
Upregulation of Receptor Tyrosine Kinase Activity and Stemness as Resistance Mechanisms to Akt Inhibitors in Breast Cancer
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Generation and Characterization of Cells Resistant to Akt Inhibitors
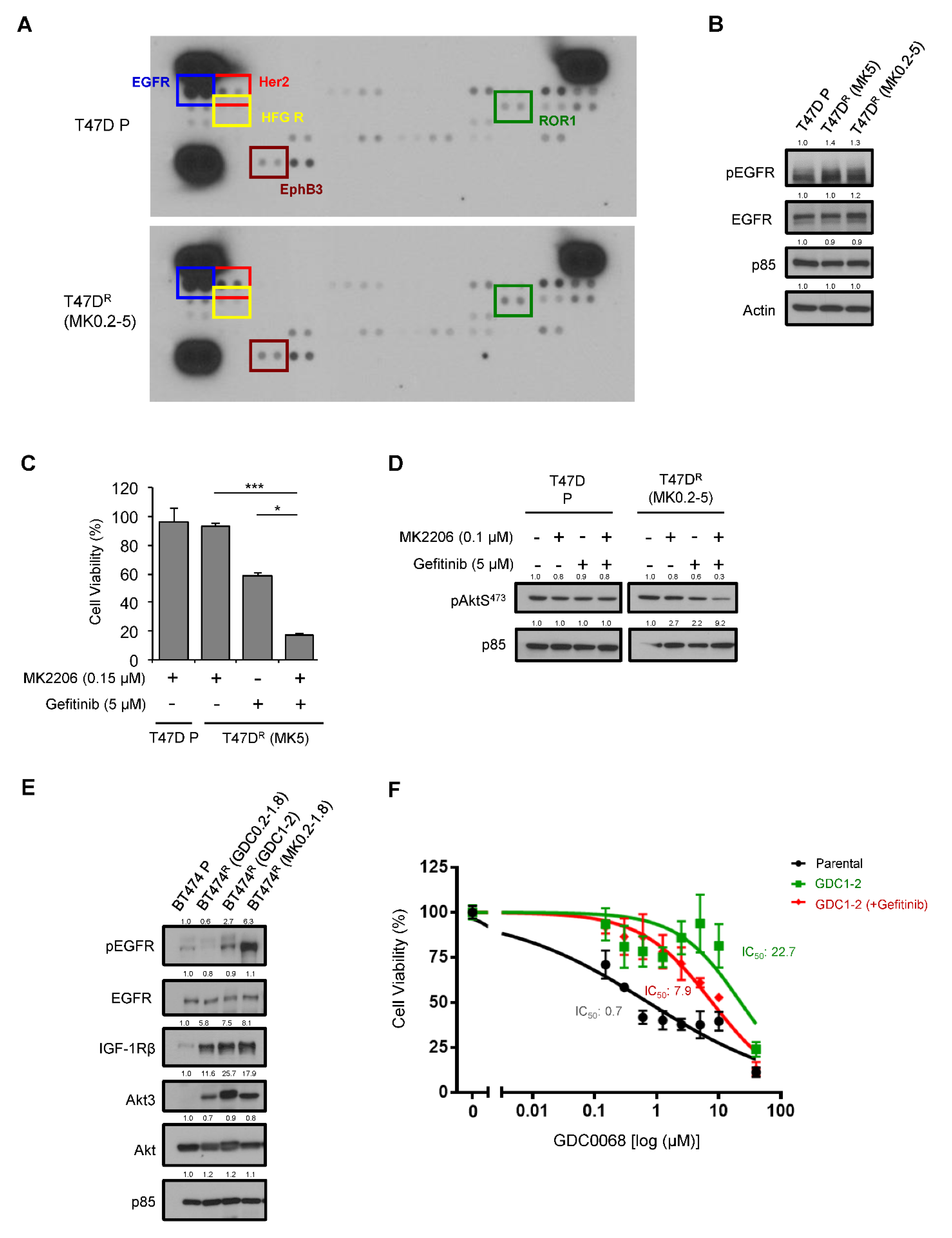
3.2. Activation of EGFR as a Mechanism of Acquired Resistance to AKT Inhibitors
3.3. Enhanced Cancer Stem Cell Properties in Akt-Inhibitor-Resistant Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, M.J.; Perou, C.M. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013, 3, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT Inhibitors: New Weapons in the Fight against Breast Cancer? Front. Pharmacol. 2021, 12, 662232. [Google Scholar] [CrossRef]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, A.M.; Nemsadze, G.; Baird, R.D.; Park, Y.H.; Hall, P.S.; Perren, T.; et al. Capivasertib Plus Paclitaxel Versus Placebo Plus Paclitaxel As First-Line Therapy for Metastatic Triple-Negative Breast Cancer: The PAKT Trial. J. Clin. Oncol. 2020, 38, 423–433. [Google Scholar] [CrossRef]
- Smyth, L.M.; Tamura, K.; Oliveira, M.; Ciruelos, E.M.; Mayer, I.A.; Sablin, M.P.; Biganzoli, L.; Ambrose, H.J.; Ashton, J.; Barnicle, A.; et al. Capivasertib, an AKT Kinase Inhibitor, as Monotherapy or in Combination with Fulvestrant in Patients with AKT1 (E17K)-Mutant, ER-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 3947–3957. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Ebi, H.; Martini, M.; Beausoleil, S.A.; Faber, A.C.; Jakubik, C.T.; Huang, A.; Wang, Y.; Nishtala, M.; Hall, B.; et al. Measurement of PIP3 levels reveals an unexpected role for p110beta in early adaptive responses to p110alpha-specific inhibitors in luminal breast cancer. Cancer Cell 2015, 27, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Leroy, C.; Ramos, P.; Cornille, K.; Bonenfant, D.; Fritsch, C.; Voshol, H.; Bentires-Alj, M. Activation of IGF1R/p110beta/AKT/mTOR confers resistance to alpha-specific PI3K inhibition. Breast Cancer Res. 2016, 18, 41. [Google Scholar] [CrossRef] [Green Version]
- Mundt, F.; Rajput, S.; Li, S.; Ruggles, K.V.; Mooradian, A.D.; Mertins, P.; Gillette, M.A.; Krug, K.; Guo, Z.; Hoog, J.; et al. Mass Spectrometry-Based Proteomics Reveals Potential Roles of NEK9 and MAP2K4 in Resistance to PI3K Inhibition in Triple-Negative Breast Cancers. Cancer Res. 2018, 78, 2732–2746. [Google Scholar] [CrossRef] [Green Version]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med. 2015, 7, 283ra251. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Santiago, S.; Raeder, M.; Zhang, F.; Isabella, A.; Yang, J.; Semaan, D.J.; Chen, C.; Fox, E.A.; et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat. Med. 2011, 17, 1116–1120. [Google Scholar] [CrossRef]
