

Molecular Characteristics of Radon Associated Lung Cancer Highlights MET Alterations
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Patient Material
2.2. Molecular Analyses
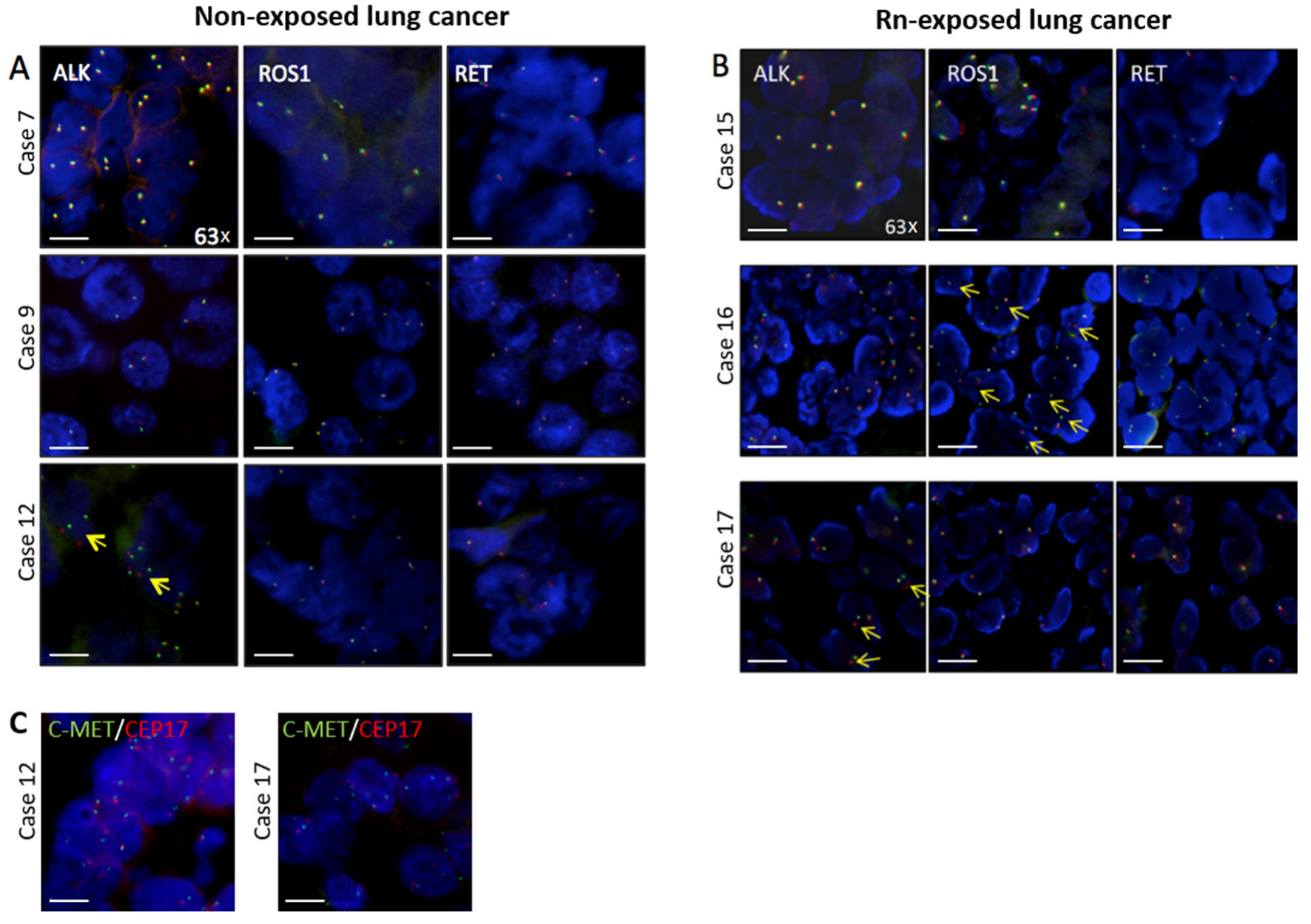
2.2.1. Fluorescence In Situ Hybridization (FISH)
2.2.2. Parallel Sequencing (Next Generation Sequencing, NGS)
2.2.3. Measurement of Indoor Radon Levels
2.3. Data/Statistical Analysis
3. Results
