of expression | Mouse Pten disrupted by homologous recombination | PIN development; formation of aberrant embryoid bodies | [45] |
| Pten x p53 | Loss of expression | Recombination of adult prostatic epithelium with embryonic rat seminal vesicle mesenchyme | HGPIN | [46] |
| Abi1 | Loss of expression | PBCre4 driver (2) | PIN development | [47] |
| EAF2 | Loss of expression | PB-CreERT2 driver (3) | Luminal epithelial hyperplasia and mPIN | [48] |
| ACSS3 | Loss of expression | Transfection of overexpressing lentivirus and sgRNA (CRISPR/Cas9) | PIN in anterior prostate; increased proliferation, migration, and invasion | [49] |
| CSF-1 | Gain of expression | PBCre4 driver | Immune cell infiltration into prostate; LGPIN | [50] |
| Model | Alteration | Driver and/or Add. Genetic Alteration | Phenotype | Reference |
|---|---|---|---|---|
| Timp3 | Loss of expression | Homozygous loss of Pten (conditional Pten allele) | HGPIN with microinvasion | [51] |
| mpAkt | Gain of expression | Myc gain of expression under control of PB driver; loss of Pten function | mPIN followed by microinvasive carcinoma, disruption of basement membrane integrity, stromal remodeling, and lymphocyte infiltration | [52] |
| Nkx3.1 | Loss of expression | Myc gain of expression under control of CMV enhancer and β-actin promoter | HGPIN with microinvasion | [53] |
| Pten | Loss of expression | Myc gain of expression under control of CMV enhancer and β-actin promoter | Microinvasive cancer with disruption of smooth muscle actin | [54] |
| Model | Alteration | Driver and/or Add. Genetic Alteration | Phenotype | Reference |
|---|---|---|---|---|
| Nkx3.1 | Loss of expression | Hemizygous loss of Pten (germline heterozygous Pten allele) | HGPIN with invasive adenocarcinoma | [55,56] |
| p27Kip1 | Loss of expression | Hemizygous loss of Pten (germline heterozygous Pten allele) | HGPIN with invasive adenocarcinoma | [57,58] |
| Aft3 | Loss of expression | Homozygous loss of Pten (conditional Pten allele) | HGPIN with invasive adenocarcinoma | [59] |
| Apcflox | Loss of expression | PBCre4 driver | HGPIN followed by local adenocarcinoma | [60] |
| Bmi1 | Gain of expression | Hemizygous loss of Pten (germline heterozygous Pten allele) | Locally invasive and highly vascularized adenocarcinoma, with frequent bladder outlet obstruction | [61] |
| Tsc2 | Loss of expression | Hemizygous loss of Pten (germline heterozygous Pten allele) | Invasive adenocarcinoma; enhanced lymphoid proliferation; development of skin cancer | [62] |
| Phlpp1 | Loss of expression | Hemizygous loss of Pten (germline heterozygous Pten allele) | Invasive adenocarcinoma at full penetrance with onset of 8 months | [63] |
| Chk1 | Loss of expression | Hemizygous loss of Pten (germline heterozygous Pten allele) | Progression of HGPIN into invasive adenocarcinoma | [64] |
| PKCε | Gain of expression | Hemizygous loss of Pten (germline heterozygous Pten allele) | Invasive adenocarcinoma, preferentially in ventral prostate | [65] |
| Gata3 | Loss of expression | Homozygous loss of Pten (conditional Pten allele) | Acceleration of invasive adenocarcinoma | [66] |
| Erg | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Foci of invasive adenocarcinoma with varying levels of Erg expression | [67,68] |
| Etv1 | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with homogenous Etv1 expression | [69] |
| Junb | Loss of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma in anterior prostate, with strong histological similarity to human PCa | [70] |
| SPOP- F133V | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive, poorly differentiated, and highly proliferative adenocarcinoma | [71] |
| PSGR | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma featuring Akt activation and extensive inflammatory cell infiltration | [72] |
| Zbtb7a | Loss of expression | Homozygous loss of Pten (conditional Pten allele) | Highly penetrant invasive adenocarcinoma at 11 weeks | [73] |
