
How Far beyond Diabetes Can the Benefits of Glucagon-like Peptide-1 Receptor Agonists Go? A Review of the Evidence on Their Effects on Hepatocellular Carcinoma

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. The Role and Effect of the Glucagon-like Peptide-1 Receptor Agonists on Liver Hepatocytes and Immune Cells
1.2. Glucagon-like Peptide-1 Receptor Agonists’ Effects on Nonalcoholic Fatty Liver Disease and Steatohepatitis
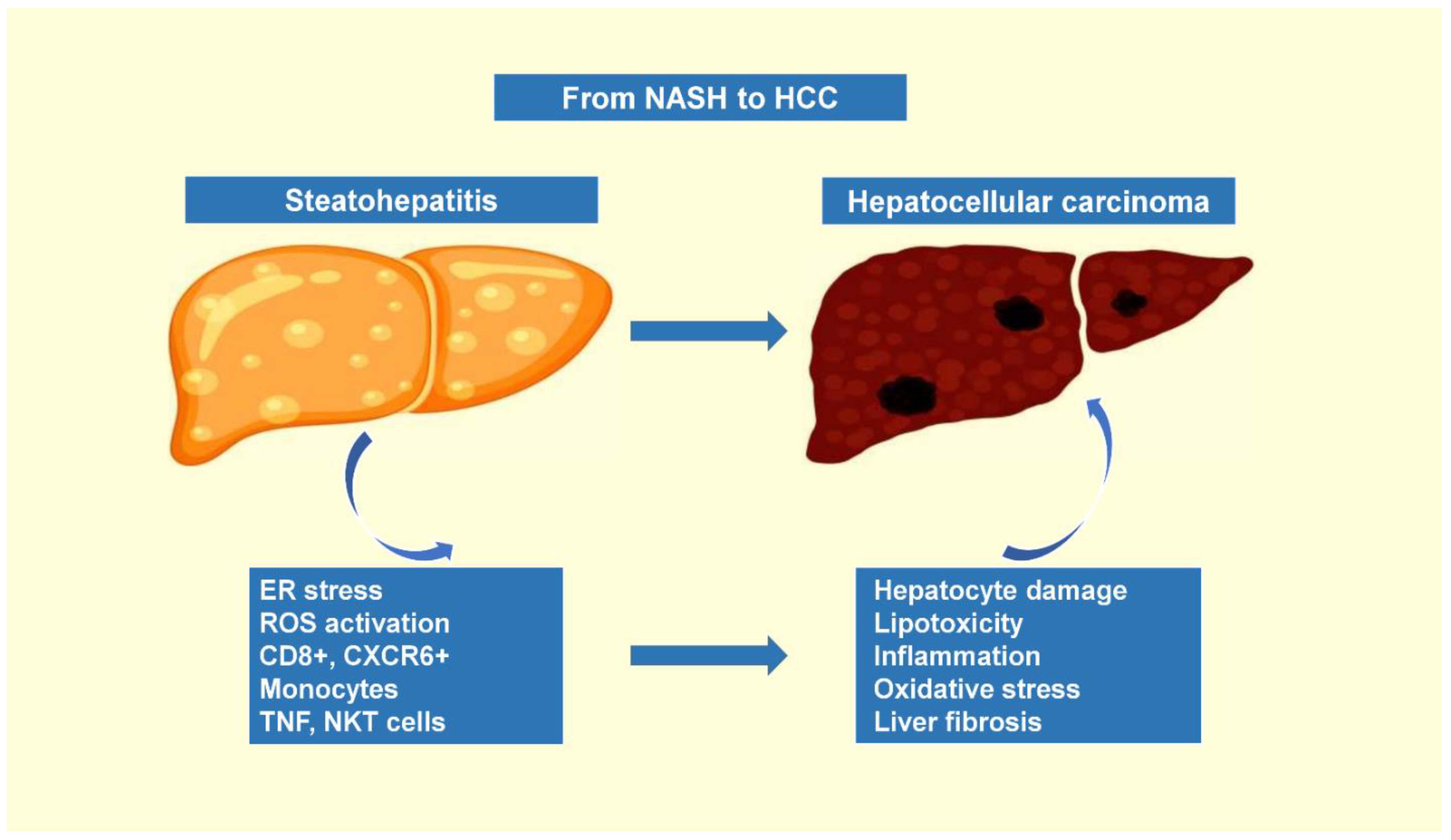
1.3. The Progression of Nonalcoholic Steatohepatitis to Hepatocellular Carcinoma
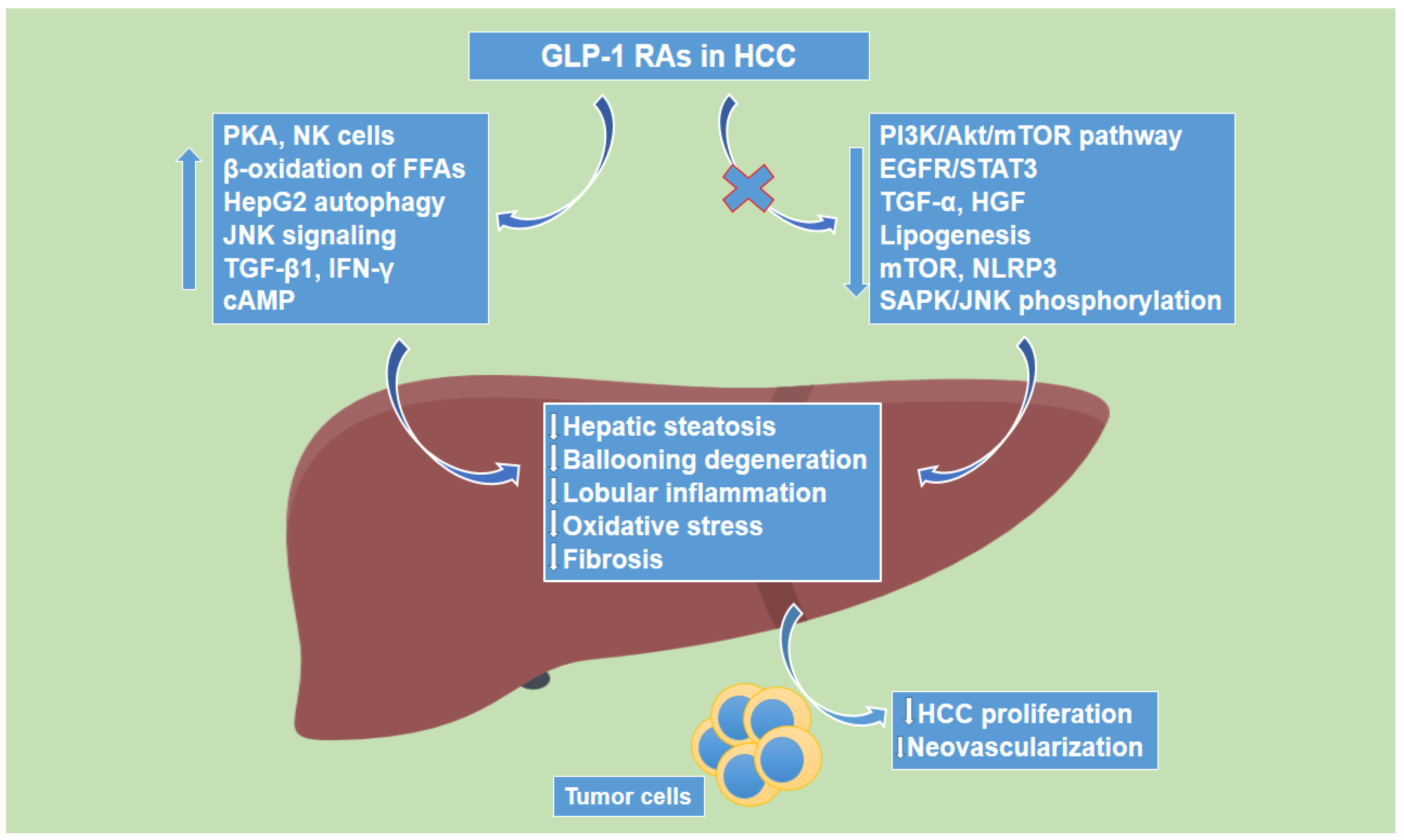
1.4. Glucagon-like Peptide-1 Receptor Agonists’ Effects on Hepatocellular Carcinoma
2. Discussion
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J.; Deacon, C.F. Glucagon-like peptide-1 mediates the therapeutic actions of DPP-IV inhibitors. Diabetologia 2005, 48, 612–615. [Google Scholar] [CrossRef]
- Alvarez, E.; Martinez, M.D.; Roncero, I.; Chowen, J.A.; Garcia-Cuartero, B.; Gispert, J.D.; Sanz, C.; Vazquez, P.; Maldonado, A.; de Caceres, J.; et al. The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. J. Neurochem. 2005, 92, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Tomas, A.; Jones, B.; Leech, C. New Insights into Beta-Cell GLP-1 Receptor and cAMP Signaling. J. Mol. Biol. 2020, 432, 1347–1366. [Google Scholar] [CrossRef]
- Bose, A.K.; Mocanu, M.M.; Carr, R.D.; Brand, C.L.; Yellon, D.M. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes 2005, 54, 146–151. [Google Scholar] [CrossRef]
- Nakatani, Y.; Maeda, M.; Matsumura, M.; Shimizu, R.; Banba, N.; Aso, Y.; Yasu, T.; Harasawa, H. Effect of GLP-1 receptor agonist on gastrointestinal tract motility and residue rates as evaluated by capsule endoscopy. Diabetes Metab. 2017, 43, 430–437. [Google Scholar] [CrossRef]
- Rehfeld, J.F.; Stadil, F. The effect of gastrin on basal- and glucose-stimulated insulin secretion in man. J. Clin. Investig. 1973, 52, 1415–1426. [Google Scholar] [CrossRef]
- Holst, J.J.; Vilsboll, T.; Deacon, C.F. The incretin system and its role in type 2 diabetes mellitus. Mol. Cell. Endocrinol. 2009, 297, 127–136. [Google Scholar] [CrossRef]
- Bagger, J.I.; Knop, F.K.; Lund, A.; Vestergaard, H.; Holst, J.J.; Vilsboll, T. Impaired regulation of the incretin effect in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2011, 96, 737–745. [Google Scholar] [CrossRef]
- Anandhakrishnan, A.; Korbonits, M. Glucagon-like peptide 1 in the pathophysiology and pharmacotherapy of clinical obesity. World J. Diabetes 2016, 7, 572–598. [Google Scholar] [CrossRef] [PubMed]
- Rentzeperi, E.; Pegiou, S.; Koufakis, T.; Grammatiki, M.; Kotsa, K. Sex Differences in Response to Treatment with Glucagon-like Peptide 1 Receptor Agonists: Opportunities for a Tailored Approach to Diabetes and Obesity Care. J. Pers. Med. 2022, 12, 454. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice Committee; Draznin, B.; Aroda, V.R.; Bakris, G.; Benson, G.; Brown, F.M.; Freeman, R.; Green, J.; Huang, E.; Isaacs, D.; et al. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45, S125–S143. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, D.; Scappaticcio, L.; Longo, M.; Caruso, P.; Maiorino, M.I.; Bellastella, G.; Ceriello, A.; Chiodini, P.; Esposito, K. GLP-1 receptor agonists and cardiorenal outcomes in type 2 diabetes: An updated meta-analysis of eight CVOTs. Cardiovasc. Diabetol. 2021, 20, 189. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.J.; Joharapurkar, A.A.; Shah, G.B.; Jain, M.R. Effect of GLP-1 based therapies on diabetic dyslipidemia. Curr. Diabetes Rev. 2014, 10, 238–250. [Google Scholar] [CrossRef]
