
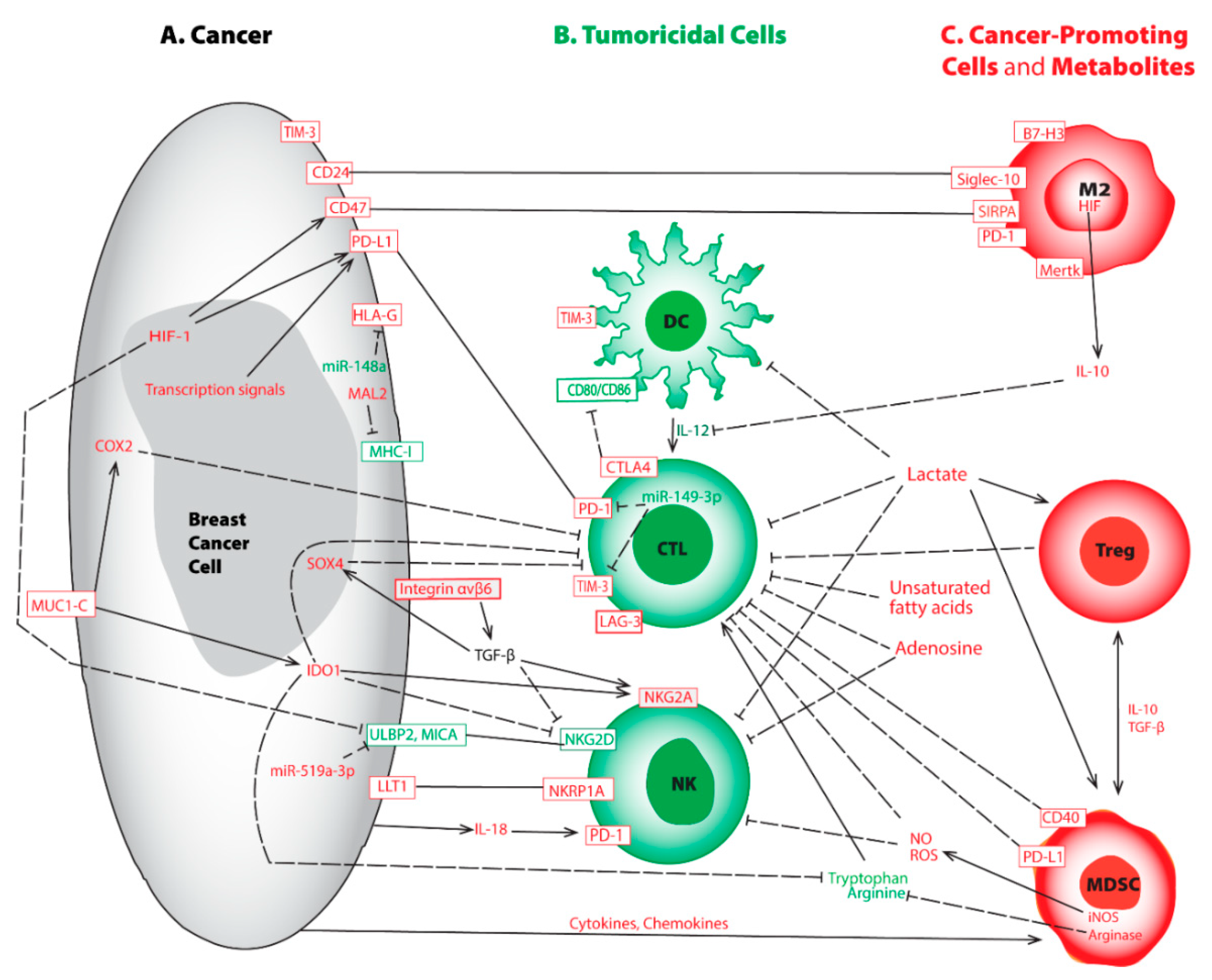
Breast Cancer Tumor Microenvironment and Molecular Aberrations Hijack Tumoricidal Immunity


Abstract
:Simple Summary
Abstract
1. Introduction
2. Initial Anti-Tumor Immunity Defeats Breast Neoplasm
3. Breast Cancer Reprograms Tumoricidal Immunity
3.1. Aberrant Presentation of Tumor-Associated Antigens (TAA)
3.2. Dysfunctional CD8+ Tumor-Infiltrating Lymphocytes (TIL)
3.3. Aberrant Immune Checkpoint Modulators
3.3.1. CTLA-4
3.3.2. PD-1 and PD-L1
3.3.3. LAG-3
3.3.4. TIM-3
3.4. Dichotomic Roles of Natural Killer (NK) Cells
3.5. Elevated Regulatory T Cells (Treg) Suppress Immune Response and Promote Tumor Growth
3.6. Polarizing Anti-Tumor Macrophage (M1) to Tumor-Promoting Macrophages (M2)
3.7. Myeloid-Derived Suppressor Cells (MDSC)
4. Tumor Stroma, Mediators, Chemical Components, and Physical Factors Constitute the Tumor Microenvironment (TME)
4.1. Dysregulated Cytokines and Chemokines
4.2. Altered Signaling Pathways
4.3. Aberrant Metabolism
4.4. Hypoxia Conveys Tumor Cell Plasticity
4.5. Extracellular Microvesicles (EVs) Modulate Immune Evasion
4.6. Radiation Influences Tumoricidal Immunity
5. Conclusions

{kind=link}
| Therapeutic Agent | Target | Rationale | Reference(s) |
| miR-148a mimic | HLA-G | Non-classical HLA promotes immune escape | [37] |
| avβ6 antibody | αvβ6-TGFβ-SOX4 pathway | SOX4 inhibits CTL | [45] |
| shRNA silences MUC1-C | MUC1-C | MUC1-C enhances IFN-γ signaling that inhibits CTL | [42,48] |
| αCTLA4 | CTLA-4 receptor | Immune checkpoint blockade | [55] |
| αPD-1 or αPD-L1 | PD-1/PDL-1 | Immune checkpoint blockade | [58] |
| PD-L1 acetyltransferase inhibitor | PD-L1 | Palmitoylation of PD-L1 stabilizes intracellular PD-L1 | [64] |
| STAT1 or STAT3 inhibitor | STAT1/STAT3 | The heterodimer activates transcription of PD-L1 | [70] |
| shRNA silences Syntenin1 | Syntenin1 | Syntenin1 upregulates PD-L1 | [71] |
| αLAG-3 | LAG-3 | Immune checkpoint blockade | [91] |
| αTIM-3 | TIM-3 | Immune checkpoint blockade | [99,100] |
| miR-149-3p mimic | TIM-3 | miR149-3p downregulates PD-1 and TIM-3 | [106] |
| αLLT1 | LLT1 | LLT1 is a tumor ligand to engage inhibitory receptors on NK cells | [116] |
| 519a-3p antagonist | Enhances ULBP2 & MICA expression | Reinstate recognition of BC by NK cells | [120] |
| αCD24, αCD47, αSIRPA, αSiglec-10 | CD24, CD47, SIRPA, Siglec-10 | Impede bindings between BC cells and M2 | [150] |
| αB7-H3 | B7-H3 | B7-H3 is an immune checkpoint modulator on tumor cells and TAMs | [155] |
| αMertk | Mertk | Mertk is an immunosuppressive receptor on TAMs | [156] |
| Syndecan 2 peptide | Syndecan-2 | Syndecan-2 secreted by the stroma is immunosuppressive | [182] |
| Galectin inhibitor | Galectin-3 | Galectin-3 produced by tumor cells inactivates multiple glycosylated cytokines and chemokines | [184] |
| MEK inhibitor | Ras/MAPK pathway | Overactivated MAPK pathway is immunosuppressive | [186] |
| GPR81 blockade | GPR81 | Lactate enhances expression of PD-L1 through the GPR81 receptor | [206] |
| A2a inhibitor | A2a | Adenosine inhibits CTL and NK through membrane receptors such as A2a and A2b | [215,216] |
| HIF-1 inhibitor | HIF | HIF has broad immunosuppressive activities | [222] |
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hammerl, D.; Smid, M.; Timmermans, A.M.; Sleijfer, S.; Martens, J.W.M.; Debets, R. Breast cancer genomics and immuno-oncological markers to guide immune therapies. Semin. Cancer Biol. 2018, 52, 178–188. [Google Scholar] [CrossRef]
- Prat, A.; Cheang, M.C.; Martin, M.; Parker, J.S.; Carrasco, E.; Caballero, R.; Tyldesley, S.; Gelmon, K.; Bernard, P.S.; Nielsen, T.O.; et al. Prognostic significance of progesterone receptor-positive tumor cells within immunohistochemically defined luminal A breast cancer. J. Clin. Oncol. 2013, 31, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, E.; Al-Gahmi, A.M.; Zeenelin, A.A.; Zekri, J.M.; Elkhodary, T.R.; Gaballa, H.E.; Fawzy, E.E.; El Sayed, M.E.; Alzahrani, M.S. Basal vs. luminal A breast cancer subtypes: A matched case-control study using estrogen receptor, progesterone receptor, and HER-2 as surrogate markers. Med. Oncol. 2009, 26, 372–378. [Google Scholar] [CrossRef]
- Mehta, R.J.; Jain, R.K.; Leung, S.; Choo, J.; Nielsen, T.; Huntsman, D.; Nakshatri, H.; Badve, S. FOXA1 is an independent prognostic marker for ER-positive breast cancer. Breast Cancer Res. Treat. 2012, 131, 881–890. [Google Scholar] [CrossRef]
- Prat, A.; Pineda, E.; Adamo, B.; Galvan, P.; Fernandez, A.; Gaba, L.; Diez, M.; Viladot, M.; Arance, A.; Munoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. 2), S26–S35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadi, V.K.; Davidson, N.E. Practical Approach to Triple-Negative Breast Cancer. J. Oncol. Pract. 2017, 13, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef]
- Fehres, C.M.; Unger, W.W.; Garcia-Vallejo, J.J.; van Kooyk, Y. Understanding the biology of antigen cross-presentation for the design of vaccines against cancer. Front. Immunol. 2014, 5, 149. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Bates, J.P.; Derakhshandeh, R.; Jones, L.; Webb, T.J. Mechanisms of immune evasion in breast cancer. BMC Cancer 2018, 18, 556. [Google Scholar] [CrossRef] [Green Version]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer immunosurveillance and immunoediting: The roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar] [PubMed]
