
Comprehensive Molecular Analysis Identified an SRSF Family-Based Score for Prognosis and Therapy Efficiency Prediction in Hepatocellular Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
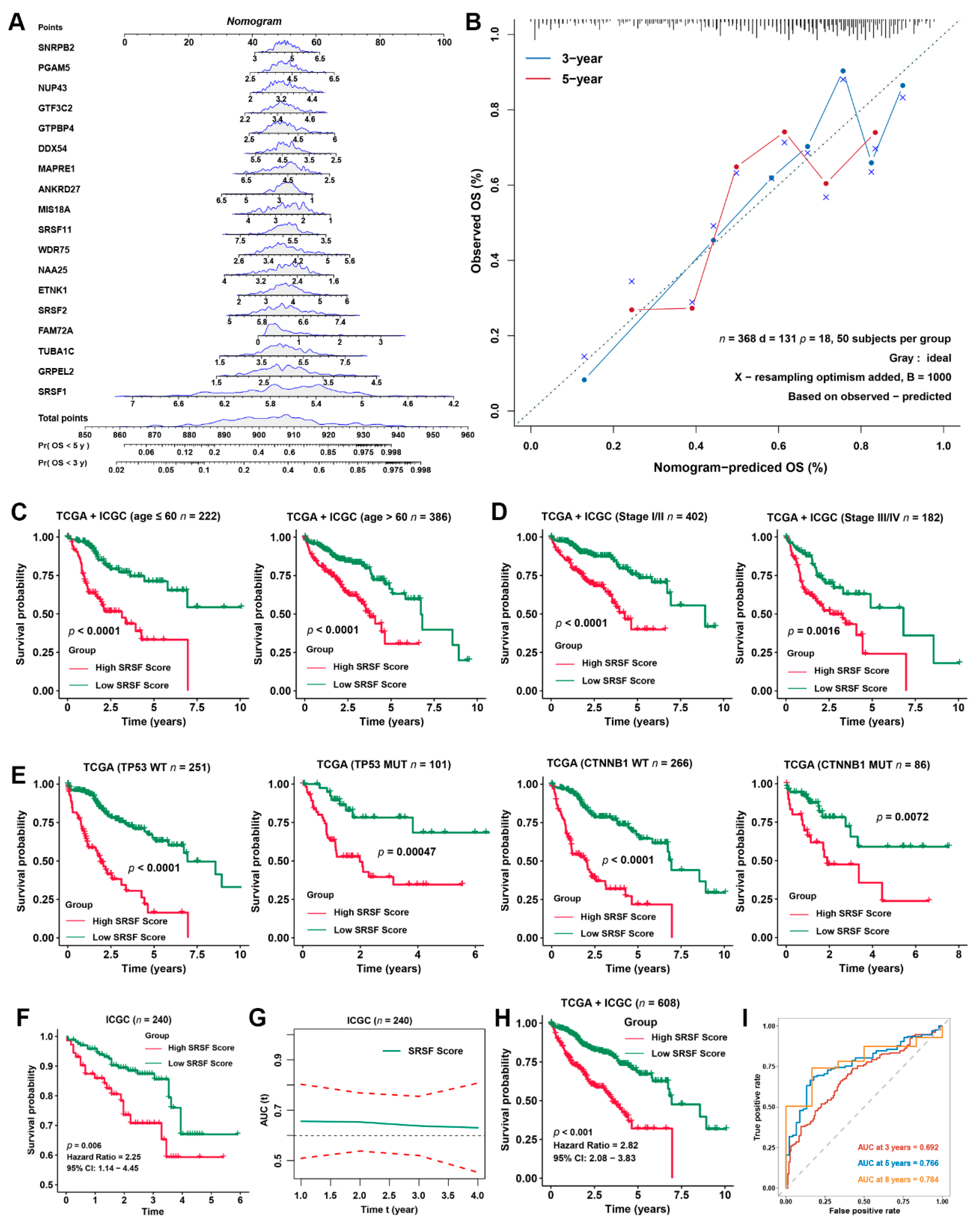{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples and Immunohistochemistry
2.2. Cell Culture and Reagents
2.3. Transfection
2.4. Western Blot Analysis
2.5. Quantitative Real-Time Polymerase Chain Reaction
2.6. Cell Counting kit-8 and EdU Assays
2.7. Data Sources and Preprocessing
2.8. Functional and Pathway Enrichment Analysis
2.9. Assessment of the Tumor Immune Microenvironment
2.10. Establishment of the SRSF-Related Score
2.11. Development and Validation of the Prognostic Nomogram
2.12. mRNA-Based Stemness Index (mRNAsi) and Therapeutic Response Prediction
2.13. Statistical Analysis
3. Results
3.1. Landscape and Prognostic Significance of the SRSF Family Genes in HCC
3.2. Identification of SRSF Family-Related Genes and Prediction of Their Functional Annotations in HCC
3.3. Construction of the SRSF score and Its Predictive Effect on the Prognosis of HCC Patients
3.4. Validation of the Prognostic Predictive Capability of the SRSF Score in Different Clinical Subgroups
3.5. Identification of SRSF Score-Related Biological Characteristics
3.6. The SRSF Score Predicts the Drug Sensitivity of HCC
3.7. SRSF11 Knockdown Inhibited the CDK1-Dependent Proliferation of HCC Cells
3.8. SRSF11 Knockdown Enhanced the Drug Sensitivity of HCC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019, 156, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (HCC): Epidemiology, etiology and molecular classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Rebouissou, S.; Nault, J.C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 2020, 72, 215–229. [Google Scholar] [CrossRef]
- Gao, Q.; Zhu, H.; Dong, L.; Shi, W.; Chen, R.; Song, Z.; Huang, C.; Li, J.; Dong, X.; Zhou, Y.; et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 2019, 179, 561–577. [Google Scholar] [CrossRef]
- Shi, J.Y.; Wang, X.; Ding, G.Y.; Dong, Z.; Han, J.; Guan, Z.; Ma, L.-J.; Zheng, Y.; Zhang, L.; Yu, G.-Z.; et al. Exploring prognostic indicators in the pathological images of hepatocellular carcinoma based on deep learning. Gut 2021, 70, 951–961. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell. 2019, 76, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Corkery, D.P.; Holly, A.C.; Lahsaee, S.; Dellaire, G. Connecting the speckles: Splicing kinases and their role in tumorigenesis and treatment response. Nucleus 2015, 6, 279–288. [Google Scholar] [CrossRef]
- Zheng, X.; Peng, Q.; Wang, L.; Zhang, X.; Huang, L.; Wang, J.; Qin, Z. Serine/arginine-rich splicing factors: The bridge linking alternative splicing and cancer. Int. J. Biol. Sci. 2020, 16, 2442–2453. [Google Scholar] [CrossRef]
- Yuan, J.; Yin, Z.; Tan, L.; Zhu, W.; Tao, K.; Wang, G.; Shi, W.; Gao, J. Interferon regulatory factor-1 reverses chemoresistance by downregulating the expression of P-glycoprotein in gastric cancer. Cancer Lett. 2019, 457, 28–39. [Google Scholar] [CrossRef]