3.1. Patients Characteristics
3.2. Molecular Profile
3.3. Number of Mutations in Correlation with Level of Rn-Exposure
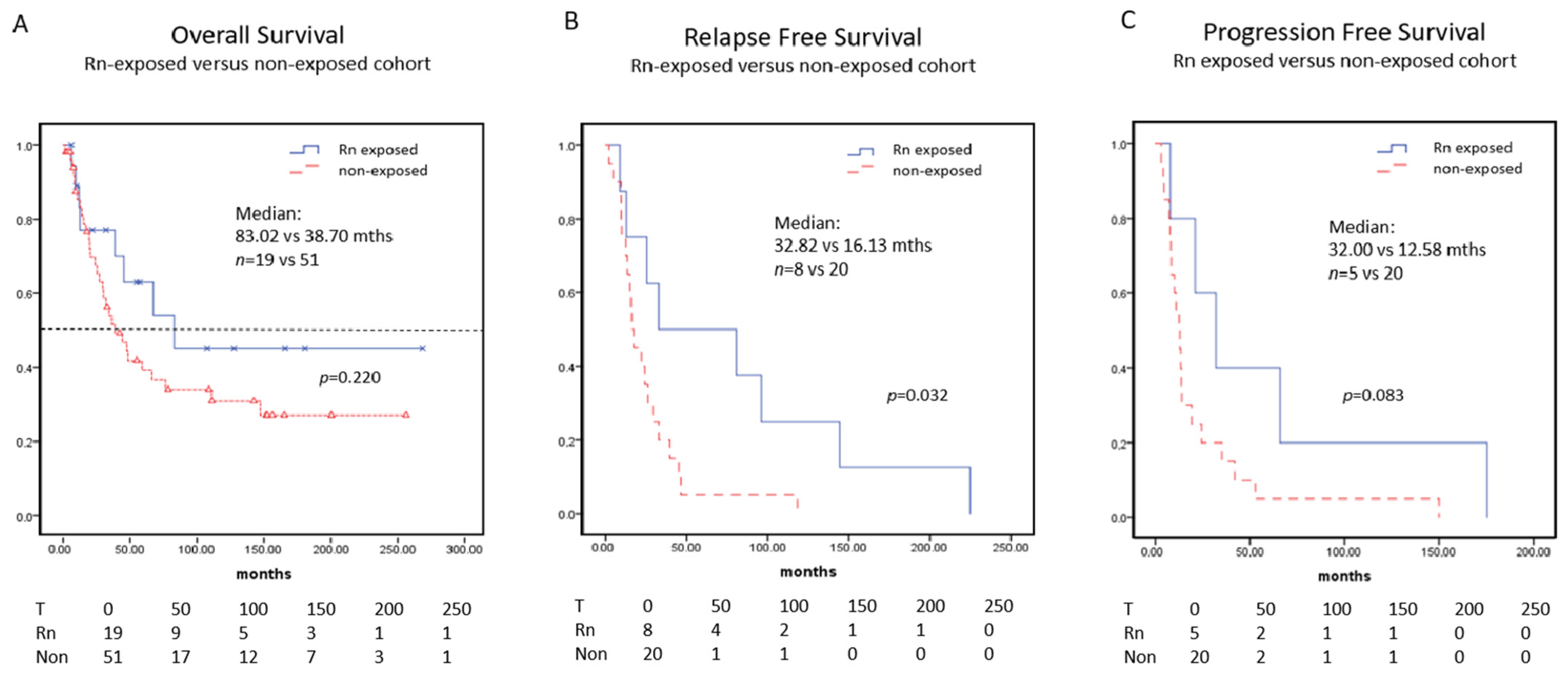
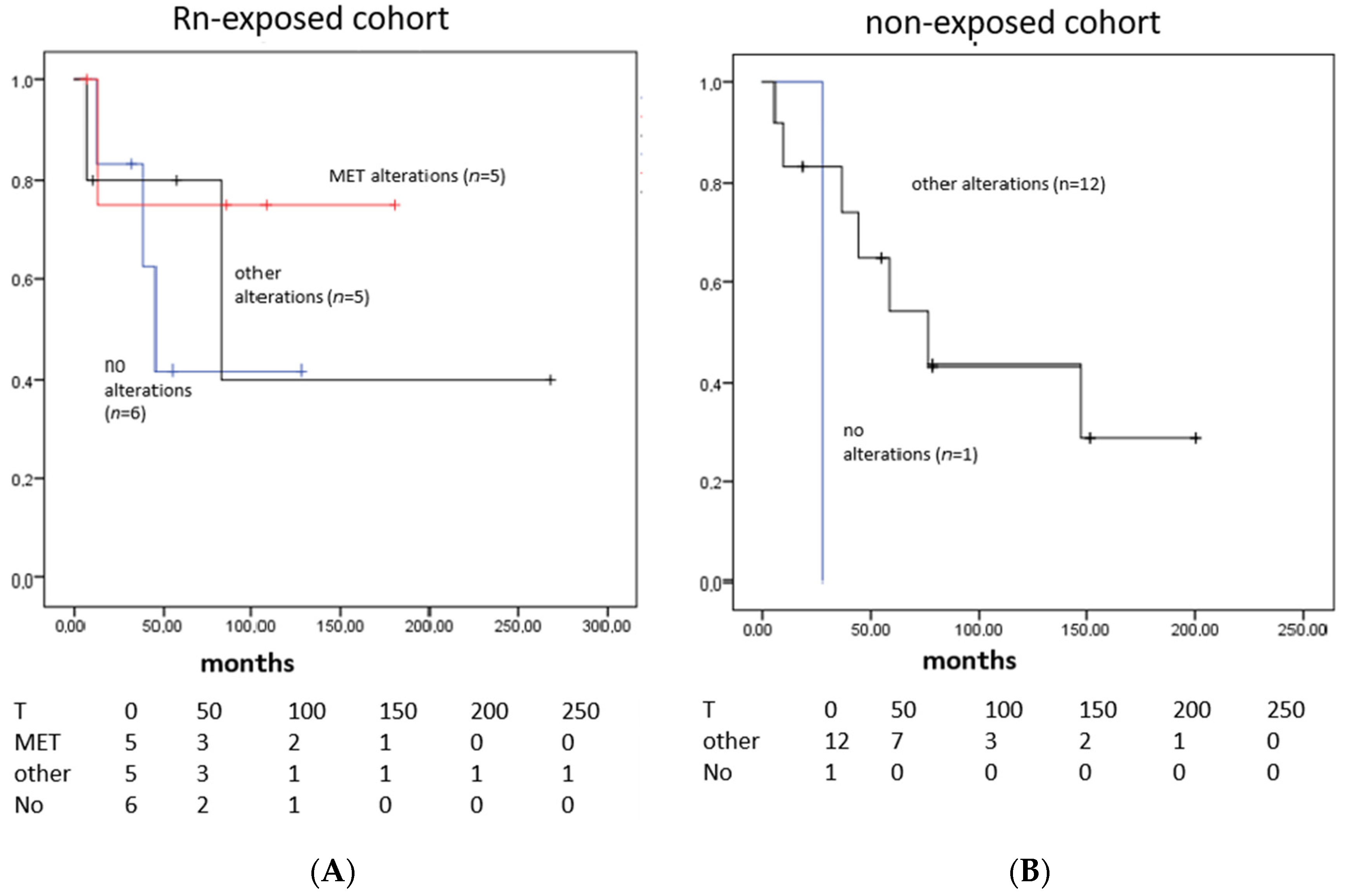
3.4. Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parkin, D.M.; Boyd, L.; Walker, L.C. The fraction of cancer attributable to lifestyle and environmental factors in the UK in 2010. Br. J. Cancer 2011, 105 (Suppl. S2), S77–S81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Couraud, S.; Zalcman, G.; Milleron, B.; Morin, F.; Souquet, P.J. Lung cancer in never smokers—A review. Eur. J. Cancer 2012, 48, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- World Health Organization. WHO Handbook on Indoor Radon: A Public Health Perspective. 2009. Available online: https://www.ncbi.nlm.nih.gov/books/NBK143216/ (accessed on 6 July 2022).