- Cai, X.; She, M.; Xu, M.; Chen, H.; Li, J.; Chen, X.; Zheng, D.; Liu, J.; Chen, S.; Zhu, J.; et al. GLP-1 treatment protects endothelial cells from oxidative stress-induced autophagy and endothelial dysfunction. Int. J. Biol. Sci. 2018, 14, 1696–1708. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, M.; Wen, Z.; Lu, Z.; Cui, L.; Fu, C.; Xue, H.; Liu, Y.; Zhang, Y. GLP-1 Receptor Agonists: Beyond Their Pancreatic Effects. Front. Endocrinol. 2021, 12, 721135. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Li, L.; Holscher, C. Semaglutide is Neuroprotective and Reduces alpha-Synuclein Levels in the Chronic MPTP Mouse Model of Parkinson’s Disease. J. Parkinsons. Dis. 2019, 9, 157–171. [Google Scholar] [CrossRef]
- Grieco, M.; Giorgi, A.; Gentile, M.C.; d’Erme, M.; Morano, S.; Maras, B.; Filardi, T. Glucagon-like Peptide-1: A Focus on Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1112. [Google Scholar] [CrossRef]
- Marsico, F.; Paolillo, S.; Gargiulo, P.; Bruzzese, D.; Dell’Aversana, S.; Esposito, I.; Renga, F.; Esposito, L.; Marciano, C.; Dellegrottaglie, S.; et al. Effects of glucagon-like peptide-1 receptor agonists on major cardiovascular events in patients with Type 2 diabetes mellitus with or without established cardiovascular disease: A meta-analysis of randomized controlled trials. Eur. Heart J. 2020, 41, 3346–3358. [Google Scholar] [CrossRef]
- Angelidi, A.M.; Belanger, M.J.; Kokkinos, A.; Koliaki, C.C.; Mantzoros, C.S. Novel Noninvasive Approaches to the Treatment of Obesity: From Pharmacotherapy to Gene Therapy. Endocr. Rev. 2022, 43, 507–557. [Google Scholar] [CrossRef] [PubMed]
- Elkind-Hirsch, K.; Marrioneaux, O.; Bhushan, M.; Vernor, D.; Bhushan, R. Comparison of single and combined treatment with exenatide and metformin on menstrual cyclicity in overweight women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 2670–2678. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Kitajima, Y.; Hyogo, H.; Takahashi, H.; Kojima, M.; Ono, M.; Araki, N.; Tanaka, K.; Yamaguchi, M.; Matsuda, Y.; et al. Pilot study of liraglutide effects in non-alcoholic steatohepatitis and non-alcoholic fatty liver disease with glucose intolerance in Japanese patients (LEAN-J). Hepatol. Res. 2015, 45, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.P.; Sanyal, A.J.; Kowdley, K.V.; Robuck, P.R.; Hoofnagle, J.; Kleiner, D.E.; Unalp, A.; Tonascia, J.; Group, N.C.R. Pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with non-alcoholic steatohepatitis: PIVENS trial design. Contemp. Clin. Trials 2009, 30, 88–96. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Goulis, D.G.; Giouleme, O.; Germanidis, G.S.; Goulas, A. Anti-obesity Medications for the Management of Nonalcoholic Fatty Liver Disease. Curr. Obes. Rep. 2022, 11, 166–179. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Koufakis, T.; Kotsa, K.; Germanidis, G. The effects of sodium-glucose cotransporter 2 inhibitors on hepatocellular carcinoma: From molecular mechanisms to potential clinical implications. Pharmacol. Res. 2022, 181, 106261. [Google Scholar] [CrossRef]
