

Targeting Microtubule-Associated Protein Tau in Chemotherapy-Resistant Models of High-Grade Serous Ovarian Carcinoma

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Mice
2.3. Antibodies and Reagents
2.4. Immunofluorescence Staining and Imaging
2.5. Duolink Proximity Ligation Assay
2.6. ELISA
2.7. Spheroid Formation
2.8. Western Blot
2.9. Cell Proliferation
2.10. Transwell Cell Migration
2.11. Proteome Profiler Human Phospho-Kinase Array
2.12. Tumor Formation and Treatments
2.13. Statistics
3. Results
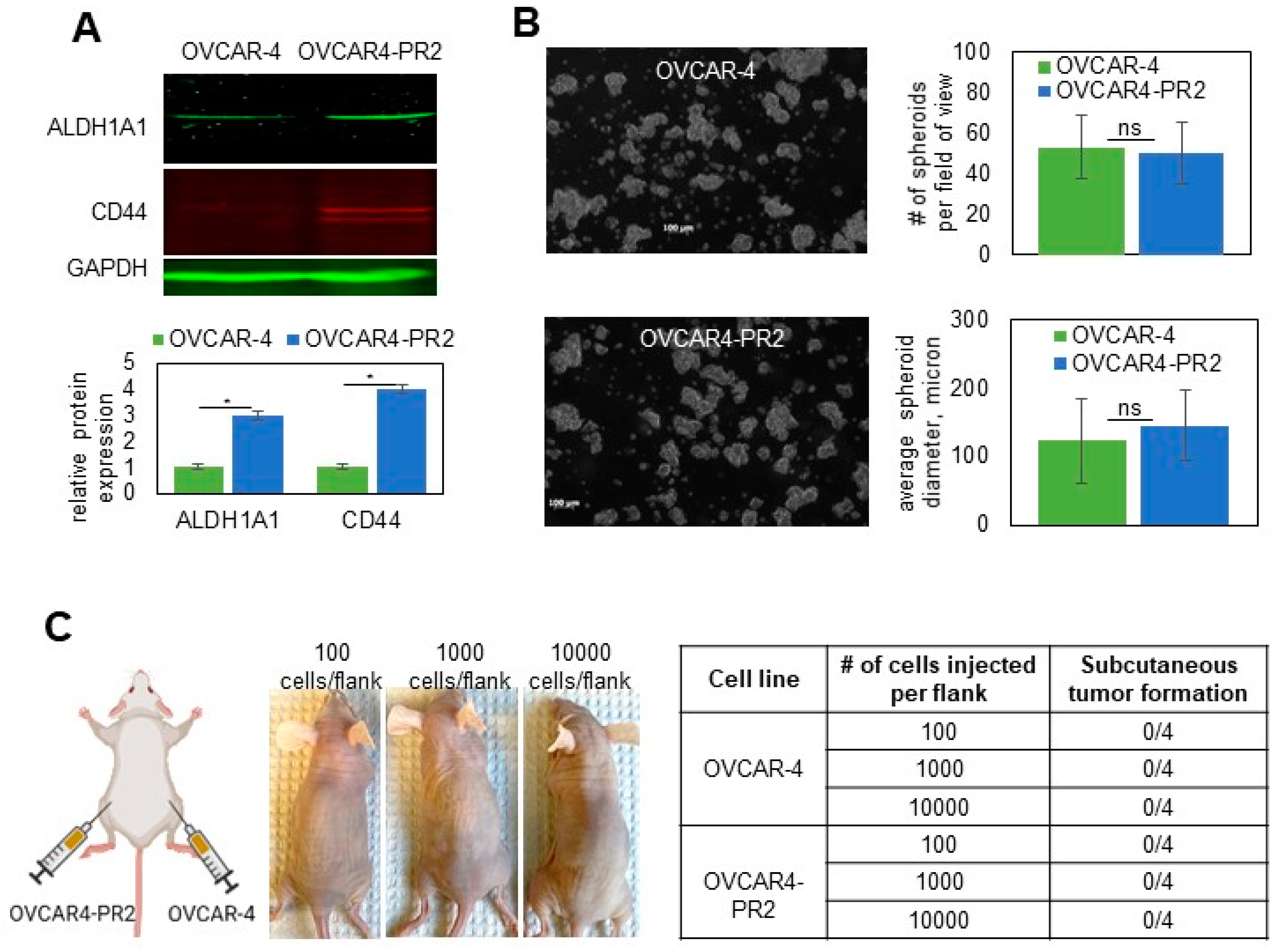
3.1. Generation of Paclitaxel-Resistant Cell Lines
3.2. Paclitaxel Resistance in Selected Subclones Is Accompanied by Upregulation of Tau
3.3. Targeting Tau with Leucomethylene Blue Is Effective in Chemotherapy-Resistant Models
3.4. A Combined Treatment with TRx0237 and Paclitaxel Results in Elimination of Tumor Burden in the Paclitaxel-Resistant Model OVCAR4-PR2
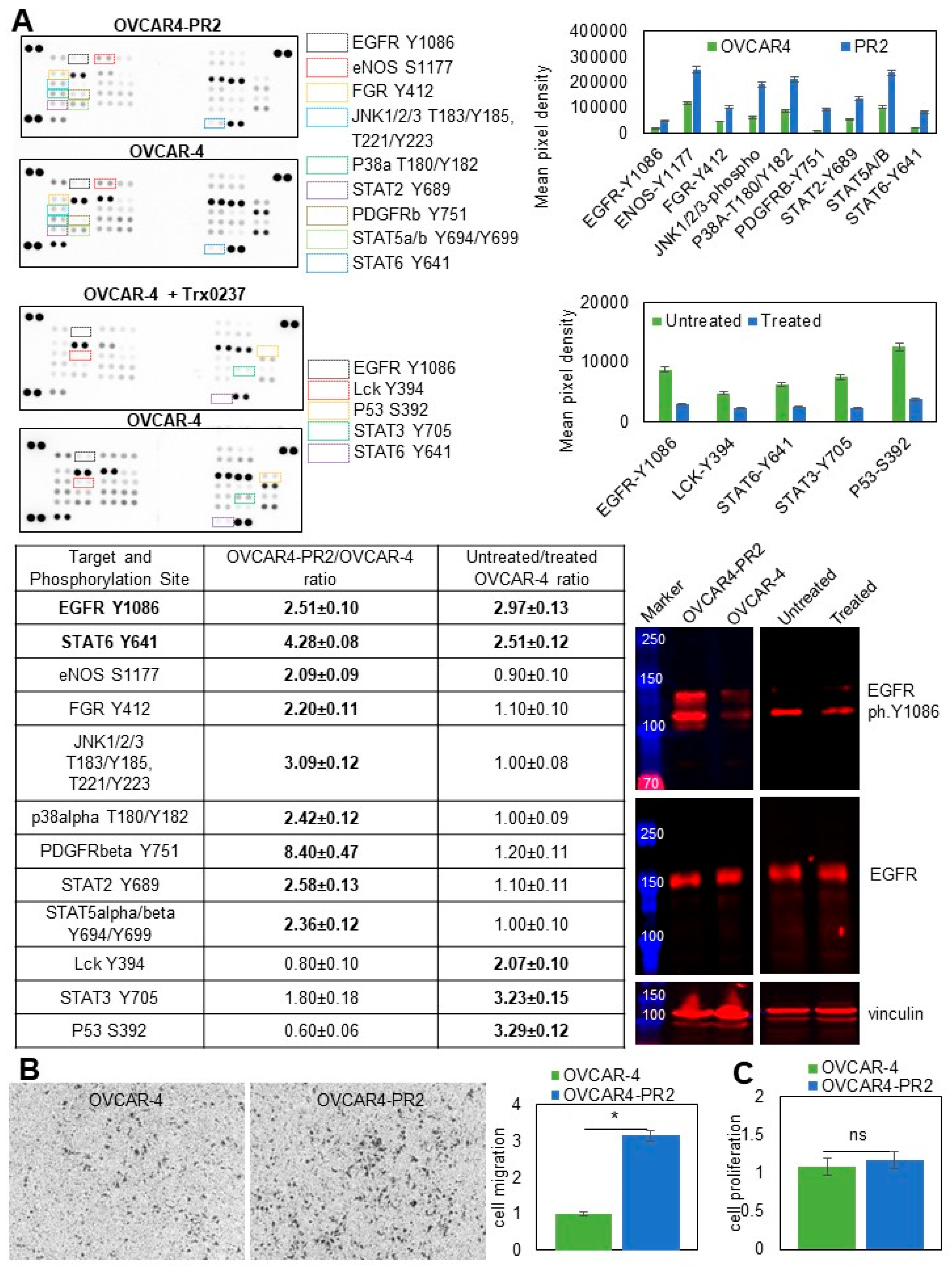
3.5. Mechanisms Regulating Drug Resistance Are Activated in Paclitaxel-Resistant Ovarian Cancer Cells
3.6. Examination of Pathways Dysregulated in OVCAR-4 after Acquisition of Paclitaxel Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Karnezis, A.N.; Cho, K.R. Of mice and women—Non-ovarian origins of “ovarian” cancer. Gynecol. Oncol. 2017, 144, 5–7. [Google Scholar] [CrossRef]
- Karnezis, A.N.; Cho, K.; Gilks, C.B.; Pearce, C.L.; Huntsman, D.G. The disparate origins of ovarian cancers: Pathogenesis and prevention strategies. Nat. Cancer 2016, 17, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef]
- Singh, N.; McCluggage, W.G.; Gilks, C.B. High-grade serous carcinoma of tubo-ovarian origin: Recent developments. Histopathology 2017, 71, 339–356. [Google Scholar] [CrossRef]
- Herzog, T.J. Update on the role of topotecan in the treatment of recurrent ovarian cancer. Oncologist 2002, 7 (Suppl. S5), 3–10. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.N.; Fleagle, J.T.; Guthrie, D.; Parkin, D.E.; Gore, M.E.; Lacave, A.J. Recurrent epithelial ovarian carcinoma: A randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J. Clin. Oncol. 2001, 19, 3312–3322. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sill, M.W.; Makker, V.; Mutch, D.G.; Carlson, J.W.; Darus, C.J.; Mannel, R.S.; Bender, D.P.; Crane, E.K.; Aghajanian, C. A randomized phase II study of cabozantinib versus weekly paclitaxel in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2019, 152, 548–553. [Google Scholar] [CrossRef]
- Obermair, A.; Beale, P.; Scott, C.L.; Beshay, V.; Kichenadasse, G.; Simcock, B.; Nicklin, J.; Lee, Y.C.; Cohen, P.; Meniawy, T. Insights into ovarian cancer care: Report from the ANZGOG Ovarian Cancer Webinar Series 2020. J. Gynecol. Oncol. 2021, 32, e95. [Google Scholar] [CrossRef]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Huang, D.; Savage, S.R.; Calinawan, A.P.; Lin, C.; Zhang, B.; Wang, P.; Starr, T.K.; Birrer, M.J.; Paulovich, A.G. A highly annotated database of genes associated with platinum resistance in cancer. Oncogene 2021, 40, 6395–6405. [Google Scholar] [CrossRef]