- Ruano-Ravina, A.; Pereyra, M.F.; Castro, M.T.; Perez-Rios, M.; Abal-Arca, J.; Barros-Dios, J.M. Genetic susceptibility, residential radon, and lung cancer in a radon prone area. J. Thorac. Oncol. 2014, 9, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darby, S.; Hill, D.; Auvinen, A.; Barros-Dios, J.M.; Baysson, H.; Bochicchio, F.; Deo, H.; Falk, R.; Forastiere, F.; Hakama, M.; et al. Radon in homes and risk of lung cancer: Collaborative analysis of individual data from 13 European case-control studies. BMJ 2005, 330, 223. [Google Scholar] [CrossRef] [Green Version]
- United Nations Scientific Committe on the Effects of Atomic Radiation. Sources and Effects of Ionizing Radiation; United Nations Pubns.: New York, NY, USA, 2000; Volume 1, Available online: https://www.unscear.org/docs/publications/2000/UNSCEAR_2000_Report_Vol.I.pdf (accessed on 6 July 2022).
- Al-Zoughool, M.; Krewski, D. Health effects of radon: A review of the literature. Int. J. Radiat. Biol. 2009, 85, 57–69. [Google Scholar] [CrossRef]
- Robertson, A.; Allen, J.; Laney, R.; Curnow, A. The cellular and molecular carcinogenic effects of radon exposure: A review. Int. J. Mol. Sci. 2013, 14, 14024–14063. [Google Scholar] [CrossRef] [Green Version]
- Appleton, J.D. Radon: Sources, health risks, and hazard mapping. Ambio 2007, 36, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Barros-Dios, J.M.; Ruano-Ravina, A.; Perez-Rios, M.; Castro-Bernardez, M.; Abal-Arca, J.; Tojo-Castro, M. Residential radon exposure, histologic types, and lung cancer risk. A case-control study in Galicia, Spain. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 951–958. [Google Scholar] [CrossRef]
- Barros-Dios, J.M.; Barreiro, M.A.; Ruano-Ravina, A.; Figueiras, A. Exposure to residential radon and lung cancer in Spain: A population-based case-control study. Am. J. Epidemiol. 2002, 156, 548–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubin, J.H.; Wang, Z.Y.; Boice, J.D., Jr.; Xu, Z.Y.; Blot, W.J.; De Wang, L.; Kleinerman, R.A. Risk of lung cancer and residential radon in China: Pooled results of two studies. Int. J. Cancer 2004, 109, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Krewski, D.; Lubin, J.H.; Zielinski, J.M.; Alavanja, M.; Catalan, V.S.; Field, R.W.; Klotz, J.B.; Létourneau, E.G.; Lynch, C.F.; Lyon, J.L. A combined analysis of North American case-control studies of residential radon and lung cancer. J. Toxicol. Environ. Health A 2006, 69, 533–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, P.; Quan, C.; Hu, C.; Zhang, J.; Xie, F.; Hu, X.; Yu, Z.; Gao, B.; Liu, Z.; Zheng, H. Nonlinear dose-response relationship between radon exposure and the risk of lung cancer: Evidence from a meta-analysis of published observational studies. Eur. J. Cancer Prev. 2015, 24, 267–277. [Google Scholar] [CrossRef]
- Vahakangas, K.H.; Metcalf, R.A.; Welsh, J.A.; Bennett, W.P.; Harris, C.C.; Samet, J.M.; Lane, D.P. Mutations of p53 and ras genes in radon-associated lung cancer from uranium miners. Lancet 1992, 339, 576–580. [Google Scholar] [CrossRef]
- Vahakangas, K.H. TP53 mutations in workers exposed to occupational carcinogens. Hum. Mutat. 2003, 21, 240–251. [Google Scholar] [CrossRef]
- Taylor, J.A.; Watson, M.A.; Devereux, T.R.; Michels, R.Y.; Saccomanno, G.; Anderson, M. p53 mutation hotspot in radon-associated lung cancer. Lancet 1994, 343, 86–87. [Google Scholar] [CrossRef]