- Wheeler, M.B.; Lu, M.I.N.G.; Dillon, J.S.; Leng, X.H.; Chen, C.H.U.A.N.; Boyd, A.E., 3rd. Functional expression of the rat glucagon-like peptide-I receptor, evidence for coupling to both adenylyl cyclase and phospholipase-C. Endocrinology 1993, 133, 57–62. [Google Scholar] [CrossRef]
- Campos, R.V.; Lee, Y.C.; Drucker, D.J. Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology 1994, 134, 2156–2164. [Google Scholar] [CrossRef]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Khalifa, O.; Al-Akl, N.S.; Errafii, K.; Arredouani, A. Exendin-4 alleviates steatosis in an in vitro cell model by lowering FABP1 and FOXA1 expression via the Wnt/-catenin signaling pathway. Sci. Rep. 2022, 12, 2226. [Google Scholar] [CrossRef] [PubMed]
- Errafii, K.; Al-Akl, N.S.; Khalifa, O.; Arredouani, A. Comprehensive analysis of LncRNAs expression profiles in an in vitro model of steatosis treated with Exendin-4. J. Transl. Med. 2021, 19, 235. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Xu, H.; Liu, J.; Zhao, Q.; Chen, H.; Yan, Z.; Yang, R.; Luo, Z.; Liu, Q.; Ouyang, J.; et al. Design of a highly potent GLP-1R and GCGR dual-agonist for recovering hepatic fibrosis. Acta Pharm. Sin. B 2022, 12, 2443–2461. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Ji, L.; Zhu, C.; Xiao, Y.; Zhang, J.; Lu, J.; Yin, J.; Wei, L. Liraglutide Alleviates Hepatic Steatosis by Activating the TFEB-Regulated Autophagy-Lysosomal Pathway. Front. Cell Dev. Biol. 2020, 8, 602574. [Google Scholar] [CrossRef]
- Lyu, J.; Imachi, H.; Fukunaga, K.; Sato, S.; Kobayashi, T.; Dong, T.; Saheki, T.; Matsumoto, M.; Iwama, H.; Zhang, H.; et al. Role of ATP-binding cassette transporter A1 in suppressing lipid accumulation by glucagon-like peptide-1 agonist in hepatocytes. Mol. Metab. 2020, 34, 16–26. [Google Scholar] [CrossRef]
- Kalavalapalli, S.; Bril, F.; Guingab, J.; Vergara, A.; Garrett, T.J.; Sunny, N.E.; Cusi, K. Impact of exenatide on mitochondrial lipid metabolism in mice with nonalcoholic steatohepatitis. J. Endocrinol. 2019, 241, 293–305. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef]
- Yokomori, H.; Ando, W. Spatial expression of glucagon-like peptide 1 receptor and caveolin-1 in hepatocytes with macrovesicular steatosis in non-alcoholic steatohepatitis. BMJ Open Gastroenterol. 2020, 7, e000370. [Google Scholar] [CrossRef]
- Zhan, J.K.; Tan, P.; Wang, Y.J.; Wang, Y.; He, J.Y.; Tang, Z.Y.; Huang, W.; Liu, Y.S. Exenatide can inhibit calcification of human VSMCs through the NF-kappaB/RANKL signaling pathway. Cardiovasc. Diabetol. 2014, 13, 153. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Ong, J.; Trimble, G.; AlQahtani, S.; Younossi, I.; Ahmed, A.; Racila, A.; Henry, L. Nonalcoholic Steatohepatitis Is the Most Rapidly Increasing Indication for Liver Transplantation in the United States. Clin. Gastroenterol. Hepatol. 2021, 19, 580–589.e5. [Google Scholar] [CrossRef]
- Nakamura, J.; Kamiya, H.; Haneda, M.; Inagaki, N.; Tanizawa, Y.; Araki, E.; Ueki, K.; Nakayama, T. Causes of death in Japanese patients with diabetes based on the results of a survey of 45,708 cases during 2001-2010: Report of the Committee on Causes of Death in Diabetes Mellitus. J. Diabetes Investig. 2017, 8, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef]