- Mora, Y.; Reyes, M.E.; Zanella, L.; Mora, B.; Buchegger, K.; Ili, C.; Brebi, P. Resistance to platinum-based cancer drugs: A special focus on epigenetic mechanisms. Pharmacogenomics 2021, 22, 777–790. [Google Scholar] [CrossRef]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Zhang, Q.-Y.; Zheng, G.-J.; Feng, B. Phytochemicals: Current strategy to sensitize cancer cells to cisplatin. Biomed. Pharmacother. 2018, 110, 518–527. [Google Scholar] [CrossRef]
- Lipinska, N.; Romaniuk-Drapała, A.; Paszel-Jaworska, A.; Totoń, E.; Kopczyński, P.; Rubis, B. Telomerase and drug resistance in cancer. Cell. Mol. Life Sci. 2017, 74, 4121–4132. [Google Scholar] [CrossRef]
- Stojanovska, V.; McQuade, R.; Rybalka, E.; Nurgali, K. Neurotoxicity Associated with Platinum-Based Anti-Cancer Agents: What are the Implications of Copper Transporters? Curr. Med. Chem. 2017, 24, 1520–1536. [Google Scholar] [CrossRef]
- Gallego-Jara, J.; Lozano-Terol, G.; Sola-Martínez, R.A.; Cánovas-Díaz, M.; de Diego Puente, T. A Compressive Review about Taxol((R)): History and Future Challenges. Molecules 2020, 25, 5986. [Google Scholar] [CrossRef]
- Bodakuntla, S.; Jijumon, A.; Villablanca, C.; Gonzalez-Billault, C.; Janke, C. Microtubule-Associated Proteins: Structuring the Cytoskeleton. Trends Cell Biol. 2019, 29, 804–819. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef]
- Devred, F.; Barbier, P.; Douillard, S.; Monasterio, O.; Andreu, J.M.; Peyrot, V. Tau induces ring and microtubule formation from alphabeta-tubulin dimers under nonassembly conditions. Biochemistry 2004, 43, 10520–10531. [Google Scholar] [CrossRef]
- Dugger, B.N.; Whiteside, C.M.; Maarouf, C.L.; Walker, D.G.; Beach, T.G.; Sue, L.I.; Garcia, A.; Dunckley, T.; Meechoovet, B.; Reiman, E.M.; et al. The Presence of Select Tau Species in Human Peripheral Tissues and Their Relation to Alzheimer’s Disease. J. Alzheimers Dis. 2016, 51, 345–356. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Crowther, R.A. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 1983–1987. [Google Scholar] [CrossRef]
- Biswas, S.; Kalil, K. The Microtubule-Associated Protein Tau Mediates the Organization of Microtubules and Their Dy-namic Exploration of Actin-Rich Lamellipodia and Filopodia of Cortical Growth Cones. J. Neurosci. 2018, 38, 291–307. [Google Scholar] [CrossRef]
- Qiang, L.; Sun, X.; Austin, T.O.; Muralidharan, H.; Jean, D.C.; Liu, M.; Yu, W.; Baas, P.W. Tau Does Not Stabilize Axonal Microtubules but Rather Enables Them to Have Long Labile Domains. Curr. Biol. 2018, 28, 2181–2189.e4. [Google Scholar] [CrossRef]
- Pagano, A.; Breuzard, G.; Parat, F.; Tchoghandjian, A.; Figarella-Branger, D.; De Bessa, T.C.; Garrouste, F.; Douence, A.; Barbier, P.; Kovacic, H. Tau Regulates Glioblastoma Progression, 3D Cell Organization, Growth and Migration via the PI3K-AKT Axis. Cancers 2021, 13, 5818. [Google Scholar] [CrossRef]
- Smoter, M.; Bodnar, L.; Grala, B.; Stec, R.; Zieniuk, K.; Kozlowski, W.; Szczylik, C. Tau protein as a potential predictive marker in epithelial ovarian cancer patients treated with paclitax-el/platinum first-line chemotherapy. J. Exp. Clin. Cancer Res. 2013, 32, 25. [Google Scholar] [CrossRef]