- Yuan, J.; Tan, L.; Yin, Z.; Zhu, W.; Tao, K.; Wang, G.; Shi, W.; Gao, J. MIR17HG-miR-18a/19a axis, regulated by interferon regulatory factor-1, promotes gastric cancer metastasis via Wnt/β-catenin signalling. Cell Death Dis. 2019, 10, 454. [Google Scholar] [CrossRef]
- Tan, L.; Yuan, J.; Zhu, W.; Tao, K.; Wang, G.; Gao, J. Interferon regulatory factor-1 suppresses DNA damage response and reverses chemotherapy resistance by downregulating the expression of RAD51 in gastric cancer. Am. J. Cancer Res. 2020, 10, 1255–1270. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Tang, W.; Wu, X.; Fu, W. A random forest approach to the detection of epistatic interactions in case-control studies. BMC Bioinform. 2009, 10 (Suppl. S1), S65. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, H.; Zhou, X.; Fang, D.; Ou, X.; Ye, J.; Peng, J.; Xu, J. Necroptosis-Related lncRNAs: Predicting Prognosis and the Distinction between the Cold and Hot Tumors in Gastric Cancer. J. Oncol. 2021, 2021, 6718443. [Google Scholar] [CrossRef]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Muñoz, Ú.; Puche, J.E.; Hannivoort, R.; Lang, U.E.; Cohen-Naftaly, M.; Friedman, S.L. Hepatocyte growth factor enhances alternative splicing of the Kruppel-like factor 6 (KLF6) tumor suppressor to promote growth through SRSF1. Mol. Cancer Res. 2012, 10, 1216–1227. [Google Scholar] [CrossRef]
- Wang, P.; Guo, L.; Li, K.; Ning, S.; Shi, W.; Liu, Z.; Chen, Y. Serine/arginine rich splicing factor 2 expression and clinic pathological features indicating a prognostic factor in human hepatocellular carcinoma patients. Cancer Biomark. 2018, 21, 681–687. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Selokar, N.L.; Saini, M.; Muzaffer, M.; Krishnakanth, G.; Saha, A.P.; Chauhan, M.S.; Manik, R.; Palta, P.; Madan, P.; Singla, S.K. Roscovitine treatment improves synchronization of donor cell cycle in G0/G1 stage and in vitro development of handmade cloned buffalo (Bubalus bubalis) embryos. Cell Reprogram. 2012, 14, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zou, X.; Yao, J. The antitumor effect of GDC-0941 alone and in combination with rapamycin in breast cancer cells. Chemotherapy 2012, 58, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xiao, Y.; Li, S.; Zhu, X.; Meng, L.; Song, C.; Yu, C.; Jiang, N.; Liu, Y. Synergistic activity of magnolin combined with B-RAF inhibitor SB590885 in hepatocellular carcinoma cells via targeting PI3K-AKT/mTOR and ERK MAPK pathway. Am. J. Transl. Res. 2019, 11, 3816–3824. [Google Scholar] [PubMed]
- Lewis, S.M.; Asselin-Labat, M.L.; Nguyen, Q.; Berthelet, J.; Tan, X.; Wimmer, V.C.; Merino, D.; Rogers, K.L.; Naik, S.H. Spatial omics and multiplexed imaging to explore cancer biology. Nat. Methods 2021, 18, 997–1012. [Google Scholar] [CrossRef]
- Wang, N.; Li, X.; Wang, R.; Ding, Z. Spatial transcriptomics and proteomics technologies for deconvoluting the tumor microenvironment. Biotechnol. J. 2021, 16, e2100041. [Google Scholar] [CrossRef]