- Muellner, M.K.; Uras, I.Z.; Gapp, B.V.; Kerzendorfer, C.; Smida, M.; Lechtermann, H.; Craig-Mueller, N.; Colinge, J.; Duernberger, G.; Nijman, S.M. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat. Chem. Biol. 2011, 7, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Serra, V.; Eichhorn, P.J.; Garcia-Garcia, C.; Ibrahim, Y.H.; Prudkin, L.; Sanchez, G.; Rodriguez, O.; Anton, P.; Parra, J.L.; Marlow, S.; et al. RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer. J. Clin. Investig. 2013, 123, 2551–2563. [Google Scholar] [CrossRef] [Green Version]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.J.; Castel, P.; Radosevic-Robin, N.; Elkabets, M.; Auricchio, N.; Aceto, N.; Weitsman, G.; Barber, P.; Vojnovic, B.; Ellis, H.; et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci. Signal. 2014, 7, ra29. [Google Scholar] [CrossRef] [Green Version]
- Stottrup, C.; Tsang, T.; Chin, Y.R. Upregulation of AKT3 Confers Resistance to the AKT Inhibitor MK2206 in Breast Cancer. Mol. Cancer Ther. 2016, 15, 1964–1974. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 2011, 29, 4688–4695. [Google Scholar] [CrossRef]
- Lin, J.; Sampath, D.; Nannini, M.A.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef] [Green Version]
- Kapeller, R.; Toker, A.; Cantley, L.C.; Carpenter, C.L. Phosphoinositide 3-kinase binds constitutively to alpha/beta-tubulin and binds to gamma-tubulin in response to insulin. J. Biol. Chem. 1995, 270, 25985–25991. [Google Scholar] [CrossRef] [Green Version]
- Shaw, F.L.; Harrison, H.; Spence, K.; Ablett, M.P.; Simoes, B.M.; Farnie, G.; Clarke, R.B. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J. Mammary Gland Biol. Neoplasia 2012, 17, 111–117. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Phillips, S.; Prat, A.; Sedic, M.; Proia, T.; Wronski, A.; Mazumdar, S.; Skibinski, A.; Shirley, S.H.; Perou, C.M.; Gill, G.; et al. Cell-state transitions regulated by SLUG are critical for tissue regeneration and tumor initiation. Stem Cell Rep. 2014, 2, 633–647. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zurrer-Hardi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Li, J.; Cheung, T.C.; Tam, V.; Lau, C.G.; Wang, X.; Chin, Y.R. Akt3 promotes cancer stemness in triple-negative breast cancer through YB1-Snail/Slug signaling axis. Genes Dis. 2022. online published. [Google Scholar] [CrossRef]
- Junankar, S.; Baker, L.A.; Roden, D.L.; Nair, R.; Elsworth, B.; Gallego-Ortega, D.; Lacaze, P.; Cazet, A.; Nikolic, I.; Teo, W.S.; et al. ID4 controls mammary stem cells and marks breast cancers with a stem cell-like phenotype. Nat. Commun. 2015, 6, 6548. [Google Scholar] [CrossRef]
- Beger, C.; Pierce, L.N.; Kruger, M.; Marcusson, E.G.; Robbins, J.M.; Welcsh, P.; Welch, P.J.; Welte, K.; King, M.C.; Barber, J.R.; et al. Identification of Id4 as a regulator of BRCA1 expression by using a ribozyme-library-based inverse genomics approach. Proc. Natl. Acad. Sci. USA 2001, 98, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Stingl, J. Mammary stem cells and the differentiation hierarchy: Current status and perspectives. Genes Dev. 2014, 28, 1143–1158. [Google Scholar] [CrossRef] [Green Version]