- Rouzier, R.; Rajan, R.; Wagner, P.; Hess, K.R.; Gold, D.L.; Stec, J.; Ayers, M.; Ross, J.S.; Zhang, P.; Buchholz, T.A.; et al. Microtubule-associated protein tau: A marker of paclitaxel sensitivity in breast cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 8315–8320. [Google Scholar] [CrossRef]
- Tanaka, S.; Nohara, T.; Iwamoto, M.; Sumiyoshi, K.; Kimura, K.; Takahashi, Y.; Tanigawa, N. Tau expression and efficacy of paclitaxel treatment in metastatic breast cancer. Cancer Chemother. Pharmacol. 2008, 64, 341–346. [Google Scholar] [CrossRef]
- Shao, Y.-Y.; Kuo, K.-T.; Hu, F.-C.; Lu, Y.-S.; Huang, C.-S.; Liau, J.-Y.; Lee, W.-C.; Hsu, C.; Kuo, W.-H.; Chang, K.-J.; et al. Predictive and Prognostic Values of Tau and ERCC1 in Advanced Breast Cancer Patients Treated with Paclitaxel and Cisplatin. Jpn. J. Clin. Oncol. 2010, 40, 286–293. [Google Scholar] [CrossRef]
- Wang, K.; Deng, Q.-T.; Liao, N.; Zhang, G.-C.; Liu, Y.-H.; Xu, F.-P.; Zu, J.; Li, X.-R.; Wu, Y.-L. Tau expression correlated with breast cancer sensitivity to taxanes-based neoadjuvant chemotherapy. Tumor Biol. 2012, 34, 33–38. [Google Scholar] [CrossRef]
- Koo, D.-H.; Lee, H.J.; Ahn, J.-H.; Yoon, D.H.; Kim, S.-B.; Gong, G.; Son, B.H.; Ahn, S.H.; Jung, K.H. Tau and PTEN status as predictive markers for response to trastuzumab and paclitaxel in patients with HER2-positive breast cancer. Tumor Biol. 2015, 36, 5865–5871. [Google Scholar] [CrossRef]
- Zhu, T.; Xu, F.; Zhang, L.; Zhang, Y.; Yang, C.; Cheng, M.; Chen, F.; Wang, K. Measurement of molecular biomarkers that predict the tumor response in estrogen receptor-positive breast cancers after dose-dense (biweekly) paclitaxel/carboplatin neoadjuvant chemotherapy. Oncotarget 2017, 8, 101087–101094. [Google Scholar] [CrossRef]
- Mimori, K.; Sadanaga, N.; Yoshikawa, Y.; Ishikawa, K.; Hashimoto, M.; Tanaka, F.; Sasaki, A.T.; Inoue, H.; Sugimachi, K.; Mori, M. Reduced tau expression in gastric cancer can identify candidates for successful Paclitaxel treatment. Br. J. Cancer 2006, 94, 1894–1897. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, N.; Shao, G.; Qian, J.; Shen, D.; Fei, Y.; Mao, W.; Wu, D. Relationship Between Gastric Cancer Tau Protein Expression and Paclitaxel Sensitivity. Pathol. Oncol. Res. 2013, 19, 429–435. [Google Scholar] [CrossRef]
- Yu, J.; Gao, J.; Lu, Z.; Gong, J.; Li, Y.; Dong, B.; Li, Z.; Zhang, X.; Shen, L. Combination of microtubule associated protein-tau and beta-tubulin III predicts chemosensitivity of paclitaxel in patients with advanced gastric cancer. Eur. J. Cancer 2014, 50, 2328–2335. [Google Scholar] [CrossRef]
- He, W.; Zhang, D.; Jiang, J.; Liu, P.; Wu, C. The relationships between the chemosensitivity of human gastric cancer to paclitaxel and the expressions of class III beta-tubulin, MAPT, and survivin. Med. Oncol. 2014, 31, 950. [Google Scholar] [CrossRef]
- Wosnitzer, M.S.; Domingo-Domenech, J.; Castillo-Martin, M.; Ritch, C.; Mansukhani, M.; Petrylack, D.P.; Benson, M.C.; McKiernan, J.M.; Cordon-Cardo, C. Predictive value of microtubule associated proteins tau and stathmin in patients with nonmuscle invasive bladder cancer receiving adjuvant intravesical taxane therapy. J. Urol. 2011, 186, 2094–2100. [Google Scholar] [CrossRef]
- Ramalingam, S.; Belani, C.P. Paclitaxel for non-small cell lung cancer. Expert Opin. Pharm. 2004, 5, 1771–1780. [Google Scholar] [CrossRef]