- Zhu, Y.; Xu, J.; Zhang, D.; Mu, X.; Shi, Y.; Chen, S.; Wu, Z.; Li, S. Efficacy and Safety of GLP-1 Receptor Agonists in Patients With Type 2 Diabetes Mellitus and Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2021, 12, 769069. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Rolin, B.; Rakipovski, G.; Skovsted, G.F.; Madsen, A.; Kolstrup, S.; Schou-Pedersen, A.M.; Skat-Rordam, J.; Lykkesfeldt, J.; Tveden-Nyborg, P. Liraglutide Decreases Hepatic Inflammation and Injury in Advanced Lean Non-Alcoholic Steatohepatitis. Basic. Clin. Pharmacol. Toxicol. 2018, 123, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Hao, M.; Liu, Y.; Ma, X.; Lin, W.; Xu, Q.; Zhou, H.; Shao, N.; Kuang, H. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 2019, 864, 172715. [Google Scholar] [CrossRef]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Csermely, A.; Lonardo, A.; Targher, G. Glucagon-like Peptide-1 Receptor Agonists for Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: An Updated Meta-Analysis of Randomized Controlled Trials. Metabolites 2021, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Lv, Q.; Li, S.; Wu, Y.; Li, L.; Li, J.; Zhang, F.; Sun, X.; Tong, N. Efficacy and safety of glucagon-like peptide-1 receptor agonists in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 284–295. [Google Scholar] [CrossRef]
- Rezaei, S.; Tabrizi, R.; Nowrouzi-Sohrabi, P.; Jalali, M.; Atkin, S.L.; Al-Rasadi, K.; Jamialahmadi, T.; Sahebkar, A. GLP-1 Receptor Agonist Effects on Lipid and Liver Profiles in Patients with Nonalcoholic Fatty Liver Disease: Systematic Review and Meta-Analysis. Can. J. Gastroenterol. Hepatol. 2021, 2021, 8936865. [Google Scholar] [CrossRef]
- Dai, Y.; He, H.; Li, S.; Yang, L.; Wang, X.; Liu, Z.; An, Z. Comparison of the Efficacy of Glucagon-like Peptide-1 Receptor Agonists in Patients With Metabolic Associated Fatty Liver Disease: Updated Systematic Review and Meta-Analysis. Front. Endocrinol. 2020, 11, 622589. [Google Scholar] [CrossRef]
- Petroni, M.L.; Brodosi, L.; Bugianesi, E.; Marchesini, G. Management of non-alcoholic fatty liver disease. BMJ 2021, 372, m4747. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Kucukoglu, O.; Sowa, J.P.; Mazzolini, G.D.; Syn, W.K.; Canbay, A. Hepatokines and adipokines in NASH-related hepatocellular carcinoma. J. Hepatol. 2021, 74, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Huby, T.; Gautier, E.L. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat. Rev. Immunol. 2022, 22, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Nunez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Fujiwara, N.; Kubota, N.; Crouchet, E.; Koneru, B.; Marquez, C.A.; Jajoriya, A.K.; Panda, G.; Qian, T.; Zhu, S.; Goossens, N.; et al. Molecular signatures of long-term hepatocellular carcinoma risk in nonalcoholic fatty liver disease. Sci. Transl. Med. 2022, 14, eabo4474. [Google Scholar] [CrossRef]
- Zhou, M.; Mok, M.T.; Sun, H.; Chan, A.W.; Huang, Y.; Cheng, A.S.; Xu, G. The anti-diabetic drug exenatide, a glucagon-like peptide-1 receptor agonist, counteracts hepatocarcinogenesis through cAMP-PKA-EGFR-STAT3 axis. Oncogene 2017, 36, 4135–4149. [Google Scholar] [CrossRef]
- Chen-Liaw, A.Y.; Hammel, G.; Gomez, G. Inhibition of exendin-4-induced steatosis by protein kinase A in cultured HepG2 human hepatoma cells. In Vitro Cell. Dev. Biol. Anim. 2017, 53, 721–727. [Google Scholar] [CrossRef]
- Krause, G.C.; Lima, K.G.; Levorse, V.; Haute, G.V.; Gassen, R.B.; Garcia, M.C.; Pedrazza, L.; Donadio, M.V.F.; Luft, C.; de Oliveira, J.R. Exenatide induces autophagy and prevents the cell regrowth in HepG2 cells. EXCLI J. 2019, 18, 540–548. [Google Scholar] [CrossRef]