- Sheng, J.; Zhao, Q.; Zhao, J.; Zhang, W.; Sun, Y.; Qin, P.; Lv, Y.; Bai, L.; Yang, Q.; Chen, L.; et al. SRSF1 modulates PTPMT1 alternative splicing to regulate lung cancer cell radioresistance. EBioMedicine 2018, 38, 113–126. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target Ther. 2021, 6, 78. [Google Scholar] [CrossRef]
- Cherry, S.; Lynch, K.W. Alternative splicing and cancer: Insights, opportunities, and challenges from an expanding view of the transcriptome. Genes Dev. 2020, 34, 1005–1016. [Google Scholar] [CrossRef]
- Frankiw, L.; Baltimore, D.; Li, G. Alternative mRNA splicing in cancer immunotherapy. Nat. Rev. Immunol. 2019, 19, 675–687. [Google Scholar] [CrossRef]
- Lei, S.; Zhang, B.; Huang, L.; Zheng, Z.; Xie, S.; Shen, L.; Breitzig, M.; Czachor, A.; Liu, H.; Luo, H.; et al. SRSF1 promotes the inclusion of exon 3 of SRA1 and the invasion of hepatocellular carcinoma cells by interacting with exon 3 of SRA1pre-mRNA. Cell Death Discov. 2021, 7, 117. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Cheng, Y.; Liu, Y.; Chen, L.; Liu, L.; Wei, N.; Xie, Z.; Wu, W.; Feng, Y. SRSF2 Regulates Alternative Splicing to Drive Hepatocellular Carcinoma Development. Cancer Res. 2017, 77, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jeong, S.A.; Khadka, P.; Hong, J.; Chung, I.K. Involvement of SRSF11 in cell cycle-specific recruitment of telomerase to telomeres at nuclear speckles. Nucleic Acids Res. 2015, 43, 8435–8451. [Google Scholar] [CrossRef] [PubMed]
- Tam, B.Y.; Chiu, K.; Chung, H.; Bossard, C.; Nguyen, J.D.; Creger, E.; Eastman, B.W.; Mak, C.C.; Ibanez, M.; Ghias, A.; et al. The CLK inhibitor SM08502 induces anti-tumor activity and reduces Wnt pathway gene expression in gastrointestinal cancer models. Cancer Lett. 2020, 473, 186–197. [Google Scholar] [CrossRef]
- Lv, Y.; Zhang, W.; Zhao, J.; Sun, B.; Qi, Y.; Ji, H.; Chen, C.; Zhang, J.; Sheng, J.; Wang, T.; et al. SRSF1 inhibits autophagy through regulating Bcl-x splicing and interacting with PIK3C3 in lung cancer. Signal Transduct. Target Ther. 2021, 6, 108. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, J.; Liu, Z.; Wu, Z.; Yang, J.; Lv, T. Comprehensive Molecular Analysis Identified an SRSF Family-Based Score for Prognosis and Therapy Efficiency Prediction in Hepatocellular Carcinoma. Cancers 2022, 14, 4727. https://doi.org/10.3390/cancers14194727
Yuan J, Liu Z, Wu Z, Yang J, Lv T. Comprehensive Molecular Analysis Identified an SRSF Family-Based Score for Prognosis and Therapy Efficiency Prediction in Hepatocellular Carcinoma. Cancers. 2022; 14(19):4727. https://doi.org/10.3390/cancers14194727
Chicago/Turabian StyleYuan, Jingsheng, Zijian Liu, Zhenru Wu, Jiayin Yang, and Tao Lv. 2022. "Comprehensive Molecular Analysis Identified an SRSF Family-Based Score for Prognosis and Therapy Efficiency Prediction in Hepatocellular Carcinoma" Cancers 14, no. 19: 4727. https://doi.org/10.3390/cancers14194727