- Gurler, H.; Yu, Y.; Choi, J.; Kajdacsy-Balla, A.A.; Barbolina, M.V. Three-Dimensional Collagen Type I Matrix Up-Regulates Nuclear Isoforms of the Microtubule Associated Protein Tau Implicated in Resistance to Paclitaxel Therapy in Ovarian Carcinoma. Int. J. Mol. Sci. 2015, 16, 3419–3433. [Google Scholar] [CrossRef] [PubMed]
- Smoter, M.; Bodnar, L.; Duchnowska, R.; Stec, R.; Grala, B.; Szczylik, C. The role of Tau protein in resistance to paclitaxel. Cancer Chemother. Pharmacol. 2011, 68, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Barbolina, M.V. Dichotomous role of microtubule associated protein tau as a biomarker of response to and a target for in-creasing efficacy of taxane treatment in cancers of epithelial origin. Pharmacol. Res. 2021, 168, 105585. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-L.; Wang, N.; Sun, F.-R.; Cao, X.-P.; Zhang, W.; Yu, J.-T. Tau in neurodegenerative disease. Ann. Transl. Med. 2018, 6, 175. [Google Scholar] [CrossRef]
- Chang, H.-Y.; Sang, T.-K.; Chiang, A.-S. Untangling the Tauopathy for Alzheimer’s disease and parkinsonism. J. Biomed. Sci. 2018, 25, 1–11. [Google Scholar] [CrossRef]
- Evans, D.B.; Rank, K.B.; Bhattacharya, K.; Thomsen, D.R.; Gurney, M.E.; Sharma, S.K. Tau phosphorylation at serine 396 and serine 404 by human recombinant tau protein kinase II inhibits tau’s ability to promote microtubule assembly. J. Biol. Chem. 2000, 275, 24977–24983. [Google Scholar] [CrossRef]
- Lee, V.M.-Y.; Brunden, K.R.; Hutton, M.; Trojanowski, J.Q. Developing Therapeutic Approaches to Tau, Selected Kinases, and Related Neuronal Protein Targets. Cold Spring Harb. Perspect. Med. 2011, 1, a006437. [Google Scholar] [CrossRef]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and con-tributes to reduced microtubule binding. Neuron 1993, 10, 1089–1099. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Kohnken, R.; Buerger, K.; Zinkowski, R.; Miller, C.; Kerkman, D.; DeBernardis, J.; Shen, J.; Möller, H.J.; Davies, P.; Hampel, H. Detection of tau phosphorylated at threonine 231 in cerebrospinal fluid of Alzheimer’s disease patients. Neurosci. Lett. 2000, 287, 187–190. [Google Scholar] [CrossRef]
- Wang, J.-Z.; Grundke-Iqbal, I.; Iqbal, K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 2007, 25, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Souter, S.; Lee, G. Microtubule-associated protein tau in human prostate cancer cells: Isoforms, phosphorylation, and interactions. J. Cell. Biochem. 2009, 108, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Bakota, L.; Brandt, R. Tau Biology and Tau-Directed Therapies for Alzheimer’s Disease. Drugs 2016, 76, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.; Avila, J.; Schöll, M.; Kovacs, G.G.; Kövari, E.; Skrabana, R.; Evans, L.D.; Kontsekova, E.; Malawska, B.; De Silva, R.; et al. A walk through tau therapeutic strategies. Acta Neuropathol. Commun. 2019, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Götz, J. Tau-based therapies in neurodegeneration: Opportunities and challenges. Nat. Rev. Drug Discov. 2017, 16, 863–883. [Google Scholar] [CrossRef]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef]