- Spranger, S.; Sivan, A.; Corrales, L.; Gajewski, T.F. Tumor and Host Factors Controlling Antitumor Immunity and Efficacy of Cancer Immunotherapy. Adv. Immunol. 2016, 130, 75–93. [Google Scholar] [PubMed] [Green Version]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Forero, A.; Li, Y.; Chen, D.; Grizzle, W.E.; Updike, K.L.; Merz, N.D.; Downs-Kelly, E.; Burwell, T.C.; Vaklavas, C.; Buchsbaum, D.J.; et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells Is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol. Res. 2016, 4, 390–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, P.; Shin, E.C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.F.; Wang, H.Y. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res. 2017, 27, 11–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Marco, M.C.; Martin-Belmonte, F.; Kremer, L.; Albar, J.P.; Correas, I.; Vaerman, J.P.; Marazuela, M.; Byrne, J.A.; Alonso, M.A. MAL2, a novel raft protein of the MAL family, is an essential component of the machinery for transcytosis in hepatoma HepG2 cells. J. Cell Biol. 2002, 159, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wang, L.; Wan, C.; Sun, Y.; Van der Jeught, K.; Zhou, Z.; Dong, T.; So, K.M.; Yu, T.; Li, Y.; et al. MAL2 drives immune evasion in breast cancer by suppressing tumor antigen presentation. J. Clin. Investig. 2021, 131, e140837. [Google Scholar] [CrossRef]
- Vitale, M.; Rezzani, R.; Rodella, L.; Zauli, G.; Grigolato, P.; Cadei, M.; Hicklin, D.J.; Ferrone, S. HLA class I antigen and transporter associated with antigen processing (TAP1 and TAP2) down-regulation in high-grade primary breast carcinoma lesions. Cancer Res. 1998, 58, 737–742. [Google Scholar] [PubMed]
- Chen, H.L.; Gabrilovich, D.; Virmani, A.; Ratnani, I.; Girgis, K.R.; Nadaf-Rahrov, S.; Fernandez-Vina, M.; Carbone, D.P. Structural and functional analysis of beta2 microglobulin abnormalities in human lung and breast cancer. Int. J. Cancer 1996, 67, 756–763. [Google Scholar] [CrossRef]
- Nomura, T.; Huang, W.C.; Zhau, H.E.; Josson, S.; Mimata, H.; Chung, L.W. beta2-Microglobulin-mediated signaling as a target for cancer therapy. Anticancer Agents Med. Chem. 2014, 14, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, K.; Ishigami, S.; Kijima, Y.; Funasako, Y.; Hirata, M.; Okumura, H.; Shinchi, H.; Koriyama, C.; Ueno, S.; Yoshinaka, H.; et al. Clinical implication of HLA class I expression in breast cancer. BMC Cancer 2011, 11, 454. [Google Scholar] [CrossRef]
- da Silva, G.B.; Silva, T.G.; Duarte, R.A.; Neto, N.L.; Carrara, H.H.; Donadi, E.A.; Goncalves, M.A.; Soares, E.G.; Soares, C.P. Expression of the Classical and Nonclassical HLA Molecules in Breast Cancer. Int. J. Breast Cancer 2013, 2013, 250435. [Google Scholar] [CrossRef] [Green Version]
- Harada, A.; Ishigami, S.; Kijima, Y.; Nakajo, A.; Arigami, T.; Kurahara, H.; Kita, Y.; Yoshinaka, H.; Natsugoe, S. Clinical implication of human leukocyte antigen (HLA)-F expression in breast cancer. Pathol. Int. 2015, 65, 569–574. [Google Scholar] [CrossRef]
- Konig, L.; Kasimir-Bauer, S.; Hoffmann, O.; Bittner, A.K.; Wagner, B.; Manvailer, L.F.; Schramm, S.; Bankfalvi, A.; Giebel, B.; Kimmig, R.; et al. The prognostic impact of soluble and vesicular HLA-G and its relationship to circulating tumor cells in neoadjuvant treated breast cancer patients. Hum. Immunol. 2016, 77, 791–799. [Google Scholar] [CrossRef]
- De Kruijf, E.M.; Engels, C.C.; van de Water, W.; Bastiaannet, E.; Smit, V.T.; van de Velde, C.J.; Liefers, G.J.; Kuppen, P.J. Tumor immune subtypes distinguish tumor subclasses with clinical implications in breast cancer patients. Breast Cancer Res. Treat. 2013, 142, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Engels, C.C.; Fontein, D.B.; Kuppen, P.J.; de Kruijf, E.M.; Smit, V.T.; Nortier, J.W.; Liefers, G.J.; van de Velde, C.J.; Bastiaannet, E. Immunological subtypes in breast cancer are prognostic for invasive ductal but not for invasive lobular breast carcinoma. Br. J. Cancer 2014, 111, 532–538. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Yan, W.H. Human Leukocyte Antigen-G (HLA-G) Expression in Cancers: Roles in Immune Evasion, Metastasis and Target for Therapy. Mol. Med. 2015, 21, 782–791. [Google Scholar] [CrossRef]
- Tao, S.; He, H.; Chen, Q.; Yue, W. GPER mediated estradiol reduces miR-148a to promote HLA-G expression in breast cancer. Biochem. Biophys. Res. Commun. 2014, 451, 74–78. [Google Scholar] [CrossRef]
- Sheu, J.; Shih Ie, M. HLA-G and immune evasion in cancer cells. J. Formos. Med. Assoc. 2010, 109, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology 2017, 6, e1386829. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Chatterjee, A.; Jana, S.; Dey, S.; Roy, H.; Das, M.K.; Alam, J.; Adhikary, A.; Chowdhury, A.; Biswas, A.; et al. Transforming growth factor beta orchestrates PD-L1 enrichment in tumor-derived exosomes and mediates CD8 T-cell dysfunction regulating early phosphorylation of TCR signalome in breast cancer. Carcinogenesis 2021, 42, 38–47. [Google Scholar] [CrossRef]
- Yamashita, N.; Long, M.; Fushimi, A.; Yamamoto, M.; Hata, T.; Hagiwara, M.; Bhattacharya, A.; Hu, Q.; Wong, K.K.; Liu, S.; et al. MUC1-C integrates activation of the IFN-gamma pathway with suppression of the tumor immune microenvironment in triple-negative breast cancer. J. Immunother. Cancer 2021, 9, e002115. [Google Scholar] [CrossRef]
- Steven, A.; Seliger, B. The Role of Immune Escape and Immune Cell Infiltration in Breast Cancer. Breast Care 2018, 13, 16–21. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [Green Version]
- Bagati, A.; Kumar, S.; Jiang, P.; Pyrdol, J.; Zou, A.E.; Godicelj, A.; Mathewson, N.D.; Cartwright, A.N.R.; Cejas, P.; Brown, M.; et al. Integrin alphavbeta6-TGFbeta-SOX4 Pathway Drives Immune Evasion in Triple-Negative Breast Cancer. Cancer Cell 2021, 39, 54–67.e59. [Google Scholar] [CrossRef]
- Mojic, M.; Takeda, K.; Hayakawa, Y. The Dark Side of IFN-gamma: Its Role in Promoting Cancer Immunoevasion. Int. J. Mol. Sci. 2017, 19, 89. [Google Scholar] [CrossRef] [Green Version]
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [Green Version]