- Choi, J.R.; Koh, S.B.; Park, S.Y.; Kim, H.R.; Lee, H.; Kang, D.R. Novel Genetic Associations between Lung Cancer and Indoor Radon Exposure. J. Cancer Prev. 2017, 22, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Wesch, H.; Mueller, K.M.; Bartsch, H.; Wegener, K.; Hollstein, M. Analysis of radon-associated squamous cell carcinomas of the lung for a p53 gene hotspot mutation. Br. J. Cancer 2000, 82, 763–766. [Google Scholar] [CrossRef] [Green Version]
- Ruano-Ravina, A.; Faraldo-Valles, M.J.; Barros-Dios, J.M. Is there a specific mutation of p53 gene due to radon exposure? A systematic review. Int. J. Radiat. Biol. 2009, 85, 614–621. [Google Scholar] [CrossRef]
- Ruano-Ravina, A.; Torres-Duran, M.; Kelsey, K.T.; Parente-Lamelas, I.; Leiro-Fernandez, V.; Abdulkader, I.; Abal-Arca, J.; Montero-Martínez, C.; Vidal-García, I.; Amendo, M. Residential radon, EGFR mutations and ALK alterations in neversmoking lung cancer cases. Eur. Respir. J. 2016, 48, 1462–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taga, M.; Mechanic, L.E.; Hagiwara, N.; Vahakangas, K.H.; Bennett, W.P.; Alavanja, M.C.; Welsh, J.A.; Khan, M.A.; Lee, A.; Diasio, R. EGFR somatic mutations in lung tumors: Radon exposure and passive smoking in former- and never-smoking U.S. women. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 988–992.21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonner, M.R.; Bennett, W.P.; Xiong, W.; Lan, Q.; Brownson, R.C.; Harris, C.C.; Field, R.W.; Lubin, J.H.; Alavanja, M.C. Radon, secondhand smoke, glutathione-S-transferase M1 and lung cancer among women. Int. J. Cancer 2006, 119, 1462–1467. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Shim, H.S.; Kim, L.; Kim, T.J.; Kwon, K.Y.; Lee, G.K.; Chung, J.H. Guideline Recommendations for Testing of ALK Gene Rearrangement in Lung Cancer: A Proposal of the Korean Cardiopulmonary Pathology Study Group. Korean J. Pathol. 2014, 48, 1–9. [Google Scholar] [CrossRef]
- Warth, A.; Muley, T.; Dienemann, H.; Goeppert, B.; Stenzinger, A.; Schnabel, P.A.; Schirmacher, P.; Penzel, R.; Weichert, W. ROS1 expression and translocations in non-small-cell lung cancer: Clinicopathological analysis of 1478 cases. Histopathology 2014, 65, 187–194. [Google Scholar] [CrossRef]
- Lee, S.E.; Lee, B.; Hong, M.; Song, J.Y.; Jung, K.; Lira, M.E.; Mao, M.; Han, J.; Kim, J.; Choi, Y.L. Comprehensive analysis of RET and ROS1 rearrangement in lung adenocarcinoma. Mod. Pathol. 2015, 28, 468–479. [Google Scholar] [CrossRef] [Green Version]
- Ach, T.; Zeitler, K.; Schwarz-Furlan, S.; Baader, K.; Agaimy, A.; Rohrmeier, C.; Zenk, J.; Gosau, M.; Reichert, T.E.; Brockhoff, G.; et al. Aberrations of MET are associated with copy number gain of EGFR and loss of PTEN and predict poor outcome in patients with salivary gland cancer. Virchows Arch. 2013, 462, 65–72. [Google Scholar] [CrossRef]
- Oberaigner, W.; Kreienbrock, L.; Schaffrath-Rosario, A.; Kreuzer, M.; Wellmann, J.; Keller, G.; Gerken, M.; Langer, B.; Wichmann, H.E. Radon und Lungenkrebs im Bezirk Imst/Österreich, Reihe Fortschritte in der Umweltmedizin; Ecomed: Landsberg am Lech, Germany, 2002. [Google Scholar]
- Ennemoser, O.; Ambach, W.; Brunner, P.; Schneider, P.; Oberaigner, W.; Purtscheller, F.; Stingl, V.; Keller, G. Unusually high indoor radon concentrations from a giant rock slide. Sci. Total Environ. 1994, 151, 235–240. [Google Scholar] [CrossRef]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef]