- Yamada, N.; Matsushima-Nishiwaki, R.; Kobayashi, K.; Tachi, J.; Kozawa, O. GLP-1 reduces the migration of hepatocellular carcinoma cells via suppression of the stress-activated protein kinase/c-Jun N-terminal kinase pathway. Arch. Biochem. Biophys. 2021, 703, 108851. [Google Scholar] [CrossRef]
- Li, Q.; Xue, A.Y.; Li, Z.L.; Yin, Z. Liraglutide promotes apoptosis of HepG2 cells by activating JNK signaling pathway. Eur Rev. Med. Pharmacol. Sci. 2019, 23, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.C.; Lima, K.G.; Dias, H.B.; da Silva, E.F.G.; Haute, G.V.; Basso, B.S.; Gassen, R.B.; Marczak, E.S.; Nunes, R.S.B.; de Oliveira, J.R. Liraglutide, a glucagon-like peptide-1 analog, induce autophagy and senescence in HepG2 cells. Eur. J. Pharmacol. 2017, 809, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Takahashi, H.; Kuwashiro, T.; Tanaka, K.; Mori, H.; Ozaki, I.; Kitajima, Y.; Matsuda, Y.; Ashida, K.; Eguchi, Y.; et al. Glucagon-like Peptide-1 Receptor Agonist Prevented the Progression of Hepatocellular Carcinoma in a Mouse Model of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 5722. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xu, C.; Dong, J.; Zuo, S.; Zhang, H.; Jiang, C.; Wu, J.; Wei, J. Liraglutide activates nature killer cell-mediated antitumor responses by inhibiting IL-6/STAT3 signaling. in hepatocellular carcinoma. Transl. Oncol. 2021, 14, 100872. [Google Scholar] [CrossRef] [PubMed]
- Tsilidis, K.K.; Kasimis, J.C.; Lopez, D.S.; Ntzani, E.E.; Ioannidis, J.P. Type 2 diabetes and cancer: Umbrella review of meta-analyses of observational studies. BMJ 2015, 350, g7607. [Google Scholar] [CrossRef]
- Shlomai, G.; Neel, B.; LeRoith, D.; Gallagher, E.J. Type 2 Diabetes Mellitus and Cancer: The Role of Pharmacotherapy. J. Clin. Oncol. 2016, 34, 4261–4269. [Google Scholar] [CrossRef]
- Pearson-Stuttard, J.; Papadimitriou, N.; Markozannes, G.; Cividini, S.; Kakourou, A.; Gill, D.; Rizos, E.C.; Monori, G.; Ward, H.A.; Kyrgiou, M.; et al. Type 2 Diabetes and Cancer: An Umbrella Review of Observational and Mendelian Randomization Studies. Cancer Epidemiol. Biomarkers Prev. 2021, 30, 1218–1228. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Kim, D.; Konyn, P.; Sandhu, K.K.; Dennis, B.B.; Cheung, A.C.; Ahmed, A. Metabolic dysfunction-associated fatty liver disease is associated with increased all-cause mortality in the United States. J. Hepatol. 2021, 75, 1284–1291. [Google Scholar] [CrossRef]
- Bjerre Knudsen, L.; Madsen, L.W.; Andersen, S.; Almholt, K.; de Boer, A.S.; Drucker, D.J.; Gotfredsen, C.; Egerod, F.L.; Hegelund, A.C.; Jacobsen, H.; et al. Glucagon-like Peptide-1 receptor agonists activate rodent thyroid C-cells causing calcitonin release and C-cell proliferation. Endocrinology 2010, 151, 1473–1486. [Google Scholar] [CrossRef] [PubMed]
- Gier, B.; Butler, P.C.; Lai, C.K.; Kirakossian, D.; DeNicola, M.M.; Yeh, M.W. Glucagon like peptide-1 receptor expression in the human thyroid gland. J. Clin. Endocrinol. Metab. 2012, 97, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Mali, G.; Ahuja, V.; Dubey, K. Glucagon-like peptide-1 analogues and thyroid cancer: An analysis of cases reported in the European pharmacovigilance database. J. Clin. Pharm. Ther. 2021, 46, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Knapen, L.M.; van Dalem, J.; Keulemans, Y.C.; van Erp, N.P.; Bazelier, M.T.; De Bruin, M.L.; Leufkens, H.G.; Croes, S.; Neef, C.; de Vries, F.; et al. Use of incretin agents and risk of pancreatic cancer: A population-based cohort study. Diabetes Obes. Metab. 2016, 18, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef]