- Chapellier, M.; Bachelard-Cascales, E.; Schmidt, X.; Clement, F.; Treilleux, I.; Delay, E.; Jammot, A.; Menetrier-Caux, C.; Pochon, G.; Besancon, R.; et al. Disequilibrium of BMP2 levels in the breast stem cell niche launches epithelial transformation by overamplifying BMPR1B cell response. Stem Cell Rep. 2015, 4, 239–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeike, M.; Spach, S.; Huber, M.; Feng, S.; Vainorius, G.; Elling, U.; Versteeg, G.A.; Buecker, C. Transient upregulation of IRF1 during exit from naive pluripotency confers viral protection. EMBO Rep. 2022, 23, e55375. [Google Scholar] [CrossRef] [PubMed]
- Roukens, M.G.; Frederiks, C.L.; Seinstra, D.; Braccioli, L.; Khalil, A.A.; Pals, C.; De Neck, S.; Bornes, L.; Beerling, E.; Mokry, M.; et al. Regulation of a progenitor gene program by SOX4 is essential for mammary tumor proliferation. Oncogene 2021, 40, 6343–6353. [Google Scholar] [CrossRef] [PubMed]
- Meeson, A.P.; Shi, X.; Alexander, M.S.; Williams, R.S.; Allen, R.E.; Jiang, N.; Adham, I.M.; Goetsch, S.C.; Hammer, R.E.; Garry, D.J. Sox15 and Fhl3 transcriptionally coactivate Foxk1 and regulate myogenic progenitor cells. EMBO J. 2007, 26, 1902–1912. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.B.; Dent, R.; Im, S.A.; Espie, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.J.; et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017, 18, 1360–1372. [Google Scholar] [CrossRef]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): A multicentre, randomised, double-blind, phase 3 trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef]
- Labrie, M.; Brugge, J.S.; Mills, G.B.; Zervantonakis, I.K. Therapy resistance: Opportunities created by adaptive responses to targeted therapies in cancer. Nat. Rev. Cancer 2022, 22, 323–339. [Google Scholar] [CrossRef]
- Juvekar, A.; Hu, H.; Yadegarynia, S.; Lyssiotis, C.A.; Ullas, S.; Lien, E.C.; Bellinger, G.; Son, J.; Hok, R.C.; Seth, P.; et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc. Natl. Acad. Sci. USA 2016, 113, E4338–E4347. [Google Scholar] [CrossRef] [Green Version]
- Arribas, J.; Baselga, J.; Pedersen, K.; Parra-Palau, J.L. p95HER2 and breast cancer. Cancer Res. 2011, 71, 1515–1519. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Li, E.; Shen, H.; Wang, X.; Tang, T.; Zhang, X.; Xu, J.; Tang, Z.; Guo, C.; Bai, X.; et al. Targeting the HGF/MET Axis in Cancer Therapy: Challenges in Resistance and Opportunities for Improvement. Front. Cell Dev. Biol. 2020, 8, 152. [Google Scholar] [CrossRef]
- Park, S.H.; Jo, M.J.; Kim, B.R.; Jeong, Y.A.; Na, Y.J.; Kim, J.L.; Jeong, S.; Yun, H.K.; Kim, D.Y.; Kim, B.G.; et al. Sonic hedgehog pathway activation is associated with cetuximab resistance and EPHB3 receptor induction in colorectal cancer. Theranostics 2019, 9, 2235–2251. [Google Scholar] [CrossRef]