- Kufe, D.W. MUC1-C oncoprotein as a target in breast cancer: Activation of signaling pathways and therapeutic approaches. Oncogene 2013, 32, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kufe, D.W. MUC1-C in chronic inflammation and carcinogenesis; emergence as a target for cancer treatment. Carcinogenesis 2020, 41, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikman, B.; Vasyanovich, S.; Lavy, R.; Habler, L.; Tolstov, G.; Kapiev, A.; Halevy, A.; Sandbank, J. COX2 expression in high-grade breast cancer: Evidence for prognostic significance in the subset of triple-negative breast cancer patients. Med. Oncol. 2014, 31, 989. [Google Scholar] [CrossRef]
- Wu, A.A.; Drake, V.; Huang, H.S.; Chiu, S.; Zheng, L. Reprogramming the tumor microenvironment: Tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology 2015, 4, e1016700. [Google Scholar] [CrossRef]
- Swoboda, A.; Nanda, R. Immune Checkpoint Blockade for Breast Cancer. Cancer Treat. Res. 2018, 173, 155–165. [Google Scholar]
- Michel, L.L.; von Au, A.; Mavratzas, A.; Smetanay, K.; Schutz, F.; Schneeweiss, A. Immune Checkpoint Blockade in Patients with Triple-Negative Breast Cancer. Target Oncol. 2020, 15, 415–428. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Van Coillie, S.; Wiernicki, B.; Xu, J. Molecular and Cellular Functions of CTLA-4. Adv. Exp. Med. Biol. 2020, 1248, 7–32. [Google Scholar]
- Urbano, A.C.; Nascimento, C.; Soares, M.; Correia, J.; Ferreira, F. Clinical Relevance of the serum CTLA-4 in Cats with Mammary Carcinoma. Sci. Rep. 2020, 10, 3822. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Muenst, S.; Soysal, S.D.; Gao, F.; Obermann, E.C.; Oertli, D.; Gillanders, W.E. The presence of programmed death 1 (PD-1)-positive tumor-infiltrating lymphocytes is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2013, 139, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, M.T.; Lenkiewicz, E.; Malasi, S.; Basu, A.; Yearley, J.H.; Annamalai, L.; McCullough, A.E.; Kosiorek, H.E.; Narang, P.; Wilson Sayres, M.A.; et al. The association of genomic lesions and PD-1/PD-L1 expression in resected triple-negative breast cancers. Breast Cancer Res. 2018, 20, 71. [Google Scholar] [CrossRef] [Green Version]
- Muenst, S.; Schaerli, A.R.; Gao, F.; Daster, S.; Trella, E.; Droeser, R.A.; Muraro, M.G.; Zajac, P.; Zanetti, R.; Gillanders, W.E.; et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MuenstMaeda, T.; Hiraki, M.; Jin, C.; Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Hu, X.; Suzuki, Y.; Miyo, M.; et al. MUC1-C Induces PD-L1 and Immune Evasion in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 205–215. [Google Scholar]
- Yao, H.; Lan, J.; Li, C.; Shi, H.; Brosseau, J.P.; Wang, H.; Lu, H.; Fang, C.; Zhang, Y.; Liang, L.; et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat. Biomed. Eng. 2019, 3, 306–317. [Google Scholar] [CrossRef]
- Nouri, K.; Azad, T.; Lightbody, E.; Khanal, P.; Nicol, C.J.; Yang, X. A kinome-wide screen using a NanoLuc LATS luminescent biosensor identifies ALK as a novel regulator of the Hippo pathway in tumorigenesis and immune evasion. FASEB J. 2019, 33, 12487–12499. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Sasidharan Nair, V.; Elkord, E. PD-L1 Expression in Human Breast Cancer Stem Cells Is Epigenetically Regulated through Posttranslational Histone Modifications. J. Oncol. 2019, 2019, 3958908. [Google Scholar] [CrossRef] [Green Version]
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.V.; Milani, M.; Volpari, T.; Chiodoni, C.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Bengsch, F.; Svoronos, N.; Rutkowski, M.R.; Bitler, B.G.; Allegrezza, M.J.; Yokoyama, Y.; Kossenkov, A.V.; Bradner, J.E.; Conejo-Garcia, J.R.; et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep. 2016, 16, 2829–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, X.; Shao, S.; Zhang, Y.; Luo, A.; Zhao, L.; Zhang, L.; Gu, S.; Zhao, X. BRD4 inhibition suppresses PD-L1 expression in triple-negative breast cancer. Exp. Cell Res. 2020, 392, 112034. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Toor, S.M.; Ali, B.R.; Elkord, E. Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD-L1 in human breast cancer cells. Expert Opin. Ther. Targets 2018, 22, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, Y.; Wang, H.; Wang, B.; Zhao, K.; Jiang, W.; Bai, W.; Liu, J.; Yin, J. Syntenin1/MDA-9 (SDCBP) induces immune evasion in triple-negative breast cancer by upregulating PD-L1. Breast Cancer Res. Treat. 2018, 171, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Davra, V.; Obr, A.E.; Geng, K.; Wood, T.L.; De Lorenzo, M.S.; Birge, R.B. Crk adaptor protein promotes PD-L1 expression, EMT and immune evasion in a murine model of triple-negative breast cancer. Oncoimmunology 2017, 7, e1376155. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.G.; O’Neill, S.; Salimu, J.; Breslin, S.; Clayton, A.; Crown, J.; O’Driscoll, L. Resistance to HER2-targeted anti-cancer drugs is associated with immune evasion in cancer cells and their derived extracellular vesicles. Oncoimmunology 2017, 6, e1362530. [Google Scholar] [CrossRef]
- Timaner, M.; Kotsofruk, R.; Raviv, Z.; Magidey, K.; Shechter, D.; Kan, T.; Nevelsky, A.; Daniel, S.; de Vries, E.G.E.; Zhang, T.; et al. Microparticles from tumors exposed to radiation promote immune evasion in part by PD-L1. Oncogene 2020, 39, 187–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zam, W.; Ali, L. Immune checkpoint inhibitors in the treatment of cancer. Curr. Clin. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, B. Immunotherapy: New insights in breast cancer treatment. Hum. Antib. 2021, 29, 193–202. [Google Scholar] [CrossRef]
- Kwapisz, D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 2021, 70, 607–617. [Google Scholar] [CrossRef]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olino, K.; Park, T.; Ahuja, N. Exposing Hidden Targets: Combining epigenetic and immunotherapy to overcome cancer resistance. Semin. Cancer Biol. 2020, 65, 114–122. [Google Scholar] [CrossRef]