- Cao, C.; Yang, S.; Zhou, Z. GLP-1 receptor agonists and pancreatic safety concerns in type 2 diabetic patients: Data from cardiovascular outcome trials. Endocrine 2020, 68, 518–525. [Google Scholar] [CrossRef]
- Iwaya, C.; Nomiyama, T.; Komatsu, S.; Kawanami, T.; Tsutsumi, Y.; Hamaguchi, Y.; Horikawa, T.; Yoshinaga, Y.; Yamashita, S.; Tanaka, T.; et al. Exendin-4, a Glucagonlike Peptide-1 Receptor Agonist, Attenuates Breast Cancer Growth by Inhibiting NF-kappaB Activation. Endocrinology 2017, 158, 4218–4232. [Google Scholar] [CrossRef]
- Nomiyama, T.; Kawanami, T.; Irie, S.; Hamaguchi, Y.; Terawaki, Y.; Murase, K.; Tsutsumi, Y.; Nagaishi, R.; Tanabe, M.; Morinaga, H.; et al. Exendin-4, a GLP-1 receptor agonist, attenuates prostate cancer growth. Diabetes 2014, 63, 3891–3905. [Google Scholar] [CrossRef]
- Natarajan, K.; Xie, Y.; Baer, M.R.; Ross, D.D. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem. Pharmacol. 2012, 83, 1084–1103. [Google Scholar] [CrossRef]
- Kanda, R.; Hiraike, H.; Wada-Hiraike, O.; Ichinose, T.; Nagasaka, K.; Sasajima, Y.; Ryo, E.; Fujii, T.; Osuga, Y.; Ayabe, T. Expression of the glucagon-like peptide-1 receptor and its role in regulating autophagy in endometrial cancer. BMC Cancer 2018, 18, 657. [Google Scholar] [CrossRef] [Green Version]
- Wenjing, H.; Shao, Y.; Yu, Y.; Huang, W.; Feng, G.; Li, J. Exendin-4 enhances the sensitivity of prostate cancer to enzalutamide by targeting Akt activation. Prostate 2020, 80, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Eftekhari, S.; Montazeri, H.; Tarighi, P. Synergistic anti-tumor effects of Liraglutide, a glucagon-like peptide-1 receptor agonist, along with Docetaxel on LNCaP prostate cancer cell line. Eur. J. Pharmacol. 2020, 878, 173102. [Google Scholar] [CrossRef] [PubMed]
- Flint, A.; Nazzal, K.; Jagielski, P.; Hindsberger, C.; Zdravkovic, M. Influence of hepatic impairment on pharmacokinetics of the human GLP-1 analogue, liraglutide. Br. J. Clin. Pharmacol. 2010, 70, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Patorno, E.; Schneeweiss, S. Glucagon-like Peptide-1 Receptor Agonists and Hepatic Decompensation Events in Patients With Cirrhosis and Diabetes. Clin. Gastroenterol. Hepatol. 2022, 20, 1382–1393.e19. [Google Scholar] [CrossRef]
- Patel Chavez, C.; Cusi, K.; Kadiyala, S. The Emerging Role of Glucagon-like Peptide-1 Receptor Agonists for the Management of NAFLD. J. Clin. Endocrinol. Metab. 2022, 107, 29–38. [Google Scholar] [CrossRef]
- Malik, I.O.; Petersen, M.C.; Klein, S. Glucagon-like peptide-1, glucose-dependent insulinotropic polypeptide, and glucagon receptor poly-agonists: A new era in obesity pharmacotherapy. Obesity 2022, 30, 1718–1721. [Google Scholar] [CrossRef]
- Frías, J.P.; Davies, M.J.; Rosenstock, J.; Pérez Manghi, F.C.; Fernández Landó, L.; Bergman, B.K.; Liu, B.; Cui, X.; Brown, K. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Bettge, K.; Kahle, M.; Abd El Aziz, M.S.; Meier, J.J.; Nauck, M.A. Occurrence of nausea, vomiting and diarrhoea reported as adverse events in clinical trials studying glucagon-like peptide-1 receptor agonists: A systematic analysis of published clinical trials. Diabetes Obes. Metab. 2017, 19, 336–347. [Google Scholar] [CrossRef]
