
CellCallEXT: Analysis of Ligand–Receptor and Transcription Factor Activities in Cell–Cell Communication of Tumor Immune Microenvironment

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Inferring Intercellular Communication
2.2. Pathway Enrichment Analysis
2.3. Data Collection and Processing of scRNA-seq Datasets
3. Results
3.1. Comparison of CellCallEXT with Other Tools
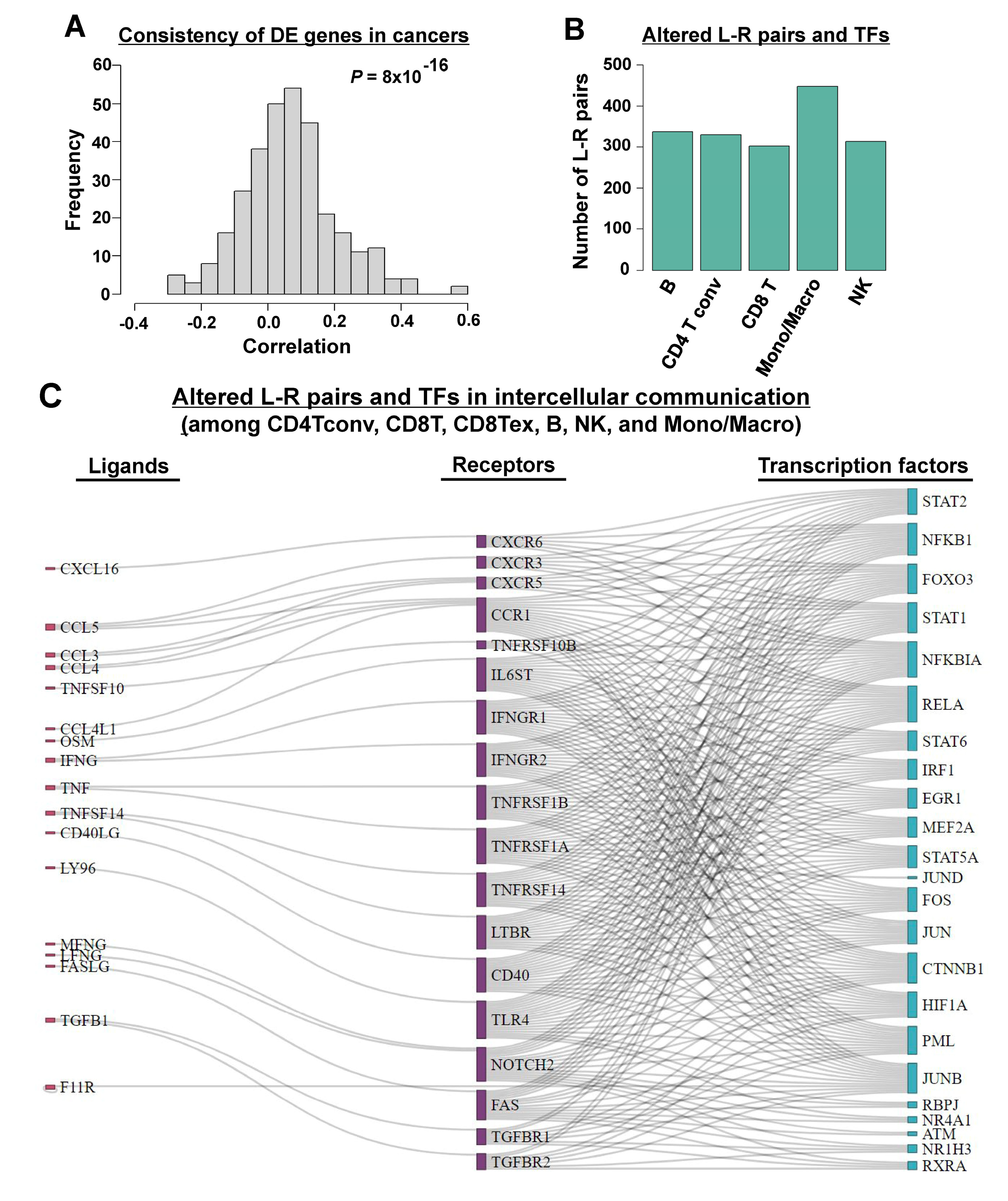
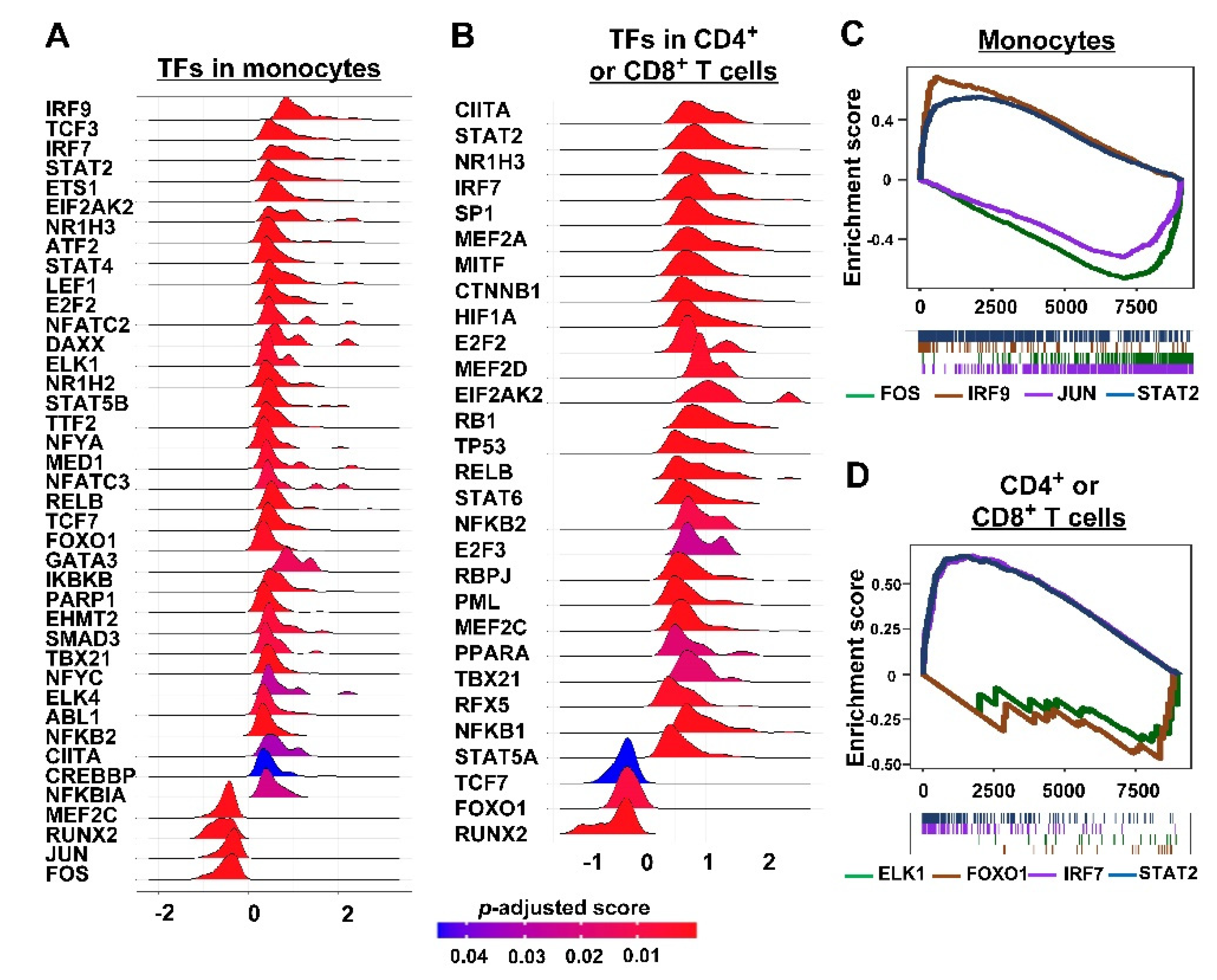
3.2. Inferring Cell–Cell Communication in TIME
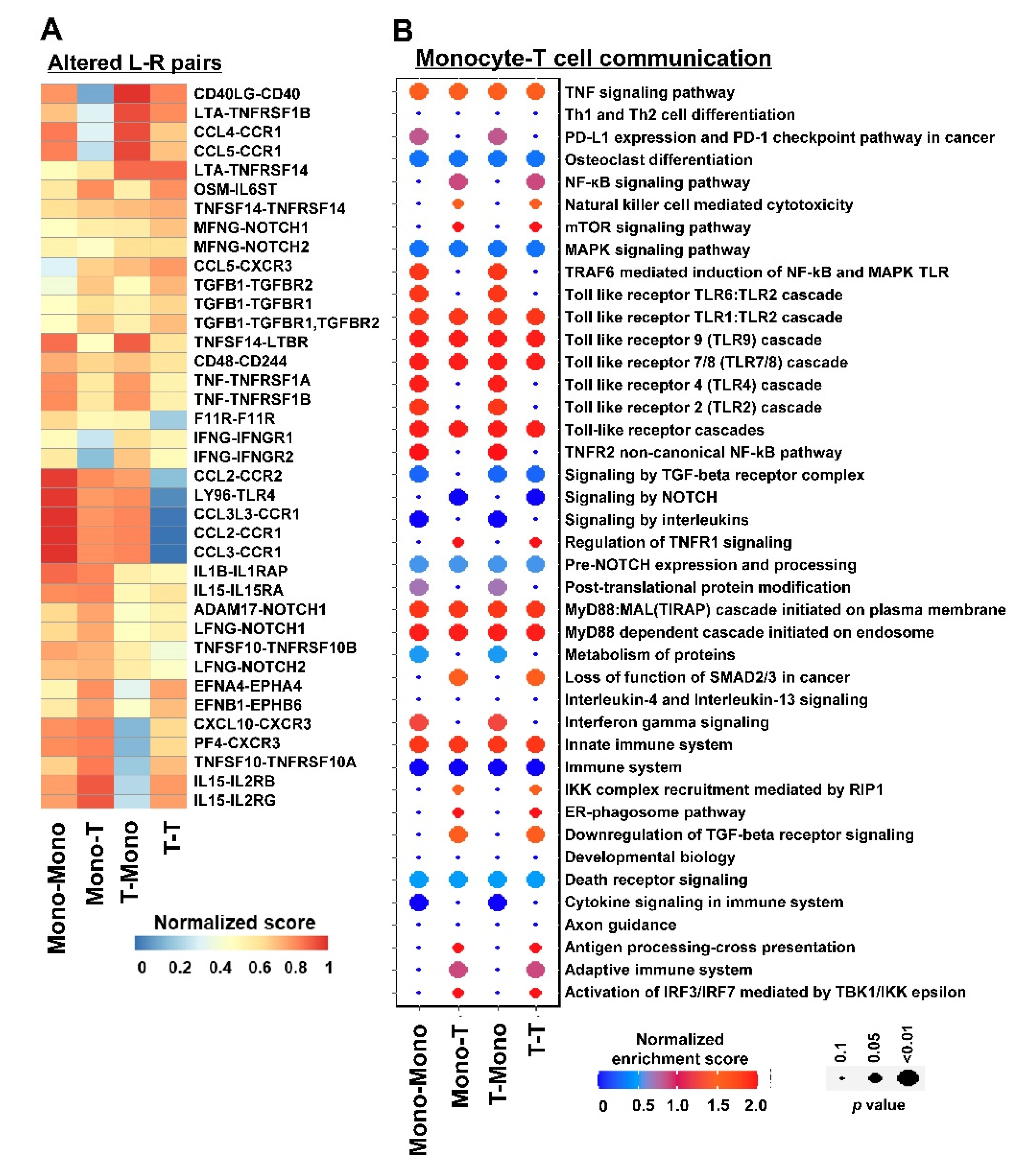
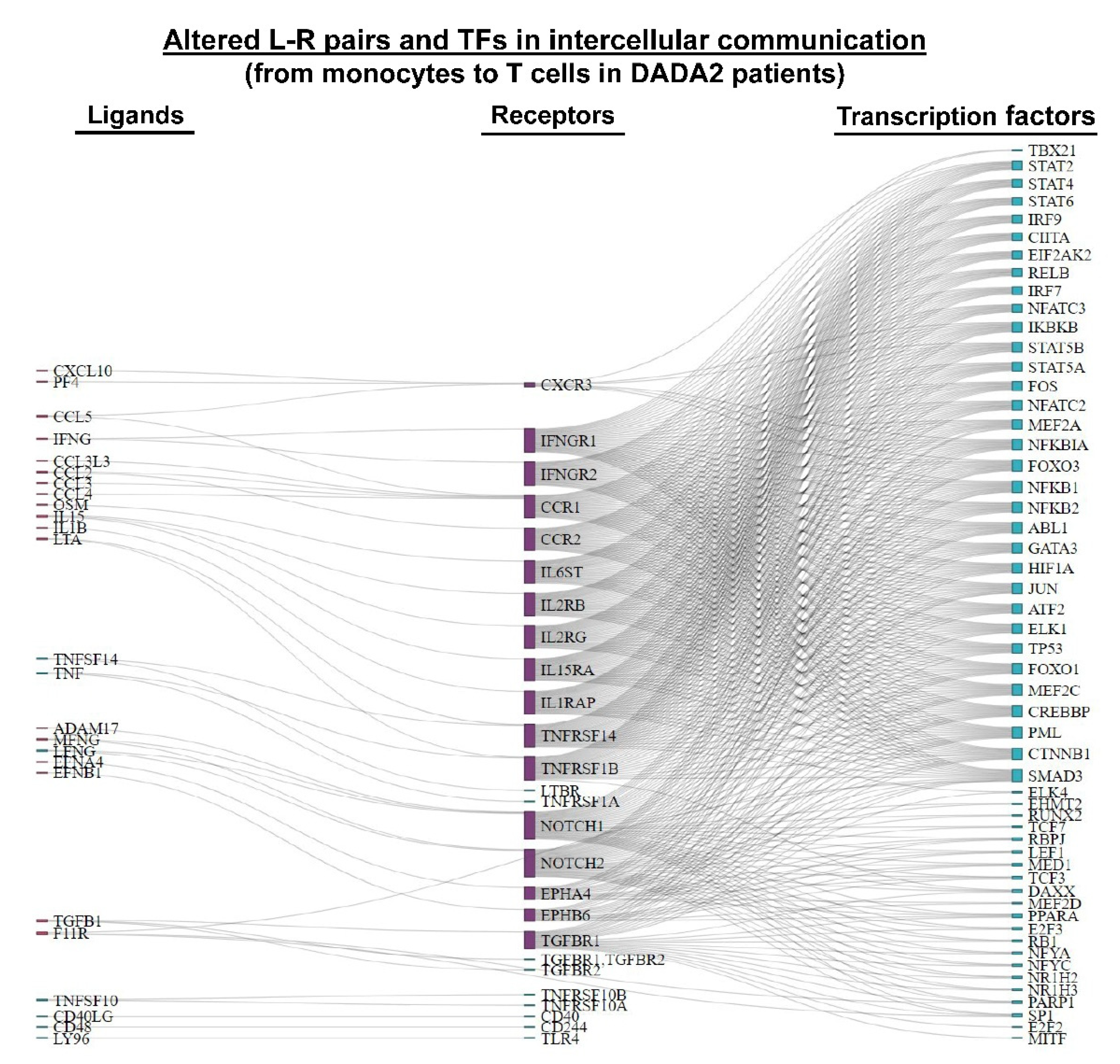
3.3. Inferring Cell–Cell Communication in DADA2