- Force, J.; Leal, J.H.S.; McArthur, H.L. Checkpoint Blockade Strategies in the Treatment of Breast Cancer: Where We Are and Where We Are Heading. Curr. Treat. Options Oncol. 2019, 20, 35. [Google Scholar] [CrossRef]
- Kang, J.; Demaria, S.; Formenti, S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J. Immunother. Cancer 2016, 4, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieper, A.A.; Rakhmilevich, A.L.; Spiegelman, D.V.; Patel, R.B.; Birstler, J.; Jin, W.J.; Carlson, P.M.; Charych, D.H.; Hank, J.A.; Erbe, A.K.; et al. Combination of radiation therapy, bempegaldesleukin, and checkpoint blockade eradicates advanced solid tumors and metastases in mice. J. Immunother. Cancer 2021, 9, e002715. [Google Scholar] [CrossRef]
- Theelen, W.; Chen, D.; Verma, V.; Hobbs, B.P.; Peulen, H.M.U.; Aerts, J.; Bahce, I.; Niemeijer, A.L.N.; Chang, J.Y.; de Groot, P.M.; et al. Pembrolizumab with or without radiotherapy for metastatic non-small-cell lung cancer: A pooled analysis of two randomised trials. Lancet Respir. Med. 2021, 9, 467–475. [Google Scholar] [CrossRef]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baixeras, E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 1992, 176, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Schnorfeil, F.M.; Lichtenegger, F.S.; Emmerig, K.; Schlueter, M.; Neitz, J.S.; Draenert, R.; Hiddemann, W.; Subklewe, M. T cells are functionally not impaired in AML: Increased PD-1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment. J. Hematol. Oncol. 2015, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Lichtenegger, F.S.; Rothe, M.; Schnorfeil, F.M.; Deiser, K.; Krupka, C.; Augsberger, C.; Schluter, M.; Neitz, J.; Subklewe, M. Targeting LAG-3 and PD-1 to Enhance T Cell Activation by Antigen-Presenting Cells. Front. Immunol. 2018, 9, 385. [Google Scholar] [CrossRef] [Green Version]
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Yasinska, I.M.; Sakhnevych, S.S.; Pavlova, L.; Teo Hansen Selno, A.; Teuscher Abeleira, A.M.; Benlaouer, O.; Goncalves Silva, I.; Mosimann, M.; Varani, L.; Bardelli, M.; et al. The Tim-3-Galectin-9 Pathway and Its Regulatory Mechanisms in Human Breast Cancer. Front. Immunol. 2019, 10, 1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Sabatos-Peyton, C.A.; Nevin, J.; Brock, A.; Venable, J.D.; Tan, D.J.; Kassam, N.; Xu, F.; Taraszka, J.; Wesemann, L.; Pertel, T.; et al. Blockade of Tim-3 binding to phosphatidylserine and CEACAM1 is a shared feature of anti-Tim-3 antibodies that have functional efficacy. Oncoimmunology 2018, 7, e1385690. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Chen, F.; Liu, D.; Gu, F.; Chen, Z.; Wang, Y. Prognostic value of immune checkpoint molecules in breast cancer. Biosci. Rep. 2020, 40, BSR20201054. [Google Scholar] [CrossRef]
- de Mingo Pulido, A.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74 e66. [Google Scholar] [CrossRef] [Green Version]
- de Mingo Pulido, A.; Hanggi, K.; Celias, D.P.; Gardner, A.; Li, J.; Batista-Bittencourt, B.; Mohamed, E.; Trillo-Tinoco, J.; Osunmakinde, O.; Pena, R.; et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 2021, 54, 1154–1167 e1157. [Google Scholar] [CrossRef]
- Gardner, A.; de Mingo Pulido, A.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef]
- Cong, Y.; Cui, Y.; Zhu, S.; Cao, J.; Zou, H.; Martin, T.A.; Qiao, G.; Jiang, W.; Yu, Z. Tim-3 promotes cell aggressiveness and paclitaxel resistance through NF-kappaB/STAT3 signalling pathway in breast cancer cells. Chin. J. Cancer Res. 2020, 32, 564–579. [Google Scholar] [CrossRef]
- Liu, Q.; Yu, S.; Li, A.; Xu, H.; Han, X.; Wu, K. Targeting interlukin-6 to relieve immunosuppression in tumor microenvironment. Tumour. Biol. 2017, 39, 1010428317712445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef]
- Cheng, S.; Han, F.; Xu, Y.; Qu, T.; Ju, Y. Expression of Tim-3 in breast cancer tissue promotes tumor progression. Int. J. Clin. Exp. Pathol. 2018, 11, 1157–1166. [Google Scholar]
- Zhang, M.; Gao, D.; Shi, Y.; Wang, Y.; Joshi, R.; Yu, Q.; Liu, D.; Alotaibi, F.; Zhang, Y.; Wang, H.; et al. miR-149-3p reverses CD8(+) T-cell exhaustion by reducing inhibitory receptors and promoting cytokine secretion in breast cancer cells. Open Biol. 2019, 9, 190061. [Google Scholar] [CrossRef] [Green Version]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef] [PubMed]
- Ngiow, S.F.; von Scheidt, B.; Akiba, H.; Yagita, H.; Teng, M.W.; Smyth, M.J. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011, 71, 3540–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.; Schaefer, W.; Regula, J.T.; Dumontet, C.; Brinkmann, U.; Bacac, M.; Umana, P. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods 2019, 154, 21–31. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Saleh, R.; Toor, S.M.; Khalaf, S.; Elkord, E. Breast Cancer Cells and PD-1/PD-L1 Blockade Upregulate the Expression of PD-1, CTLA-4, TIM-3 and LAG-3 Immune Checkpoints in CD4(+) T Cells. Vaccines 2019, 7, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, R.; Elkord, E. Treg-mediated acquired resistance to immune checkpoint inhibitors. Cancer Lett. 2019, 457, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Soto, A.; Huergo-Zapico, L.; Acebes-Huerta, A.; Villa-Alvarez, M.; Gonzalez, S. NKG2D signaling in cancer immunosurveillance. Int. J. Cancer 2015, 136, 1741–1750. [Google Scholar] [CrossRef]
- Schmiedel, D.; Mandelboim, O. NKG2D Ligands-Critical Targets for Cancer Immune Escape and Therapy. Front. Immunol. 2018, 9, 2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.B.; Bettadapura, J.; Alsharifi, M.; Mathew, P.A.; Warren, H.S.; Lanier, L.L. Cutting edge: Lectin-like transcript-1 is a ligand for the inhibitory human NKR-P1A receptor. J. Immunol. 2005, 175, 7796–7799. [Google Scholar] [CrossRef] [PubMed]