- Lee, S.Y.; Na, Y.J.; Jeong, Y.A.; Kim, J.L.; Oh, S.C.; Lee, D.H. Upregulation of EphB3 in gastric cancer with acquired resistance to a FGFR inhibitor. Int. J. Biochem. Cell Biol. 2018, 102, 128–137. [Google Scholar] [CrossRef]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehn, M.; Cho, R.W.; Clarke, M.F. Therapeutic implications of the cancer stem cell hypothesis. Semin. Radiat. Oncol. 2009, 19, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Dylla, S.J.; Beviglia, L.; Park, I.K.; Chartier, C.; Raval, J.; Ngan, L.; Pickell, K.; Aguilar, J.; Lazetic, S.; Smith-Berdan, S.; et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS ONE 2008, 3, e2428. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 2007, 25, 1315–1321. [Google Scholar] [CrossRef]
- Yan, Y.; Serra, V.; Prudkin, L.; Scaltriti, M.; Murli, S.; Rodriguez, O.; Guzman, M.; Sampath, D.; Nannini, M.; Xiao, Y.; et al. Evaluation and clinical analyses of downstream targets of the Akt inhibitor GDC-0068. Clin. Cancer Res. 2013, 19, 6976–6986. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.M.; Sohn, Y.W.; Oh, S.Y.; Kim, S.H.; Beck, S.; Kim, S.; Kim, H. ID4 imparts chemoresistance and cancer stemness to glioma cells by derepressing miR-9*-mediated suppression of SOX2. Cancer Res. 2011, 71, 3410–3421. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.R.; Toker, A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell. Signal. 2009, 21, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.R.; Yoshida, T.; Marusyk, A.; Beck, A.H.; Polyak, K.; Toker, A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014, 74, 964–973. [Google Scholar] [CrossRef] [Green Version]
- Savill, K.M.Z.; Lee, B.B.; Oeh, J.; Lin, J.; Lin, E.; Chung, W.J.; Young, A.; Chen, W.; Mis, M.; Mesh, K.; et al. Distinct resistance mechanisms arise to allosteric vs. ATP-competitive AKT inhibitors. Nat. Commun. 2022, 13, 2057. [Google Scholar] [CrossRef]
- Liu, J.; Duan, Z.; Guo, W.; Zeng, L.; Wu, Y.; Chen, Y.; Tai, F.; Wang, Y.; Lin, Y.; Zhang, Q.; et al. Targeting the BRD4/FOXO3a/CDK6 axis sensitizes AKT inhibition in luminal breast cancer. Nat. Commun. 2018, 9, 5200. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsang, T.; He, Q.; Cohen, E.B.; Stottrup, C.; Lien, E.C.; Zhang, H.; Lau, C.G.; Chin, Y.R. Upregulation of Receptor Tyrosine Kinase Activity and Stemness as Resistance Mechanisms to Akt Inhibitors in Breast Cancer. Cancers 2022, 14, 5006. https://doi.org/10.3390/cancers14205006
Tsang T, He Q, Cohen EB, Stottrup C, Lien EC, Zhang H, Lau CG, Chin YR. Upregulation of Receptor Tyrosine Kinase Activity and Stemness as Resistance Mechanisms to Akt Inhibitors in Breast Cancer. Cancers. 2022; 14(20):5006. https://doi.org/10.3390/cancers14205006
Chicago/Turabian StyleTsang, Tiffany, Qingling He, Emily B. Cohen, Casey Stottrup, Evan C. Lien, Huiqi Zhang, C. Geoffrey Lau, and Y. Rebecca Chin. 2022. "Upregulation of Receptor Tyrosine Kinase Activity and Stemness as Resistance Mechanisms to Akt Inhibitors in Breast Cancer" Cancers 14, no. 20: 5006. https://doi.org/10.3390/cancers14205006