3.4. Comparison of DADA2 Results between CellCallEXT and NicheNet
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling intercellular communication by linking ligands to target genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Brucher, B.L.; Jamall, I.S. Cell-cell communication in the tumor microenvironment, carcinogenesis, and anticancer treatment. Cell Physiol. Biochem. 2014, 34, 213–243. [Google Scholar] [CrossRef]
- Cabello-Aguilar, S.; Alame, M.; Kon-Sun-Tack, F.; Fau, C.; Lacroix, M.; Colinge, J. SingleCellSignalR: Inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 2020, 48, e55. [Google Scholar] [CrossRef] [Green Version]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlicev, M.; Wagner, G.P.; Chavan, A.R.; Owens, K.; Maziarz, J.; Dunn-Fletcher, C.; Kallapur, S.G.; Muglia, L.; Jones, H. Single-cell transcriptomics of the human placenta: Inferring the cell communication network of the maternal-fetal interface. Genome Res. 2017, 27, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, N.; Gao, S.; Wu, Z.; Batchu, S.; Kajigaya, S.; Diamond, C.; Alemu, L.; Raffo, D.Q.; Hoffmann, P.; Stone, D.; et al. Analysis of deficiency of adenosine deaminase 2 pathogenesis based on single-cell RNA sequencing of monocytes. J. Leukoc. Biol. 2021, 110, 409–424. