
Validation of MRI-Based Models to Predict MGMT Promoter Methylation in Gliomas: BraTS 2021 Radiogenomics Challenge
 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods and Materials
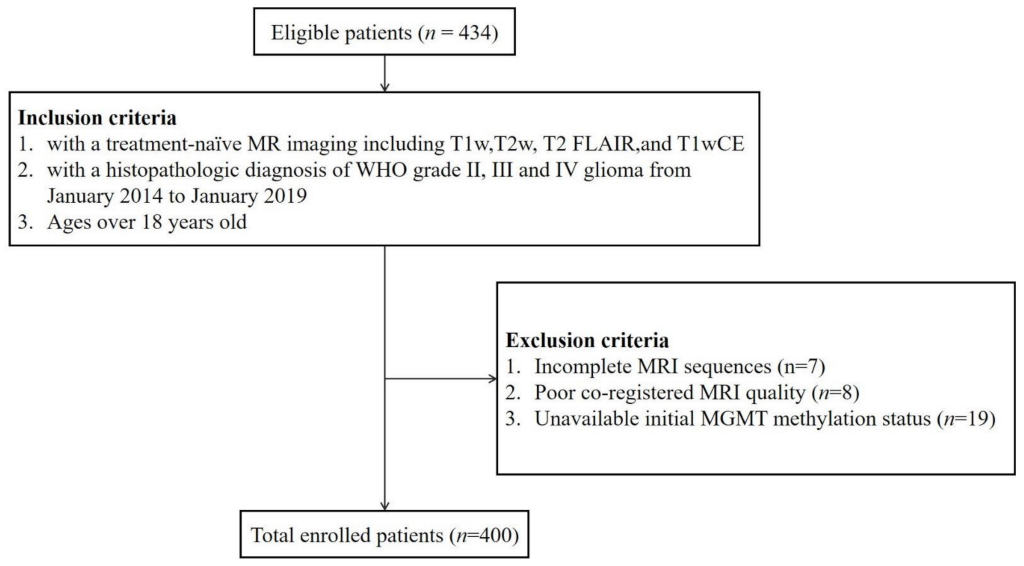
2.1. Datasets
2.2. Data Preprocessing and Model Implementation
2.3. Experiments
3. Results
3.1. Patient Characteristics
3.2. Model Performance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MGMT | O6-methylguanine-DNA methyltransferase |
| IDH | isocitrate dehydrogenase |
| MRI | magnetic resonance imaging |
| CNN | Convolutional neural network |
| T1w | T1-weighted imaging |
| T2w | T2-weighted imaging |
| T2 FLAIR | T2-weighted fluid-attenuated inversion recovery |
| T1wCE | contrast-enhanced T1-weighted imaging |
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Esteller, M.; Herman, J.G. Generating mutations but providing chemosensitivity: The role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene 2004, 23, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, A.; Hachem, L.D.; Mansouri, S.; Nassiri, F.; Laperriere, N.J.; Xia, D.; Lindeman, N.I.; Wen, P.Y.; Chakravarti, A.; Mehta, M.P.; et al. MGMT promoter methylation status testing to guide therapy for glioblastoma: Refining the approach based on emerging evidence and current challenges. Neuro-Oncol. 2019, 21, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Rudie, J.D.; Rauschecker, A.M.; Bryan, R.N.; Davatzikos, C.; Mohan, S. Emerging Applications of Artificial Intelligence in Neuro-Oncology. Radiology 2019, 290, 607–618. [Google Scholar] [CrossRef]
- Suh, C.H.; Kim, H.S.; Jung, S.C.; Choi, C.G.; Kim, S.J. Clinically Relevant Imaging Features for MGMT Promoter Methylation in Multiple Glioblastoma Studies: A Systematic Review and Meta-Analysis. AJNR Am. J. Neuroradiol. 2018, 39, 1439–1445. [Google Scholar] [CrossRef]
- Han, L.; Kamdar, M.R. MRI to MGMT: Predicting methylation status in glioblastoma patients using convolutional recurrent neural networks. Pac. Symp. Biocomputing. Pac. Symp. Biocomput. 2018, 23, 331–342. [Google Scholar]
- Baid, U.; Ghodasara, S.; Mohan, S.; Bilello, M.; Calabrese, E.; Colak, E.; Farahani, K.; Kalpathy-Cramer, J.; Kitamura, F.C.; Pati, S.; et al. The RSNA-ASNR-MICCAI BraTS 2021 Benchmark on Brain Tumor Segmentation and Radiogenomic Classification. arXiv 2021, arXiv:2107.02314. [Google Scholar]
- Levner, I.; Drabycz, S.; Roldan, G.; De Robles, P.; Cairncross, J.G.; Mitchell, R. Predicting MGMT methylation status of glioblastomas from MRI texture. In Medical Image Computing and Computer-Assisted Intervention: MICCAI ... International Conference on Medical Image Computing and Computer-Assisted Intervention; Springer: Berlin/Heidelberg, Germany, 2009; Volume 12, pp. 522–530. [Google Scholar] [CrossRef]
- Sasaki, T.; Kinoshita, M.; Fujita, K.; Fukai, J.; Hayashi, N.; Uematsu, Y.; Okita, Y.; Nonaka, M.; Moriuchi, S.; Uda, T.; et al. Radiomics and MGMT promoter methylation for prognostication of newly diagnosed glioblastoma. Sci. Rep. 2019, 9, 14435. [Google Scholar] [CrossRef] [Green Version]