- Marrufo, A.M.; Mathew, S.O.; Chaudhary, P.; Malaer, J.D.; Vishwanatha, J.K.; Mathew, P.A. Blocking LLT1 (CLEC2D, OCIL)-NKRP1A (CD161) interaction enhances natural killer cell-mediated lysis of triple-negative breast cancer cells. Am. J. Cancer Res. 2018, 8, 1050–1063. [Google Scholar]
- Mamessier, E.; Sylvain, A.; Thibult, M.L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Goncalves, A.; Andre, P.; Romagne, F.; Thibault, G.; et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Investig. 2011, 121, 3609–3622. [Google Scholar] [CrossRef] [Green Version]
- Berrien-Elliott, M.M.; Romee, R.; Fehniger, T.A. Improving natural killer cell cancer immunotherapy. Curr. Opin. Organ. Transplant. 2015, 20, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Park, I.H.; Yang, H.N.; Lee, K.J.; Kim, T.S.; Lee, E.S.; Jung, S.Y.; Kwon, Y.; Kong, S.Y. Tumor-derived IL-18 induces PD-1 expression on immunosuppressive NK cells in triple-negative breast cancer. Oncotarget 2017, 8, 32722–32730. [Google Scholar] [CrossRef] [Green Version]
- Breunig, C.; Pahl, J.; Kublbeck, M.; Miller, M.; Antonelli, D.; Erdem, N.; Wirth, C.; Will, R.; Bott, A.; Cerwenka, A.; et al. MicroRNA-519a-3p mediates apoptosis resistance in breast cancer cells and their escape from recognition by natural killer cells. Cell Death Dis. 2017, 8, e2973. [Google Scholar] [CrossRef]
- Fang, D.; Zhu, J. Dynamic balance between master transcription factors determines the fates and functions of CD4 T cell and innate lymphoid cell subsets. J. Exp. Med. 2017, 214, 1861–1876. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Dong, P.; Ren, M.; Song, Y.; Qian, X.; Yang, Y.; Li, S.; Zhang, X.; Liu, F. PD-L1 Expression Is Associated with Tumor FOXP3(+) Regulatory T-Cell Infiltration of Breast Cancer and Poor Prognosis of Patient. J. Cancer 2016, 7, 784–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, Y.; Nishikawa, H. Roles of regulatory T cells in cancer immunity. Int. Immunol. 2016, 28, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. 2006, 24, 5373–5380. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Zhang, Z.; Lai, Y.; Chen, Z.; Huang, J. Worse outcome in breast cancer with higher tumor-infiltrating FOXP3+ Tregs: A systematic review and meta-analysis. BMC Cancer 2016, 16, 687. [Google Scholar] [CrossRef] [Green Version]
- Chuang, L.S.; Ito, Y. RUNX3 is multifunctional in carcinogenesis of multiple solid tumors. Oncogene 2010, 29, 2605–2615. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Panda, A.K.; Bose, S.; Roy, D.; Kajal, K.; Guha, D.; Sa, G. Transcriptional regulation of FOXP3 requires integrated activation of both promoter and CNS regions in tumor-induced CD8(+) Treg cells. Sci. Rep. 2017, 7, 1628. [Google Scholar] [CrossRef] [Green Version]
- Manandhar, S.; Lee, Y.M. Emerging role of RUNX3 in the regulation of tumor microenvironment. BMB. Rep. 2018, 51, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohling, S.D.; Allison, K.H. Immunosuppressive regulatory T cells are associated with aggressive breast cancer phenotypes: A potential therapeutic target. Mod. Pathol. 2008, 21, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Ohara, M.; Yamaguchi, Y.; Matsuura, K.; Murakami, S.; Arihiro, K.; Okada, M. Possible involvement of regulatory T cells in tumor onset and progression in primary breast cancer. Cancer Immunol. Immunother. 2009, 58, 441–447. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, M.; Sasano, H.; Tamaki, K.; Chan, M.; Hirakawa, H.; Suzuki, A.; Tada, H.; Watanabe, G.; Nemoto, N.; Nakagawa, S.; et al. Tumor-infiltrating CD8+ and FOXP3+ lymphocytes in triple-negative breast cancer: Its correlation with pathological complete response to neoadjuvant chemotherapy. Breast Cancer Res. Treat. 2014, 148, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Kashiwagi, S.; Goto, W.; Kurata, K.; Noda, S.; Takashima, T.; Onoda, N.; Tanaka, S.; Ohsawa, M.; Hirakawa, K. Tumour-infiltrating CD8 to FOXP3 lymphocyte ratio in predicting treatment responses to neoadjuvant chemotherapy of aggressive breast cancer. Br. J. Surg. 2016, 103, 845–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladoire, S.; Arnould, L.; Apetoh, L.; Coudert, B.; Martin, F.; Chauffert, B.; Fumoleau, P.; Ghiringhelli, F. Pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of tumor-infiltrating foxp3+ regulatory T cells. Clin. Cancer Res. 2008, 14, 2413–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Ostling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [Green Version]
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, N.B.; Lu, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Ham, S.; Lima, L.G.; Chai, E.P.Z.; Muller, A.; Lobb, R.J.; Krumeich, S.; Wen, S.W.; Wiegmans, A.P.; Moller, A. Breast Cancer-Derived Exosomes Alter Macrophage Polarization via gp130/STAT3 Signaling. Front. Immunol. 2018, 9, 871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leek, R.D.; Talks, K.L.; Pezzella, F.; Turley, H.; Campo, L.; Brown, N.S.; Bicknell, R.; Taylor, M.; Gatter, K.C.; Harris, A.L. Relation of hypoxia-inducible factor-2 alpha (HIF-2 alpha) expression in tumor-infiltrative macrophages to tumor angiogenesis and the oxidative thymidine phosphorylase pathway in Human breast cancer. Cancer Res. 2002, 62, 1326–1329. [Google Scholar]