- Lorenzo-Gonzalez, M.; Ruano-Ravina, A.; Peon, J.; Pineiro, M.; Barros-Dios, J.M. Residential radon in Galicia: A cross-sectional study in a radon-prone area. J. Radiol. Prot. 2017, 37, 728–741. [Google Scholar] [CrossRef]
- Jiao, X.D.; Qin, B.D.; You, P.; Cai, J.; Zang, Y.S. The prognostic value of TP53 and its correlation with EGFR mutation in advanced non-small cell lung cancer, an analysis based on cBioPortal data base. Lung Cancer 2018, 123, 70–75. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, Z.; Yin, H.; Yang, G. Interaction between p53 and Ras signaling controls cisplatin resistance via HDAC4- and HIF-1α-mediated regulation of apoptosis and autophagy. Theranostics 2019, 9, 1096–1114. [Google Scholar] [CrossRef] [PubMed]
- AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, J.W.; Taylor, J.A.; Watson, M.A.; Saccomanno, G.; Devereux, T.R. p53 and K-ras in radon-associated lung adenocarcinoma. Cancer Epidemiol. Biomarkers Prev. 1995, 4, 791–793. [Google Scholar] [PubMed]
- Jeanson, A.; Tomasini, P.; Souquet-Bressand, M.; Brandone, N.; Boucekine, M.; Grangeon, M.; Chaleat, S.; Khobta, N.; Milia, J.; Mhanna, L.; et al. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC). J. Thorac. Oncol. 2019, 14, 1095–1101. [Google Scholar] [CrossRef]
- Hayama, T.; Hashiguchi, Y.; Okamoto, K.; Okada, Y.; Ono, K.; Shimada, R.; Ozawa, T.; Toyoda, T.; Tsuchiya, T.; Iinuma, H.; et al. G12V and G12C mutations in the gene KRAS are associated with a poorer prognosis in primary colorectal cancer. Int. J. Color. Dis. 2019, 34, 1491–1496. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Reference SNP Cluster Report: rs56391007. 2022. Available online: https://www.ncbi.nlm.nih.gov/snp/rs56391007?horizontal_tab=true (accessed on 6 July 2022).
- Wasenius, V.M.; Hemmer, S.; Karjalainen-Lindsberg, M.L.; Nupponen, N.N.; Franssila, K.; Joensuu, H. MET receptor tyrosine kinase sequence alterations in differentiated thyroid carcinoma. Am. J. Surg. Pathol. 2005, 29, 544–549. [Google Scholar] [CrossRef]
- Ma, P.C.; Kijima, T.; Maulik, G.; Fox, E.A.; Sattler, M.; Griffin, J.D.; Johnsen, B.E.; Salgia, R. c-MET mutational analysis in small cell lung cancer: Novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003, 63, 6272–6281. [Google Scholar]
- Voortman, J.; Harada, T.; Chang, R.P.; Killian, J.K.; Suuriniemi, M.; Smith, W.I.; Meltzer, P.S.; Lucchi, M.; Wang, Y.; Giaconne, G. Detection and therapeutic implications of c-Met mutations in small cell lung cancer and neuroendocrine tumors. Curr. Pharm. Des. 2013, 19, 833–840. [Google Scholar] [CrossRef]
- Vansteenkiste, J.F.; Van De Kerkhove, C.; Wauters, E.; Van Mol, P. Capmatinib for the treatment of non-small cell lung cancer. Expert Rev. Anticancer Ther. 2019, 19, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.X.; Li, J.; Dong, S.; Lin, L.; Zou, C.; Chen, Z.S. Tepotinib hydrochloride for the treatment of non-small cell lung cancer. Drugs. Today 2021, 57, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zou, Q.; Liu, H.; Qiu, B.; Li, Q.; Lin, Y.; Liang, Y. Management of Non-small Cell Lung Cancer Patients with MET Exon 14 Skipping Mutations. Curr. Treat. Options Oncol. 2020, 21, 33. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.E.; Camidge, D.R.; Ou, S.-H.I.; Clark, J.W.; Socinski, M.A.; Weiss, J.; Riely, G.J.; Winter, M.; Wang, S.C.; Monti, K.; et al. Efficacy and safety of crizotinib in patients (pts) with advanced MET exon 14-altered non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2016, 34 (Suppl. 15), 108. [Google Scholar] [CrossRef]