- Yogananda, C.G.B.; Shah, B.R.; Nalawade, S.S.; Murugesan, G.K.; Yu, F.F.; Pinho, M.C.; Wagner, B.C.; Mickey, B.; Patel, T.R.; Fei, B.; et al. MRI-Based Deep-Learning Method for Determining Glioma MGMT Promoter Methylation Status. AJNR Am. J. Neuroradiol. 2021, 42, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Moons, K.G.; Altman, D.G.; Reitsma, J.B.; Ioannidis, J.P.; Macaskill, P.; Steyerberg, E.W.; Vickers, A.J.; Ransohoff, D.F.; Collins, G.S. Transparent Reporting of a multivariable prediction model for Individual Prognosis or Diagnosis (TRIPOD): Explanation and elaboration. Ann. Intern. Med. 2015, 162, W1–W73. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Le, Q. Efficientnet: Rethinking model scaling for convolutional neural networks. In Proceedings of the International Conference on Machine Learning, Long Beach, CA, USA, 9–15 June 2019; pp. 6105–6114. [Google Scholar]
- Hu, J.; Shen, L.; Sun, G. Squeeze-and-Excitation Networks. In Proceedings of the 2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition, Salt Lake City, UT, USA, 18–23 June 2018; pp. 7132–7141. [Google Scholar]
- Xie, S.; Girshick, R.; Dollár, P.; Tu, Z.; He, K. Aggregated residual transformations for deep neural networks. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017; pp. 1492–1500. [Google Scholar]
- Huang, G.; Chen, D.; Li, T.; Wu, F.; Van Der Maaten, L.; Weinberger, K.Q. Multi-scale dense networks for resource efficient image classification. arXiv Prepr. 2017, arXiv:1703.09844. [Google Scholar]
- Drabycz, S.; Roldán, G.; de Robles, P.; Adler, D.; McIntyre, J.B.; Magliocco, A.M.; Cairncross, J.G.; Mitchell, J.R. An analysis of image texture, tumor location, and MGMT promoter methylation in glioblastoma using magnetic resonance imaging. NeuroImage 2010, 49, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yang, G.; Hao, X.; Gu, D.; Tan, Y.; Wang, X.; Dong, D.; Zhang, S.; Wang, L.; Zhang, H.; et al. A multi-sequence and habitat-based MRI radiomics signature for preoperative prediction of MGMT promoter methylation in astrocytomas with prognostic implication. Eur. Radiol. 2019, 29, 877–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korfiatis, P.; Kline, T.L.; Coufalova, L.; Lachance, D.H.; Parney, I.F.; Carter, R.E.; Buckner, J.C.; Erickson, B.J. MRI texture features as biomarkers to predict MGMT methylation status in glioblastomas. Med. Phys. 2016, 43, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Eoli, M.; Menghi, F.; Bruzzone, M.G.; De Simone, T.; Valletta, L.; Pollo, B.; Bissola, L.; Silvani, A.; Bianchessi, D.; Incerti, L.; et al. Methylation of O6-Methylguanine DNA Methyltransferase and Loss of Heterozygosity on 19q and/or 17p Are Overlapping Features of Secondary Glioblastomas with Prolonged Survival. Clin. Cancer Res. 2007, 13, 2606. [Google Scholar] [CrossRef] [Green Version]
- An, C.; Park, Y.W.; Ahn, S.S.; Han, K.; Kim, H.; Lee, S.-K. Radiomics machine learning study with a small sample size: Single random training-test set split may lead to unreliable results. PLoS ONE 2021, 16, e0256152. [Google Scholar] [CrossRef]