| Hepsin | Gain of expression | Myc gain of expression under control of PB driver | Invasive adenocarcinoma lacking glandular prostate differentiation and clear basement membrane contour | [74] |
| MMP7 | Gain of expression | Loss of Pten function | Invasive adenocarcinoma through induction of epithelial-to-mesenchymal transition (EMT) | [75] |
| Ptenadcre+(1) | Loss of expression | Cre-expressing adenovirus via intraductal injection into anterior- posterior prostate | Invasive adenocarcinoma with onset of 8–16 weeks | [76] |
| Kindlin-3 | Loss of expression | Xenograft | Subcutaneous prostate cancer tumor growth | [77] |
| Model | Alteration | Driver and/or Add. Genetic Alteration | Phenotype | Reference |
|---|---|---|---|---|
| Ptenflox/flox (exon 5) | Loss of expression | PBCre4 driver | Invasive adenocarcinoma with metastasis to lungs, rarely to lymph nodes | [91] |
| Nr2f2 (COUP-TFII) | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with metastasis to lymph nodes | [92] |
| NCoA2 | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with metastasis to lymph nodes, lungs | [93] |
| NSD2 (Whsc-1) | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with metastasis to lymph nodes, lungs, bone | [94] |
| Trp53 | Loss of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with metastasis to lymph nodes, spleen, liver, organs near GU tract excluding bladder | [95,96,97] |
| Rb | Loss of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with metastasis to lymph nodes, lungs, liver that resembles NEPC | [98] |
| Jnk1/2 | Loss of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with metastasis to lymph nodes | [99] |
| Stat3 and IL-6 | Loss of expression | Homozygous loss of Pten (conditional Pten allele) | Poorly differentiated cancer with metastasis to liver, lungs | [100] |
| NICD | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with metastasis to liver, lungs | [101] |
| Smad4 | Loss of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with metastasis | [102] |
| Smad4/p53 | Loss of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with metastasis to bone | [103] |
| HoxB13/Myc | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with metastasis to lymph nodes, liver, lungs | [104] |
| Braf V600E (1) | Gain of expression | Homozygous loss of Pten (conditional Pten allele) | Invasive adenocarcinoma with metastasis to lymph nodes, bone marrow, lungs | [105] |
| p53floxRbflox | Loss of expression | Homozygous loss of Pten (conditional Pten allele) PBCre4 driver (2) | Metastatic carcinoma, with distant metastases | [106] |
| * NPKEYFP | Nkx3.1 loss of expression Pten loss of expression Kras gain of expression | Nkx3.1CreERT2/+ (3) Ptenflox/flox KrasLSL-G12D/+ (4) | Invasive adenocarcinoma with metastasis to bone | [107] |
| SIRT-6 | Gain of expression | Luciferase expressing PC3M cells in an orthotopic xenograft mouse model | Metastasis to liver; upregulation of N-cadherin and vimentin, downregulation of E-cadherin in vitro | [108] |
| RIPK2 | Gain of expression | Injection of RIPK2-KO 22Rv1 cells into male SCID/Beige mice | Invasive adenocarcinoma with metastasis to bone | [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamiecki, R.; Hryniewicz-Jankowska, A.; Ortiz, M.A.; Li, X.; Porter-Hansen, B.A.; Nsouli, I.; Bratslavsky, G.; Kotula, L. In Vivo Models for Prostate Cancer Research. Cancers 2022, 14, 5321. https://doi.org/10.3390/cancers14215321
Adamiecki R, Hryniewicz-Jankowska A, Ortiz MA, Li X, Porter-Hansen BA, Nsouli I, Bratslavsky G, Kotula L. In Vivo Models for Prostate Cancer Research. Cancers. 2022; 14(21):5321. https://doi.org/10.3390/cancers14215321
Chicago/Turabian StyleAdamiecki, Robert, Anita Hryniewicz-Jankowska, Maria A. Ortiz, Xiang Li, Baylee A. Porter-Hansen, Imad Nsouli, Gennady Bratslavsky, and Leszek Kotula. 2022. "In Vivo Models for Prostate Cancer Research" Cancers 14, no. 21: 5321. https://doi.org/10.3390/cancers14215321