- Wischik, C.M.; Staff, R.T.; Wischik, D.J.; Bentham, P.; Murray, A.D.; Storey, J.; Kook, K.A.; Harrington, C.R. Tau aggregation inhibitor therapy: An exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 705–720. [Google Scholar] [CrossRef]
- Medina, D.X.; Caccamo, A.; Oddo, S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011, 21, 140–149. [Google Scholar] [CrossRef]
- Harrington, C.R.; Storey, J.M.; Clunas, S.; Harrington, K.A.; Horsley, D.; Ishaq, A.; Kemp, S.J.; Larch, C.P.; Marshall, C.; Nicoll, S.L.; et al. Cellular Models of Aggregation-dependent Template-directed Proteolysis to Characterize Tau Aggregation Inhibitors for Treatment of Alzheimer Disease. J. Biol. Chem. 2015, 290, 10862–10875. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef]
- Burleson, K.M.; Casey, R.C.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Skubitz, A.P.N. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol. Oncol. 2004, 93, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Burleson, K.M.; Hansen, L.K.; Skubitz, A.P.N. Ovarian carcinoma spheroids disaggregate on type I collagen and invade live human mesothelial cell monolayers. Clin. Exp. Metastasis 2005, 21, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, M.; Xie, J.; Gurler, H.; Muralidhar, G.G.; Sacks, J.D.; Burdette, J.E.; Barbolina, M.V. Versican regulates metastasis of epithelial ovarian carcinoma cells and spheroids. J. Ovarian Res. 2014, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Barbolina, M.V.; Adley, B.P.; Ariztia, E.V.; Liu, Y.; Stack, M.S. Microenvironmental Regulation of Membrane Type 1 Matrix Metalloproteinase Activity in Ovarian Carcinoma Cells via Collagen-induced EGR1 Expression. J. Biol. Chem. 2007, 282, 4924–4931. [Google Scholar] [CrossRef] [PubMed]
- Barbolina, M.V.; Adley, B.P.; Kelly, D.L.; Fought, A.J.; Scholtens, D.M.; Shea, L.D.; Stack, M.S. Motility-related actinin alpha-4 is associated with advanced and metastatic ovarian carcinoma. Lab. Investig. 2008, 88, 602–614. [Google Scholar] [CrossRef]
- Barbolina, M.V.; Kim, M.; Liu, Y.; Shepard, J.; Belmadani, A.; Miller, R.J.; Shea, L.D.; Stack, M.S. Microenvironmental Regulation of Chemokine (C-X-C-Motif) Receptor 4 in Ovarian Carcinoma. Mol. Cancer Res. 2010, 8, 653–664. [Google Scholar] [CrossRef]
- Kim, M.; Rooper, L.; Xie, J.; Kajdacsy-Balla, A.A.; Barbolina, M.V. Fractalkine Receptor CX3CR1 Is Expressed in Epithelial Ovarian Carcinoma Cells and Required for Motility and Adhesion to Peritoneal Mesothelial Cells. Mol. Cancer Res. 2012, 10, 11–24. [Google Scholar] [CrossRef]
- Kim, M.; Rooper, L.; Xie, J.; Rayahin, J.; Burdette, J.E.; Kajdacsy-Balla, A.A.; Barbolina, M.V. The Lymphotactin Receptor Is Expressed in Epithelial Ovarian Carcinoma and Contributes to Cell Migration and Proliferation. Mol. Cancer Res. 2012, 10, 1419–1429. [Google Scholar] [CrossRef]
- Gurler Main, H.; Xie, J.; Muralidhar, G.G.; Elfituri, O.; Xu, H.; Kajdacsy-Balla, A.A.; Barbolina, M.V. Emergent role of the fractalkine axis in dissemination of peritoneal metastasis from epithelial ovarian carcinoma. Oncogene 2017, 36, 3025–3036. [Google Scholar] [CrossRef]