- Refaeilzadeh, P.; Tang, L.; Liu, H. Cross-Validation. In Encyclopedia of Database Systems; Liu, L., ÖZsu, M.T., Eds.; Springer: Boston, MA, USA, 2009; pp. 532–538. [Google Scholar]
- Liu, X.; Faes, L.; Kale, A.U.; Wagner, S.K.; Fu, D.J.; Bruynseels, A.; Mahendiran, T.; Moraes, G.; Shamdas, M.; Kern, C.; et al. A comparison of deep learning performance against health-care professionals in detecting diseases from medical imaging: A systematic review and meta-analysis. Lancet Digit. Health 2019, 1, e271–e297. [Google Scholar] [CrossRef]
- Chang, K.; Bai, H.X.; Zhou, H.; Su, C.; Bi, W.L.; Agbodza, E.; Kavouridis, V.K.; Senders, J.T.; Boaro, A.; Beers, A.; et al. Residual Convolutional Neural Network for the Determination of IDH Status in Low- and High-Grade Gliomas from MR Imaging. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.S.; Bae, S.; Chang, J.H.; Kang, S.G.; Kim, S.H.; Kim, J.; Rim, T.H.; Choi, S.H.; Jain, R.; Lee, S.K. Fully automated hybrid approach to predict the IDH mutation status of gliomas via deep learning and radiomics. Neuro-Oncol. 2021, 23, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Omuro, A.M.P.; Shah, A.D.; Graber, J.J.; Shi, W.; Zhang, Z.; Young, R.J. Continuing the search for MR imaging biomarkers for MGMT promoter methylation status: Conventional and perfusion MRI revisited. Neuroradiology 2012, 54, 641–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, W.J.; Choi, J.W.; Roh, H.G.; Lim, S.D.; Koh, Y.C. Imaging parameters of high grade gliomas in relation to the MGMT promoter methylation status: The CT, diffusion tensor imaging, and perfusion MR imaging. Neuroradiology 2012, 54, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Calabria, L.F.; Tavanti, F.; Minniti, G.; Rossi-Espagnet, M.C.; Coppola, V.; Pugliese, S.; Guida, D.; Francione, G.; Colonnese, C.; et al. Apparent diffusion coefficient obtained by magnetic resonance imaging as a prognostic marker in glioblastomas: Correlation with MGMT promoter methylation status. Eur. Radiol. 2013, 23, 513–520. [Google Scholar] [CrossRef]
- Rundle-Thiele, D.; Day, B.; Stringer, B.; Fay, M.; Martin, J.; Jeffree, R.L.; Thomas, P.; Bell, C.; Salvado, O.; Gal, Y.; et al. Using the apparent diffusion coefficient to identifying MGMT promoter methylation status early in glioblastoma: Importance of analytical method. J. Med. Radiat. Sci. 2015, 62, 92–98. [Google Scholar] [CrossRef]
- Ryoo, I.; Choi, S.H.; Kim, J.H.; Sohn, C.H.; Kim, S.C.; Shin, H.S.; Yeom, J.A.; Jung, S.C.; Lee, A.L.; Yun, T.J.; et al. Cerebral blood volume calculated by dynamic susceptibility contrast-enhanced perfusion MR imaging: Preliminary correlation study with glioblastoma genetic profiles. PLoS ONE 2013, 8, e71704. [Google Scholar] [CrossRef] [Green Version]
- Smits, M.; van den Bent, M.J. Imaging Correlates of Adult Glioma Genotypes. Radiology 2017, 284, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Pak, E.; Choi, K.S.; Choi, S.H.; Park, C.-K.; Kim, T.M.; Park, S.-H.; Lee, J.H.; Lee, S.-T.; Hwang, I.; Yoo, R.-E.; et al. Prediction of Prognosis in Glioblastoma Using Radiomics Features of Dynamic Contrast-Enhanced MRI. Korean J. Radiol. 2021, 22, 1514–1524. [Google Scholar] [CrossRef]