- Medrek, C.; Ponten, F.; Jirstrom, K.; Leandersson, K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 2012, 12, 306. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Yeh, E.S.; Soloff, A.C. Tumor-associated macrophages: Unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2016, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Gok Yavuz, B.; Gunaydin, G.; Gedik, M.E.; Kosemehmetoglu, K.; Karakoc, D.; Ozgur, F.; Guc, D. Cancer associated fibroblasts sculpt tumour microenvironment by recruiting monocytes and inducing immunosuppressive PD-1(+) TAMs. Sci. Rep. 2019, 9, 3172. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- McCracken, M.N.; Cha, A.C.; Weissman, I.L. Molecular Pathways: Activating T Cells after Cancer Cell Phagocytosis from Blockade of CD47 “Don’t Eat Me” Signals. Clin. Cancer Res. 2015, 21, 3597–3601. [Google Scholar] [CrossRef] [Green Version]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yan, L.; Kim, J.A. Modulating mammary tumor growth, metastasis and immunosuppression by siRNA-induced MIF reduction in tumor microenvironment. Cancer Gene Ther. 2015, 22, 463–474. [Google Scholar] [CrossRef]
- Charan, M.; Das, S.; Mishra, S.; Chatterjee, N.; Varikuti, S.; Kaul, K.; Misri, S.; Ahirwar, D.K.; Satoskar, A.R.; Ganju, R.K. Macrophage migration inhibitory factor inhibition as a novel therapeutic approach against triple-negative breast cancer. Cell Death Dis. 2020, 11, 774. [Google Scholar] [CrossRef] [PubMed]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, N.; Bei, Y.; Song, Y.; Zhang, W.; Xu, L.; Zhang, W.; Yang, N.; Bai, X.; Shu, Y.; Shen, P. B7-H3 augments the pro-angiogenic function of tumor-associated macrophages and acts as a novel adjuvant target for triple-negative breast cancer therapy. Biochem. Pharmacol. 2021, 183, 114298. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Davra, V.; Kumar, S.; Geng, K.; Calianese, D.; Mehta, D.; Gadiyar, V.; Kasikara, C.; Lahey, K.C.; Chang, Y.J.; Wichroski, M.; et al. Axl and Mertk Receptors Cooperate to Promote Breast Cancer Progression by Combined Oncogenic Signaling and Evasion of Host Antitumor Immunity. Cancer Res. 2021, 81, 698–712. [Google Scholar] [CrossRef]
- De Cicco, P.; Ercolano, G.; Ianaro, A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front. Immunol. 2020, 11, 1680. [Google Scholar] [CrossRef] [PubMed]
- Bergenfelz, C.; Larsson, A.M.; von Stedingk, K.; Gruvberger-Saal, S.; Aaltonen, K.; Jansson, S.; Jernstrom, H.; Janols, H.; Wullt, M.; Bredberg, A.; et al. Systemic Monocytic-MDSCs Are Generated from Monocytes and Correlate with Disease Progression in Breast Cancer Patients. PLoS ONE 2015, 10, e0127028. [Google Scholar] [CrossRef] [Green Version]
- Morales, J.K.; Kmieciak, M.; Knutson, K.L.; Bear, H.D.; Manjili, M.H. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat. 2010, 123, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Shou, D.; Wen, L.; Song, Z.; Yin, J.; Sun, Q.; Gong, W. Suppressive role of myeloid-derived suppressor cells (MDSCs) in the microenvironment of breast cancer and targeted immunotherapies. Oncotarget 2016, 7, 64505–64511. [Google Scholar] [CrossRef] [Green Version]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Gantt, S.; Gervassi, A.; Jaspan, H.; Horton, H. The role of myeloid-derived suppressor cells in immune ontogeny. Front. Immunol. 2014, 5, 387. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Lv, Z.; Fu, Q.; Jiang, C.; Liu, Y.; Lai, L.; Chen, Q.; Shen, J.; Wang, Q. IFN-gamma producing T cells contribute to the increase of myeloid derived suppressor cells in tumor-bearing mice after cyclophosphamide treatment. Int. Immunopharmacol. 2012, 12, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jochems, C.; Talaie, T.; Anderson, A.; Jales, A.; Tsang, K.Y.; Madan, R.A.; Gulley, J.L.; Schlom, J. Elevated serum soluble CD40 ligand in cancer patients may play an immunosuppressive role. Blood 2012, 120, 3030–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Ma, H.S.; Poudel, B.; Torres, E.R.; Sidhom, J.W.; Robinson, T.M.; Christmas, B.; Scott, B.; Cruz, K.; Woolman, S.; Wall, V.Z.; et al. A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell-Mediated Anticancer Activity. Cancer Immunol. Res. 2019, 7, 428–442. [Google Scholar] [CrossRef]
- Song, X.; Krelin, Y.; Dvorkin, T.; Bjorkdahl, O.; Segal, S.; Dinarello, C.A.; Voronov, E.; Apte, R.N. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J. Immunol. 2005, 175, 8200–8208. [Google Scholar] [CrossRef] [Green Version]
- Bunt, S.K.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol. 2006, 176, 284–290. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.; Billet, S.; Liu, C.; Haldar, S.; Choudhury, D.; Tripathi, M.; Hav, M.; Merchant, A.; Hu, T.; Huang, H.; et al. Periodontal inflammation recruits distant metastatic breast cancer cells by increasing myeloid-derived suppressor cells. Oncogene 2020, 39, 1543–1556. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.D.; Lu, C.; Payne, D.; Paschall, A.V.; Klement, J.D.; Redd, P.S.; Ibrahim, M.L.; Yang, D.; Han, Q.; Liu, Z.; et al. Autocrine IL6-Mediated Activation of the STAT3-DNMT Axis Silences the TNFalpha-RIP1 Necroptosis Pathway to Sustain Survival and Accumulation of Myeloid-Derived Suppressor Cells. Cancer Res. 2020, 80, 3145–3156. [Google Scholar] [CrossRef]
- Weber, R.; Groth, C.; Lasser, S.; Arkhypov, I.; Petrova, V.; Altevogt, P.; Utikal, J.; Umansky, V. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell Immunol. 2021, 359, 104254. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H. Blocking IDO activity to enhance anti-tumor immunity. Front. Biosci. 2012, 4, 734–745. [Google Scholar] [CrossRef]
- Shojaei, F.; Wu, X.; Qu, X.; Kowanetz, M.; Yu, L.; Tan, M.; Meng, Y.G.; Ferrara, N. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc. Natl. Acad. Sci. USA 2009, 106, 6742–6747. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103 e106. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Wilkes, D.W.; Samuel, N.; Blanco, M.A.; Nayak, A.; Alicea-Torres, K.; Gluck, C.; Sinha, S.; Gabrilovich, D.; Chakrabarti, R. DeltaNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J. Clin. Investig. 2018, 128, 5095–5109. [Google Scholar] [CrossRef] [Green Version]