- Wharton, S.; Davies, M.; Dicker, D.; Lingvay, I.; Mosenzon, O.; Rubino, D.M.; Pedersen, S.D. Managing the gastrointestinal side effects of GLP-1 receptor agonists in obesity: Recommendations for clinical practice. Postgrad. Med. 2022, 134, 14–19. [Google Scholar] [CrossRef]
- Pratley, R.; Amod, A.; Hoff, S.T.; Kadowaki, T.; Lingvay, I.; Nauck, M.; Pedersen, K.B.; Saugstrup, T.; Meier, J.J. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): A randomised, double-blind, phase 3a trial. Lancet 2019, 394, 39–50. [Google Scholar] [CrossRef]
- Trujillo, J.M.; Nuffer, W.; Smith, B.A. GLP-1 receptor agonists: An updated review of head-to-head clinical studies. Ther. Adv. Endocrinol. Metab. 2021, 12, 2042018821997320. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidou, A.; Kyriazou, A.V.; Koufakis, T.; Vasilopoulos, Y.; Grammatiki, M.; Tsekmekidou, X.; Avramidis, I.; Baltagiannis, S.; Goulis, D.G.; Zebekakis, P.; et al. Clinical and Genetic Predictors of Glycemic Control and Weight Loss Response to Liraglutide in Patients with Type 2 Diabetes. J. Pers. Med. 2022, 12, 424. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidou, A.; Koufakis, T.; Goulis, D.G.; Vasilopoulos, Y.; Zebekakis, P.; Kotsa, K. Pharmacogenetics of the Glucagon-like Peptide-1 Receptor Agonist Liraglutide: A Step Towards Personalized Type 2 Diabetes Management. Curr. Pharm. Des. 2021, 27, 1025–1034. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study (Year) | Primary Outcome | Secondary Outcome |
|---|---|---|
| Zhu et al. [43] (2021) | GLP-1 RAs reduce intrahepatic adipose tissue, weight mean difference, subcutaneous adipose tissue and visceral adipose tissue | GLP-1 RAs decrease the levels of ALT, AST, body weight, body mass index, waist circumference, fasting blood glucose, HbA1c, TC and TG |
| Armstrong [44] (2016) | Liraglutide leads to histological resolution of non-alcoholic steatohepatitis without the worsening of fibrosis | Liraglutide treatment is associated with reductions in body weight, BMI and HbA1c |
| Eguchi et al. [23] (2015) | Liraglutide decreases liver fat deposition, BMI, visceral fat accumulation, AST, ALT, γGT, HbA1c and FPG | Liraglutide reduces the histological features of steatohepatitis, fibrotic stage and NAS score |
| Ipsen et al. [45] (2018) | Liraglutide reduces NASH progression by diminishing hepatocyte inflammation and ballooning, while it increases hepatic α-tocopherol | Combined liraglutide and chow diet decreases liver weight, TC, LDL-C, VLDL-C and hepatic cholesterol, and increases hepatic vitamin C |
| Yu et al. [46] (2019) | Liraglutide ameliorates NASH by inhibiting NOD-, LRR- and NLRP3-inflammasome and pyroptosis activation via mitophagy | Mitophagy inhibition with 3-methyladenine/PINK1-directed siRNA weakens the liraglutide-mediated suppression of inflammatory injury |
| Mantovani et al. [47] (2021) | GLP-1 RAs reduce the absolute percentage of liver fat content and serum liver enzyme levels and leads to histological resolution of NASH without the worsening of liver fibrosis | Treatment with GLP-1 RAs is associated with significant reductions in body weight HbA1c levels |
| Dong et al. [48] (2017) | GLP-1 RA therapy reduces liver histology scores for steatosis, lobular inflammation, hepatocellular ballooning and fibrosis | GLP-1 RAs treatment significantly reduces the levels of γGT |