- Derrien, A.; Gouard, S.; Maurel, C.; Gaugler, M.H.; Bruchertseifer, F.; Morgenstern, A.; Faivre-Chauvet, A.; Classe, J.M.; Chérel, M. Therapeutic Efficacy of Alpha-RIT Using a (213)Bi-Anti-hCD138 Antibody in a Mouse Model of Ovarian Peritoneal Carcinomatosis. Front. Med. 2015, 2, 88. [Google Scholar] [CrossRef] [Green Version]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Ikediobi, O.N.; Davies, H.; Bignell, G.; Edkins, S.; Stevens, C.; O’Meara, S.; Santarius, T.; Avis, T.; Barthorpe, S.; Brackenbury, L.; et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol. Cancer Ther. 2006, 5, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.W.; Laub, P.B.; Beesley, J.S.; Ozols, R.F.; Hamilton, T.C. Increased platinum-DNA damage tolerance is associated with cisplatin resistance and cross-resistance to various chemotherapeutic agents in unrelated human ovarian cancer cell lines. Cancer Res. 1997, 57, 850–856. [Google Scholar] [PubMed]
- Hernandez, L.; Kim, M.K.; Lyle, L.T.; Bunch, K.P.; House, C.D.; Ning, F.; Noonan, A.M.; Annunziata, C.M. Characterization of ovarian cancer cell lines as in vivo models for preclinical studies. Gynecol. Oncol. 2016, 142, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Davis, D.A.; Tomar, S.; Roy, L.; Gurler, H.; Xie, J.; Lantvit, D.D.; Cardenas, H.; Fang, F.; Liu, Y.; et al. In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecol. Oncol. 2015, 138, 372–377. [Google Scholar] [CrossRef]
- McCloskey, C.W.; Goldberg, R.L.; Carter, L.E.; Gamwell, L.F.; Al-Hujaily, E.M.; Collins, O.; Macdonald, E.A.; Garson, K.; Daneshmand, M.; Carmona, E.; et al. A New Spontaneously Transformed Syngeneic Model of High-Grade Serous Ovarian Cancer with a Tumor-Initiating Cell Population. Front. Oncol. 2014, 4, 53. [Google Scholar] [CrossRef]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B.; O’Donovan, N. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef]
- Barbolina, M.V.; Moss, N.M.; Westfall, S.D.; Liu, Y.; Burkhalter, R.J.; Marga, F.; Forgacs, G.; Hudson, L.G.; Stack, M.S. Microenvironmental Regulation of Ovarian Cancer Metastasis. Cancer Treat Res. 2009, 149, 319–334. [Google Scholar]
- Witz, C.; Montoya-Rodriguez, I.A.; Cho, S.; Centonze, V.E.; Bonewald, L.F.; Schenken, R.S. Composition of the extracellular matrix of the peritoneum. J. Soc. Gynecol. Investig. 2001, 8, 299–304. [Google Scholar] [CrossRef]
- Sudo, H.; Nakajima, K. The mitotic tensegrity guardian tau protects mammary epithelia from katanin-like1-induced aneuploidy. Oncotarget 2016, 7, 53712–53734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, A.; Kobayashi, A.; Oikiri, H.; Yokoyama, Y. Functional role of the Tau protein in epithelial ovarian cancer cells. Reprod. Med. Biol. 2017, 16, 143–151. [Google Scholar] [PubMed]
- Gyorffy, B.; Lánczky, A.; Szallasi, Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr. Relat. Cancer 2012, 19, 197–208. [Google Scholar] [CrossRef]
- Spitzer, M.; Wildenhain, J.; Rappsilber, J.; Tyers, M. BoxPlotR: A web tool for generation of box plots. Nat. Methods 2014, 11, 121–122. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Raha, D.; Wilson, T.R.; Peng, J.; Peterson, D.; Yue, P.; Evangelista, M.; Wilson, C.; Merchant, M.; Settleman, J. The Cancer Stem Cell Marker Aldehyde Dehydrogenase Is Required to Maintain a Drug-Tolerant Tumor Cell Subpopulation. Cancer Res. 2014, 74, 3579–3590. [Google Scholar] [CrossRef]