- Chouaib, S.; Janji, B.; Tittarelli, A.; Eggermont, A.; Thiery, J.P. Tumor plasticity interferes with anti-tumor immunity. Crit. Rev. Immunol. 2014, 34, 91–102. [Google Scholar] [CrossRef]
- You, F.P.; Zhang, J.; Cui, T.; Zhu, R.; Lv, C.Q.; Tang, H.T.; Sun, D.W. Th9 cells promote antitumor immunity via IL-9 and IL-21 and demonstrate atypical cytokine expression in breast cancer. Int. Immunopharmacol. 2017, 52, 163–167. [Google Scholar] [CrossRef]
- Wu, S.Z.; Roden, D.L.; Wang, C.; Holliday, H.; Harvey, K.; Cazet, A.S.; Murphy, K.J.; Pereira, B.; Al-Eryani, G.; Bartonicek, N.; et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. 2020, 39, e104063. [Google Scholar] [CrossRef]
- Loftus, P.G.; Watson, L.; Deedigan, L.M.; Camarillo-Retamosa, E.; Dwyer, R.M.; O’Flynn, L.; Alagesan, S.; Griffin, M.; O’Brien, T.; Kerin, M.J.; et al. Targeting stromal cell Syndecan-2 reduces breast tumour growth, metastasis and limits immune evasion. Int. J. Cancer 2021, 148, 1245–1259. [Google Scholar] [CrossRef]
- Gal, P.; Varinska, L.; Faber, L.; Novak, S.; Szabo, P.; Mitrengova, P.; Mirossay, A.; Mucaji, P.; Smetana, K. How Signaling Molecules Regulate Tumor Microenvironment: Parallels to Wound Repair. Molecules 2017, 22, 1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon-Alonso, M.; Hirsch, T.; Wildmann, C.; van der Bruggen, P. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat. Commun. 2017, 8, 793. [Google Scholar] [CrossRef]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, D.A.; James, J.L.; Axelrod, M.L.; Balko, J.M. MEK inhibition activates STAT signaling to increase breast cancer immunogenicity via MHC-I expression. Cancer Drug. Resist. 2020, 3, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Campesato, L.F.; Silva, A.P.M.; Cordeiro, L.; Correa, B.R.; Navarro, F.C.P.; Zanin, R.F.; Marcola, M.; Inoue, L.T.; Duarte, M.L.; Molgora, M.; et al. High IL-1R8 expression in breast tumors promotes tumor growth and contributes to impaired antitumor immunity. Oncotarget 2017, 8, 49470–49483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [Green Version]
- Fuertes, M.B.; Woo, S.R.; Burnett, B.; Fu, Y.X.; Gajewski, T.F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013, 34, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Garlanda, C.; Anders, H.J.; Mantovani, A. TIR8/SIGIRR: An IL-1R/TLR family member with regulatory functions in inflammation and T cell polarization. Trends Immunol. 2009, 30, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Wald, D.; Qin, J.; Zhao, Z.; Qian, Y.; Naramura, M.; Tian, L.; Towne, J.; Sims, J.E.; Stark, G.R.; Li, X. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 2003, 4, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Qian, Y.; Yao, J.; Grace, C.; Li, X. SIGIRR inhibits interleukin-1 receptor- and toll-like receptor 4-mediated signaling through different mechanisms. J. Biol. Chem. 2005, 280, 25233–25241. [Google Scholar] [CrossRef]
- Wegiel, B.; Vuerich, M.; Daneshmandi, S.; Seth, P. Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anti-cancer Therapy. Front. Oncol. 2018, 8, 284. [Google Scholar] [CrossRef] [Green Version]
- Hayes, C.; Donohoe, C.L.; Davern, M.; Donlon, N.E. The oncogenic and clinical implications of lactate induced immunosuppression in the tumour microenvironment. Cancer Lett. 2021, 500, 75–86. [Google Scholar] [CrossRef]
- Singer, K.; Kastenberger, M.; Gottfried, E.; Hammerschmied, C.G.; Buttner, M.; Aigner, M.; Seliger, B.; Walter, B.; Schlosser, H.; Hartmann, A.; et al. Warburg phenotype in renal cell carcinoma: High expression of glucose-transporter 1 (GLUT-1) correlates with low CD8(+) T-cell infiltration in the tumor. Int. J. Cancer 2011, 128, 2085–2095. [Google Scholar] [CrossRef]
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2017, 8, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillies, R.J.; Gatenby, R.A. Metabolism and its sequelae in cancer evolution and therapy. Cancer J. 2015, 21, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Yuan, C.; Zhang, J.; Lou, J.; Wang, S.; Jiang, Y.; Wu, F.; Wang, S. Comprehensive Analysis of Monocarboxylate Transporter 4 (MCT4) expression in breast cancer prognosis and immune infiltration via integrated bioinformatics analysis. Bioengineered 2021, 12, 3850–3863. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Chen, S.; Chen, X.; Wu, T.; Yang, Y.; Li, S.; Ma, J.; Zhao, J.; Lin, W.; Li, W.; et al. M2TAM subsets altered by lactic acid promote Tcell apoptosis through the PDL1/PD1 pathway. Oncol. Rep. 2020, 44, 1885–1894. [Google Scholar] [PubMed]
- Zhou, H.C.; Xin-Yan, Y.; Yu, W.W.; Liang, X.Q.; Du, X.Y.; Liu, Z.C.; Long, J.P.; Zhao, G.H.; Liu, H.B. Lactic acid in macrophage polarization: The significant role in inflammation and cancer. Int. Rev. Immunol. 2021, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.P.; Bhattacharjee, P.; Ramachandran, S.; Sivaprakasam, S.; Ristic, B.; Sikder, M.O.F.; Ganapathy, V. The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene 2020, 39, 3292–3304. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, D.; Bhattacharya, R.; Sinha, B.P.; Liu, C.S.C.; Ghosh, A.R.; Rahaman, O.; Bandopadhyay, P.; Sarif, J.; D’Rozario, R.; Paul, S.; et al. Lactate Induces Pro-tumor Reprogramming in Intratumoral Plasmacytoid Dendritic Cells. Front. Immunol. 2019, 10, 1878. [Google Scholar] [CrossRef] [Green Version]
- Kleinfeld, A.M.; Okada, C. Free fatty acid release from human breast cancer tissue inhibits cytotoxic T-lymphocyte-mediated killing. J. Lipid Res. 2005, 46, 1983–1990. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased level of extracellular ATP at tumor sites: In vivo imaging with plasma membrane luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourdin, N.; Bossennec, M.; Rodriguez, C.; Vigano, S.; Machon, C.; Jandus, C.; Bauche, D.; Faget, J.; Durand, I.; Chopin, N.; et al. Autocrine Adenosine Regulates Tumor Polyfunctional CD73(+)CD4(+) Effector T Cells Devoid of Immune Checkpoints. Cancer Res. 2018, 78, 3604–3618. [Google Scholar] [CrossRef] [Green Version]