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.; Stephenson, E.; Polanski, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell. 2019, 75, 644–660.e5. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Hu, X.; Wang, M.; Wang, J.; Zou, B.; Tan, P.; Cui, T.; Dou, Y.; Ning, L.; et al. CellCall: Integrating paired ligand-receptor and transcription factor activities for cell-cell communication. Nucleic Acids Res. 2021, 49, 8520–8534. [Google Scholar] [CrossRef]
- Wu, Z.; Gao, S.; Watanabe, N.; Batchu, S.; Kajigaya, S.; Diamond, C.; Alemu, L.; Raffo, D.Q.; Feng, X.; Hoffmann, P.; et al. Single-cell profiling of T lymphocytes in deficiency of adenosine deaminase 2. J. Leukoc. Biol. 2021, 111, 301–312. [Google Scholar] [CrossRef]
- Sun, D.; Wang, J.; Han, Y.; Dong, X.; Ge, J.; Zheng, R.; Shi, X.; Wang, B.; Li, Z.; Ren, P.; et al. TISCH: A comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 2021, 49, D1420–D1430. [Google Scholar] [CrossRef]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [Green Version]
- Nichenetr/faq.md at Master. FAQ NicheNet Github. Available online: https://github.com/saeyslab/nichenetr/blob/master/vignettes/faq.md (accessed on 29 September 2022).
- GitHub—Shouguog/cellcallEXT: Package for Cell Cell Interactions Analysis. GitHub. Available online: https://github.com/shouguog/CellcallEXT (accessed on 29 September 2022).