- Meng, E.; Mitra, A.; Tripathi, K.; Finan, M.A.; Scalici, J.; McClellan, S.; da Silva, L.M.; Reed, E.; Shevde, L.A.; Palle, K.; et al. ALDH1A1 Maintains Ovarian Cancer Stem Cell-Like Properties by Altered Regulation of Cell Cycle Checkpoint and DNA Repair Network Signaling. PLoS ONE 2014, 9, e107142. [Google Scholar] [CrossRef]
- Meng, E.; Long, B.; Sullivan, P.; McClellan, S.; Finan, M.A.; Reed, E.; Shevde, L.; Rocconi, R.P. CD44+/CD24- ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin. Exp. Metastasis 2012, 29, 939–948. [Google Scholar] [CrossRef]
- Montgomery, R.B.; Guzman, J.; O’Rourke, D.M.; Stahl, W.L. Expression of oncogenic epidermal growth factor receptor family kinases induces paclitaxel re-sistance and alters beta-tubulin isotype expression. J. Biol. Chem. 2000, 275, 17358–17363. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Jiang, X.; Xie, H.; He, J.; Xiao, S. The Jun N-terminal kinases signaling pathway plays a “seesaw” role in ovarian carcinoma: A molecular aspect. J. Ovarian Res. 2019, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Bacus, S.S.; Gudkov, A.V.; Lowe, M.; Lyass, L.; Yung, Y.; Komarov, A.P.; Keyomarsi, K.; Yarden, Y.; Seger, R. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene 2001, 20, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, L.; Zhou, C.; Sun, Y.; Di, W.; Scheffler, E.; Healey, S.; Kouttab, N.; Chu, W.; Wan, Y. Crosstalk between EGFR and TrkB enhances ovarian cancer cell migration and proliferation. Int. J. Oncol. 2006, 29, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Rajfur, Z.; Borchers, C.; Schaller, M.D.; Jacobson, K. JNK phosphorylates paxillin and regulates cell migration. Nature 2003, 424, 219–223. [Google Scholar] [CrossRef]
- Hedges, J.C.; Dechert, M.A.; Yamboliev, I.A.; Martin, J.L.; Hickey, E.; Weber, L.A.; Gerthoffer, W.T. A role for p38(MAPK)/HSP27 pathway in smooth muscle cell migration. J. Biol. Chem. 1999, 274, 24211–24219. [Google Scholar] [CrossRef] [PubMed]
- Kawada, K.; Upadhyay, G.; Ferandon, S.; Janarthanan, S.; Hall, M.; Vilardaga, J.-P.; Yajnik, V. Cell Migration Is Regulated by Platelet-Derived Growth Factor Receptor Endocytosis. Mol. Cell. Biol. 2009, 29, 4508–4518. [Google Scholar] [CrossRef] [PubMed]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Fichou, Y.; Al-Hilaly, Y.K.; Devred, F.; Smet-Nocca, C.; Tsvetkov, P.O.; Verelst, J.; Winderickx, J.; Geukens, N.; Vanmechelen, E.; Perrotin, A.; et al. The elusive tau molecular structures: Can we translate the recent breakthroughs into new targets for inter-vention? Acta Neuropathol. Commun. 2019, 7, 31. [Google Scholar] [CrossRef]
- Maina, M.B.; Al-Hilaly, Y.K.; Serpell, L.C. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 2016, 6, 9. [Google Scholar] [CrossRef]
- Derisbourg, M.; Leghay, C.; Chiappetta, G.; Fernandez-Gomez, F.J.; Laurent, C.; Demeyer, D.; Carrier, S.; Buée-Scherrer, V.; Blum, D.; Vinh, J.; et al. Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncat-ed forms. Sci. Rep. 2015, 5, 9659. [Google Scholar] [CrossRef]