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.W.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011, 71, 2892–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [Green Version]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, D.; Young, A.; Stannard, K.; Yong, M.; Teng, M.W.; Allard, B.; Stagg, J.; Smyth, M.J. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res. 2014, 74, 3652–3658. [Google Scholar] [CrossRef] [Green Version]
- Ohta, A. A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol. 2016, 7, 109. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Hockel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal. 2007, 9, 1221–1235. [Google Scholar] [CrossRef]
- Terry, S.; Faouzi Zaarour, R.; Hassan Venkatesh, G.; Francis, A.; El-Sayed, W.; Buart, S.; Bravo, P.; Thiery, J.; Chouaib, S. Role of Hypoxic Stress in Regulating Tumor Immunogenicity, Resistance and Plasticity. Int. J. Mol. Sci. 2018, 19, 3044. [Google Scholar] [CrossRef] [Green Version]
- Barsoum, I.B.; Hamilton, T.K.; Li, X.; Cotechini, T.; Miles, E.A.; Siemens, D.R.; Graham, C.H. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: Role of nitric oxide. Cancer Res. 2011, 71, 7433–7441. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47(+)/CD73(+)/PDL1(+) immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Belikoff, B.; Schreiber, T.H.; Sethumadhavan, S.; Abbott, R.; Philbrook, P.; Thayer, M.; Shujia, D.; et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1alpha-dependent and extracellular adenosine-mediated tumor protection. J. Mol. Med. 2014, 92, 1283–1292. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Jene, N.; Byrne, D.; Millar, E.K.; O’Toole, S.A.; McNeil, C.M.; Bates, G.J.; Harris, A.L.; Banham, A.H.; Sutherland, R.L.; et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Res. 2011, 13, R47. [Google Scholar] [CrossRef] [Green Version]
- Voloshin, T.; Fremder, E.; Shaked, Y. Small but mighty: Microparticles as mediators of tumor progression. Cancer Microenviron. 2014, 7, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Paolillo, M.; Comincini, S.; Schinelli, S. Fostering “Education”: Do Extracellular Vesicles Exploit Their Own Delivery Code? Cells 2021, 10, 1741. [Google Scholar] [CrossRef]
- Giordano, C.; La Camera, G.; Gelsomino, L.; Barone, I.; Bonofiglio, D.; Ando, S.; Catalano, S. The Biology of Exosomes in Breast Cancer Progression: Dissemination, Immune Evasion and Metastatic Colonization. Cancers 2020, 12, 2179. [Google Scholar] [CrossRef]
- Rong, L.; Li, R.; Li, S.; Luo, R. Immunosuppression of breast cancer cells mediated by transforming growth factor-beta in exosomes from cancer cells. Oncol. Lett. 2016, 11, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Liu, Y.; Wu, S.Y.; Wu, K.; Sharma, S.; Mo, Y.Y.; Feng, J.; Sanders, S.; Jin, G.; Singh, R.; et al. Loss of XIST in Breast Cancer Activates MSN-c-Met and Reprograms Microglia via Exosomal miRNA to Promote Brain Metastasis. Cancer Res. 2018, 78, 4316–4330. [Google Scholar] [CrossRef] [Green Version]
- Cereghetti, D.M.; Lee, P.P. Tumor-Derived Exosomes Contain microRNAs with Immunological Function: Implications for a Novel Immunosuppression Mechanism. Microrna 2014, 2, 194–204. [Google Scholar] [CrossRef]
- Wen, S.W.; Sceneay, J.; Lima, L.G.; Wong, C.S.; Becker, M.; Krumeich, S.; Lobb, R.J.; Castillo, V.; Wong, K.N.; Ellis, S.; et al. The Biodistribution and Immune Suppressive Effects of Breast Cancer-Derived Exosomes. Cancer Res. 2016, 76, 6816–6827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H. Integrative radiation carcinogenesis: Interactions between cell and tissue responses to DNA damage. Semin. Cancer Biol. 2005, 15, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Systemic effects of local radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Wargo, J.A.; Reuben, A.; Cooper, Z.A.; Oh, K.S.; Sullivan, R.J. Immune Effects of Chemotherapy, Radiation, and Targeted Therapy and Opportunities for Combination With Immunotherapy. Semin. Oncol. 2015, 42, 601–616. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef]
- Timaner, M.; Bril, R.; Kaidar-Person, O.; Rachman-Tzemah, C.; Alishekevitz, D.; Kotsofruk, R.; Miller, V.; Nevelsky, A.; Daniel, S.; Raviv, Z.; et al. Dequalinium bl.locks macrophage-induced metastasis following local radiation. Oncotarget 2015, 6, 27537–27554. [Google Scholar] [CrossRef] [Green Version]
- Ahn, G.O.; Brown, J.M. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: Role of bone marrow-derived myelomonocytic cells. Cancer Cell 2008, 13, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.I.; Han, M.G.; Kang, M.H.; Park, J.M.; Kim, E.E.; Bae, J.; Ahn, S.; Kim, I.A. PI3Kalphadelta Inhibitor Combined With Radiation Enhances the Antitumor Immune Effect of Anti-PD1 in a Syngeneic Murine Triple-Negative Breast Cancer Model. Int. J. Radiat. Oncol. Biol. Phys. 2021, 110, 845–858. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.-J.; Liu, Y.; Lofland, D.; Lin, J. Breast Cancer Tumor Microenvironment and Molecular Aberrations Hijack Tumoricidal Immunity. Cancers 2022, 14, 285. https://doi.org/10.3390/cancers14020285
Lin H-J, Liu Y, Lofland D, Lin J. Breast Cancer Tumor Microenvironment and Molecular Aberrations Hijack Tumoricidal Immunity. Cancers. 2022; 14(2):285. https://doi.org/10.3390/cancers14020285
Chicago/Turabian StyleLin, Huey-Jen, Yingguang Liu, Denene Lofland, and Jiayuh Lin. 2022. "Breast Cancer Tumor Microenvironment and Molecular Aberrations Hijack Tumoricidal Immunity" Cancers 14, no. 2: 285. https://doi.org/10.3390/cancers14020285