- Landi, L.; Chiari, R.; Tiseo, M.; D’Inca, F.; Dazzi, C.; Chella, A.; Delmonte, A.; Bonanno, L.; Giannarelli, D.; Cortinovis, D.L.; et al. Crizotinib in MET deregulated or ROS1 rearranged pretreated non-small-cell lung cancer (METROS): A phase II, prospective, multicentre, two-arms trial. Clin. Cancer Res. 2019, 25, 7312–7319. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Author (Ref.) | Year | Region | Subjects | Gene |
|---|---|---|---|---|
| S. Darby et al. [7] | 2005 | Europa | 7142 patients + 14,208 controls | - |
| K. Vahakangas et al. [17] | 1992 | Europa | 19 uranium miners | TP53 |
| J.A. Taylor et al. [19] | 1994 | America | 52 uranium miners | TP53 |
| Q. Yang et al. [21] | 2000 | Germany | 79 uranium miners | TP53 |
| A. Ruano-Ravina et al. [22] | 2009 | Worldwide | 578 individuals | TP53 |
| A. Ruano-Ravina et al. [23] | 2016 | Spain | 323 Patients | EGFR/ALK |
| M. Taga et al. [24] | 2012 | USA | 70 women | EGFR |
| Bonner et al. [25] | 2006 | USA | 270 individuals | GSTM1 |
| J.R. Choi et al. [20] | 2017 | Asia | 19 patients | EGFR/TP53/NKX 2.1/PTEN/CHD7/DDR2/MLL3/CHD5/FAT1/DUSP27 |
| Rn-Exposed LC Cohort n = 20 | Non-Exposed LC Cohort n = 52 | |
|---|---|---|
| Mean age at diagnosis | 63.0 years | 63.4 years |
| Gender (female/male) | 9/11 | 21/31 |
| Alive/dead | 12/8 | 20/31 * |
| UICC | ||
| I | 3 (20.0%) | 12 (23.1%) |
| II | 3 (20.0%) | 9 (17.3%) |
| III | 4 (26.7%) | 17 (32.7%) |
| IV | 5 (33.3%) * | 14 (26.9%) |
| ECOG | ||
| 0–1 | 10 (76.9%) | 39 (86.7%) |
| 2 | 1 (7.7%) | 5 (11.1%) |
| >2 | 2 (15.4%) * | 1 (2.2%) * |
| Histology | ||
| Adenocarcinoma | 8 (40.0%) | 27 (52.0%) |
| Squamous cell carcinoma | 10 (50.0%) | 22 (42.3%) |
| Adenosquamous cell carcinoma | 1 (5.0%) | 0 (0.0%) |
| Large cell carcinoma | 0 (0.0%) | 2 (3.8%) |
| Large cell neuroendocrine carcinoma | 1 (5.0%) | 0 (0.0%) |
| NOS | 0 (0.0%) | 1 (1.9%) |
| Smoking | ||
| Smoker or former smoker | 6 (30.0%) | 34 (65.5%) |
| Never smoker | 4 (20.0%) | 10 (19.2%) |
| Unknown | 10 (50.0%) | 8 (15.3%) |
| Cohort | Frequency (n) | Percent (%) | |
|---|---|---|---|
| Non-exposed | no alterations | 1 | 7 |
| KRAS_mut | 1 | 7 | |
| TP53_mut | 8 | 57 | |
| TP53/ERBB2mut | 1 | 7 | |
| PTEN mut | 1 | 7 | |
| ALK-rearr | 2 | 14 | |
| Rn-exposed | no alterations | 6 | 35 |
| KRAS_mut | 2 | 12 | |
| TP53_mut | 3 | 18 | |
| ROS-rearr | 1 | 6 | |
| MET-amplification | 1 | 6 | |
| MET_mutation | 2 | 12 | |
| MET T1010I, ALK rearr | 1 | 6 | |
| MET T1010I, TP53_mut | 1 | 6 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamerith, G.; Kloppenburg, M.; Mildner, F.; Amann, A.; Merkelbach-Bruse, S.; Heydt, C.; Siemanowski, J.; Buettner, R.; Fiegl, M.; Manzl, C.; et al. Molecular Characteristics of Radon Associated Lung Cancer Highlights MET Alterations. Cancers 2022, 14, 5113. https://doi.org/10.3390/cancers14205113
Gamerith G, Kloppenburg M, Mildner F, Amann A, Merkelbach-Bruse S, Heydt C, Siemanowski J, Buettner R, Fiegl M, Manzl C, et al. Molecular Characteristics of Radon Associated Lung Cancer Highlights MET Alterations. Cancers. 2022; 14(20):5113. https://doi.org/10.3390/cancers14205113
Chicago/Turabian StyleGamerith, Gabriele, Marcel Kloppenburg, Finn Mildner, Arno Amann, Sabine Merkelbach-Bruse, Carina Heydt, Janna Siemanowski, Reinhard Buettner, Michael Fiegl, Claudia Manzl, and et al. 2022. "Molecular Characteristics of Radon Associated Lung Cancer Highlights MET Alterations" Cancers 14, no. 20: 5113. https://doi.org/10.3390/cancers14205113