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Jing, F.; Wang, J.; Zhou, L.; Ning, Y.; Xu, S.; Zhu, Y. Bioinformatics analysis of the role of CXC ligands in the microenvironment of head and neck tumor. Aging 2021, 13, 17789–17817. [Google Scholar] [CrossRef]
- Zaidi, M.R. The Interferon-Gamma Paradox in Cancer. J. Interferon Cytokine Res. 2019, 39, 30–38. [Google Scholar] [CrossRef]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Segui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Wang, C.; Wang, B.; Yang, J.; Wang, Y.; Luo, F.; Xu, J.; Zhao, C.; Liu, R.; Chu, Y. The IFN-gamma/PD-L1 axis between T cells and tumor microenvironment: Hints for glioma anti-PD-1/PD-L1 therapy. J. Neuroinflamm. 2018, 15, 290. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Deuitch, N.T.; Yang, D.; Lee, P.Y.; Yu, X.; Moura, N.S.; Schnappauf, O.; Ombrello, A.K.; Stone, D.; Kuehn, H.S.; Rosenzweig, S.; et al. TNF inhibition in vasculitis management in adenosine deaminase 2 deficiency (DADA2). J. Allergy Clin. Immunol. 2021, 149, 1812–1816. [Google Scholar] [CrossRef]
- Pinto, B.; Deo, P.; Sharma, S.; Syal, A.; Sharma, A. Expanding spectrum of DADA2: A review of phenotypes, genetics, pathogenesis and treatment. Clin. Rheumatol. 2021, 40, 3883–3896. [Google Scholar] [CrossRef] [PubMed]
- Nihira, H.; Izawa, K.; Ito, M.; Umebayashi, H.; Okano, T.; Kajikawa, S.; Nanishi, E.; Keino, D.; Murakami, K.; Isa-Nishitani, M.; et al. Detailed analysis of Japanese patients with adenosine deaminase 2 deficiency reveals characteristic elevation of type II interferon signature and STAT1 hyperactivation. J. Allergy Clin. Immunol. 2021, 148, 550–562. [Google Scholar] [CrossRef]
- Schena, F.; Penco, F.; Volpi, S.; Pastorino, C.; Caorsi, R.; Kalli, F.; Fenoglio, D.; Salis, A.; Bertoni, A.; Prigione, I.; et al. Dysregulation in B-cell responses and T follicular helper cell function in ADA2 deficiency patients. Eur. J. Immunol. 2021, 51, 206–219. [Google Scholar] [CrossRef]
- Ma, F.; Zhang, S.; Song, L.; Wang, B.; Wei, L.; Zhang, F. Applications and analytical tools of cell communication based on ligand-receptor interactions at single cell level. Cell Biosci. 2021, 11, 121. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Liao, J.; Li, C.; Lu, X.; Cheng, J.; Fan, X. CellTalkDB: A manually curated database of ligand-receptor interactions in humans and mice. Brief. Bioinform. 2021, 22, bbaa269. [Google Scholar] [CrossRef]
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N. Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet 2021, 22, 71–88. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title 1 | CellCallEXT | CellCall | NicheNet | CellPhoneDB | SingleCellSignalR |
|---|---|---|---|---|---|
| Ligand | Expression value | Expression value | If expressed (Boolean) | Expression value | Expression value |
| Receptor | Expression value | Expression value | If expressed (Boolean) | Expression value | Expression value |
| Target genes | Expression alteration by disease | Expression abundance | Expression alteration by disease | Not considered | Not considered |
| Data size | 43,793 L–R–TF | 19,144 L–R–TF | 12,019 | 1396 | 3251 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, S.; Feng, X.; Wu, Z.; Kajigaya, S.; Young, N.S. CellCallEXT: Analysis of Ligand–Receptor and Transcription Factor Activities in Cell–Cell Communication of Tumor Immune Microenvironment. Cancers 2022, 14, 4957. https://doi.org/10.3390/cancers14194957
Gao S, Feng X, Wu Z, Kajigaya S, Young NS. CellCallEXT: Analysis of Ligand–Receptor and Transcription Factor Activities in Cell–Cell Communication of Tumor Immune Microenvironment. Cancers. 2022; 14(19):4957. https://doi.org/10.3390/cancers14194957
Chicago/Turabian StyleGao, Shouguo, Xingmin Feng, Zhijie Wu, Sachiko Kajigaya, and Neal S. Young. 2022. "CellCallEXT: Analysis of Ligand–Receptor and Transcription Factor Activities in Cell–Cell Communication of Tumor Immune Microenvironment" Cancers 14, no. 19: 4957. https://doi.org/10.3390/cancers14194957