| Rezaei et al. [49] (2021) | GLP-1 RAs therapy reduces ALT, γGT and ALP concentrations | GLP-1 therapy does not alter TG, TC, HDL-C and LDL-C concentrations |
| Dai et al. [50] (2020) | GLP-1 RAs treatment reduces the liver fat content body weight, waist circumference, ALT and γGT | GLP-1 RAs therapy reduces fasting blood glucose and HbA1c |
| Study (Year) | Primary Outcome | Secondary Outcome |
|---|---|---|
| Zhou et al. [57] (2017) | Ex-4 inhibits obesity-dependent and -independent hepatocarcinogenesis, downregulating EGFR-STAT3 signaling in dose- and time-dependent manners | PKA and EGFR signaling potentiates the tumor-suppressing effect of Ex-4, while GLP-1R expression is enhanced in HCC |
| Chen-Liaw [58] (2017) | Ex-4 causes partial reduction in triglycerides in steatotic hepatocytes via GLP-1R-mediated activation of protein kinase A | The reduction in hepatocyte triglyceride content is mediated by downregulation of lipogenesis and upregulation of β-oxidation of FFAs |
| Krause et al. [59] (2019) | Ex-4 induces autophagy and prevents HepG2 cell regrowth via the modulation of mTOR signaling in HCC | Ex-4 decreases HepG2 cells viability and inhibits mTOR expression in a more significant way than liraglutide |
| Yamada et al. [60] (2021) | GLP-1 attenuates the phosphorylation of SAPK/JNK by TGF-α and HGF in HCC | GLP-1 suppresses both TGF-α- and HGF-induced migration of HuH7 cells |
| Li et al. [61] (2019) | Liraglutide promotes apoptosis of HCC HepG2 cells by activating the JNK signaling pathway | The proliferation inhibition rate of HepG2 cells increases with time and with the increase in the concentration of liraglutide |
| Krause et al. [62] (2017) | Liraglutide inhibits cell proliferation in HepG2 HCC cells and induces their autophagy via the inhibition of the PI3K/Akt/mTOR pathway | Liraglutide induces cell cycle arrest and senescence, significantly increasing TGF-β production of HepG2 cells |
| Kojima et al. [63] (2020) | Liraglutide ameliorates NASH and suppresses HCC formation in diabetic mice | Liraglutide ameliorates steatosis, inflammation, and hepatocyte ballooning of non-tumorous lesions in the liver |
| Lu et al. [64] (2021) | Liraglutide enhances NK cell-mediated oncolytic activity by suppressing the IL-6/STAT3 signaling pathway in HCC | Liraglutide increases IFN-γ-producing cells in HCC, activating antitumor immunity both in vivo and in vitro |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arvanitakis, K.; Koufakis, T.; Kotsa, K.; Germanidis, G. How Far beyond Diabetes Can the Benefits of Glucagon-like Peptide-1 Receptor Agonists Go? A Review of the Evidence on Their Effects on Hepatocellular Carcinoma. Cancers 2022, 14, 4651. https://doi.org/10.3390/cancers14194651
Arvanitakis K, Koufakis T, Kotsa K, Germanidis G. How Far beyond Diabetes Can the Benefits of Glucagon-like Peptide-1 Receptor Agonists Go? A Review of the Evidence on Their Effects on Hepatocellular Carcinoma. Cancers. 2022; 14(19):4651. https://doi.org/10.3390/cancers14194651
Chicago/Turabian StyleArvanitakis, Konstantinos, Theocharis Koufakis, Kalliopi Kotsa, and Georgios Germanidis. 2022. "How Far beyond Diabetes Can the Benefits of Glucagon-like Peptide-1 Receptor Agonists Go? A Review of the Evidence on Their Effects on Hepatocellular Carcinoma" Cancers 14, no. 19: 4651. https://doi.org/10.3390/cancers14194651