- Choi, K.S.; Choi, S.H.; Jeong, B. Prediction of IDH genotype in gliomas with dynamic susceptibility contrast perfusion MR imaging using an explainable recurrent neural network. Neuro-Oncol. 2019, 21, 1197–1209. [Google Scholar] [CrossRef]
- Choi, H.J.; Choi, S.H.; You, S.H.; Yoo, R.E.; Kang, K.M.; Yun, T.J.; Kim, J.H.; Sohn, C.H.; Park, C.K.; Park, S.H. MGMT Promoter Methylation Status in Initial and Recurrent Glioblastoma: Correlation Study with DWI and DSC PWI Features. AJNR Am. J. Neuroradiol. 2021, 42, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Forum. RSNA MICCAI Brain Tumor Radiogenomic Classification Discussion 1. Available online: https://www.kaggle.com/c/rsna-miccai-brain-tumor-radiogenomic-classification/discussion/279820 (accessed on 18 December 2021).
- Forum. RSNA MICCAI Brain Tumor Radiogenomic Classification Discussion 2. Available online: https://www.kaggle.com/c/rsna-miccai-brain-tumor-radiogenomic-classification/discussion/279777 (accessed on 18 December 2021).
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar] [CrossRef]
- Szegedy, C.; Vanhoucke, V.; Ioffe, S.; Shlens, J.; Wojna, Z. Rethinking the Inception Architecture for Computer Vision. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016; pp. 2818–2826. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Liu, Z.; Van Der Maaten, L.; Weinberger, K.Q. Densely Connected Convolutional Networks. In Proceedings of the 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Honolulu, HI, USA, 21–26 July 2017; pp. 2261–2269. [Google Scholar] [CrossRef] [Green Version]
- Consortium. MONAI: Medical Open Network for AI, Version 0.8.0; Zenodo, 2021. [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Number of Patients | Age, Mean ± SD (Years) | PFS, Median (95% CI) (Days) | p Value | |
|---|---|---|---|---|
| Sex | ||||
| Male | 240 (60%) | 52.6 ± 15.7 | 327 (287–372) | 0.655 |
| Female | 160 (40%) | 51.9 ± 14.7 | 362 (301–481) | |
| MGMT | ||||
| Unmethylated | 203 (50.8%) | 52.6 ± 15.6 | 396 (328–526) | <0.0001 * |
| Methylated | 197 (49.2%) | 52.0 ± 14.9 | 974 (698–1302) |
| Previous Study | Dataset | MR Sequence | Input Feature | Model Architecture | Dimension | Diagnostic Performance |
|---|---|---|---|---|---|---|
| Han et al. [8] | TCIA (n = 262): Grade IV glioblastoma | T1w, T2w, FLAIR | Raw images | CRNN | 2D axial CNN with RNN in slice-direction (z-axis) | Acc 67% (validation), 62% (test), precision (67%), recall (67%) |
| Sasaki et al. [11] | Osaka International Cancer Institute (n = 201): Grade IV glioblastoma | T1w, T2w, FLAIR, T1wCE | Radiomics | Supervised principal component analysis | 3D VOI of 1 mm isotropic resampled image | Acc 67% (mean by 10-fold cross-validation) |
| Levner et al. [10] | Tom Baker Cancer Centre (n = 59): Grade IV glioblastoma | T2w, FLAIR, T1wCE | Texture analysis | L1-regularized neural network | 2D axial | Acc 87.7% |
| Drabycz et al. [18] | Tom Baker Cancer Centre (n = 59): Grade IV glioblastoma | T2w, FLAIR, T1wCE | Texture analysis | Linear discriminant analysis | 2D axial | Acc 71% |
| Yogananda et al. [12] | TCIA (n = 247); Grade II-IV gliomas | T2w | Raw images | 3D-DenseUNet | 3D patch | Acc 94.7% (mean by 3-fold cross-validation) |
| Wei et al. [19] | Shanxi Medical University (n = 105); Grade II-IV astrocytoma | T1wCE, FLAIR, ADC | Radiomics | Logistic regression | 3D VOI | Acc 77% (validation; n = 31) |
| Korfiatis et al. [20] | Mayo Clinic (n = 155); Grade IV glioblastoma | T2w, T1wCE | Texture analysis | Support vector machines, random forest classifiers | 2D ROI | AUC 0.85 |
| Dataset | CNN Architecture | MR Sequence | Metrics † | |||
|---|---|---|---|---|---|---|
| Best AUROC (%) | Accuracy (%) | Precision (%) | Recall (%) | |||
| Experiment 1 (Train/valid BraTS, Test SNUH) | EfficientNet-B0 | FLAIR-T1wCE-T2w-T1w | 46.4 4.7 (39.1–52.0) | 55.9 1.2 (54.2–57.6) | 55.9 2.8 (53.4–60.7) | 83.2 18.7 (54.8–100.0) |
| Experiment 2 (Train/valid SNUH, Test BraTS) | SEResNeXt50 | FLAIR-T1wCE | 57.8 8.3 (46.0–67.5) | 55.5 5.4 (50.0–62.5) | 36.6 34.0 (0.0–71.4) | 44.0 49.9 (0.0–100.0) |
| Experiment 3 (Train/valid SNUH + BraTS, Test SNUH + BraTS) | SEResNet50 | T2w | 54.9 5.4 (48.8–63.1) | 57.1 3.1 (53.9–61.8) | 61.6 10.3 (53.2–75.0) | 68.7 33.4 (26.1–97.8) |
| Dataset | CNN Architecture | MR Sequence | Metrics † | |||
|---|---|---|---|---|---|---|
| Best AUROC (%) | Accuracy (%) | Precision (%) | Recall (%) | |||
| Experiment 1 (Train/valid BraTS, Test SNUH) | EfficientNet-B0 | FLAIR-T1wCE-T2w-T1w | 51.6 3.8 (47.0–57.2) | 49.8 1.3 (48.5–51.5) | 49.0 1.8 (45.9–50.5) | 80.4 31.5 (25.9–100.0) |
| Experiment 2 (Train/valid SNUH, Test BraTS) | SEResNeXt50 | FLAIR-T1wCE | 51.7 7.7 (45.9–64.5) | 51.9 3.4 (47.5–55.6) | 54.6 5.7 (49.4–64.3) | 62.0 36.5 (17.6–94.1) |
| Experiment 3 (Train/valid SNUH + BraTS, Test SNUH + BraTS) | SEResNet50 | T2w | 51.5 2.7 (48.5–55.5) | 50.4 3.3 (47.3–54.6) | 32.6 29.8 (0–55.6) | 38.2 43.7 (0–90.2) |
| Experiment 4 (BraTS winner codes, Test SNUH) | 3D-ResNet | T1wCE | 56.2 | 54.8 | 53.6 | 59.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.-H.; Lee, H.; Choi, K.S.; Nam, J.G.; Park, C.-K.; Park, S.-H.; Chung, J.W.; Choi, S.H. Validation of MRI-Based Models to Predict MGMT Promoter Methylation in Gliomas: BraTS 2021 Radiogenomics Challenge. Cancers 2022, 14, 4827. https://doi.org/10.3390/cancers14194827
Kim B-H, Lee H, Choi KS, Nam JG, Park C-K, Park S-H, Chung JW, Choi SH. Validation of MRI-Based Models to Predict MGMT Promoter Methylation in Gliomas: BraTS 2021 Radiogenomics Challenge. Cancers. 2022; 14(19):4827. https://doi.org/10.3390/cancers14194827
Chicago/Turabian StyleKim, Byung-Hoon, Hyeonhoon Lee, Kyu Sung Choi, Ju Gang Nam, Chul-Kee Park, Sung-Hye Park, Jin Wook Chung, and Seung Hong Choi. 2022. "Validation of MRI-Based Models to Predict MGMT Promoter Methylation in Gliomas: BraTS 2021 Radiogenomics Challenge" Cancers 14, no. 19: 4827. https://doi.org/10.3390/cancers14194827