- Amadoro, G.; Serafino, A.L.; Barbato, C.; Ciotti, M.T.; Sacco, A.; Calissano, P.; Canu, N. Role of N-terminal tau domain integrity on the survival of cerebellar granule neurons. Cell Death Differ. 2003, 11, 217–230. [Google Scholar] [CrossRef]
- Sayas, C.L.; Medina, M.; Cuadros, R.; Ollá, I.; García, E.; Pérez, M.; Ferrer, I.; Hernández, F.; Avila, J. Role of tau N-terminal motif in the secretion of human tau by End Binding proteins. PLoS ONE 2019, 14, e0210864. [Google Scholar]
- Amadoro, G.; Latina, V.; Corsetti, V.; Calissano, P. N-terminal tau truncation in the pathogenesis of Alzheimer’s disease (AD): Developing a novel diag-nostic and therapeutic approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165584. [Google Scholar] [CrossRef] [PubMed]
- McMillan, P.; Korvatska, E.; Poorkaj, P.; Evstafjeva, Z.; Robinson, L.; Greenup, L.; Leverenz, J.; Schellenberg, G.D.; D’Souza, I. Tau isoform regulation is region- and cell-specific in mouse brain. J. Comp. Neurol. 2008, 511, 788–803. [Google Scholar] [CrossRef] [PubMed]
- Sealey, M.A.; Vourkou, E.; Cowan, C.M.; Bossing, T.; Quraishe, S.; Grammenoudi, S.; Skoulakis, E.M.; Mudher, A. Distinct phenotypes of three-repeat and four-repeat human tau in a transgenic model of tauopathy. Neurobiol. Dis. 2017, 105, 74–83. [Google Scholar] [CrossRef]
- Schoch, K.M.; DeVos, S.L.; Miller, R.L.; Chun, S.J.; Norrbom, M.; Wozniak, D.F.; Dawson, H.N.; Bennett, C.F.; Rigo, F.; Miller, T.M. Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model. Neuron 2016, 90, 941–947. [Google Scholar] [CrossRef]
- Panda, D.; Samuel, J.C.; Massie, M.; Feinstein, S.C.; Wilson, L. Differential regulation of microtubule dynamics by three- and four-repeat tau: Implications for the onset of neurodegenerative disease. Proc. Natl. Acad. Sci. USA 2003, 100, 9548–9553. [Google Scholar] [CrossRef]
- Bachmann, S.; Bell, M.; Klimek, J.; Zempel, H. Differential Effects of the Six Human TAU Isoforms: Somatic Retention of 2N-TAU and Increased Mi-crotubule Number Induced by 4R-TAU. Front. Neurosci. 2021, 15, 643115. [Google Scholar] [CrossRef]
- Dregni, A.J.; Wang, H.K.; Wu, H.; Duan, P.; Jin, J.; DeGrado, W.F.; Hong, M. Inclusion of the C-Terminal Domain in the beta-Sheet Core of Heparin-Fibrillized Three-Repeat Tau Protein Revealed by Solid-State Nuclear Magnetic Resonance Spectroscopy. J. Am. Chem. Soc. 2021, 143, 7839–7851. [Google Scholar] [CrossRef]
- Abraha, A.; Ghoshal, N.; Gamblin, T.C.; Cryns, V.; Berry, R.W.; Kuret, J.; Binder, L.I. C-terminal inhibition of tau assembly in vitro and in Alzheimer’s disease. J. Cell. Sci. 2000, 113 Pt 21, 3737–3745. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Von Bergen, M.; Biernat, J.; Fischer, D.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. The “jaws” of the tau-microtubule interaction. J. Biol. Chem. 2007, 282, 12230–12239. [Google Scholar] [CrossRef]
- Yu, Y.; Run, X.; Liang, Z.; Li, Y.; Liu, F.; Liu, Y.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J. Neurochem. 2009, 108, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. Pathogenesis of ovarian cancer: Lessons from morphology and molecular biology and their clinical implications. Int. J. Gynecol. Pathol. 2008, 27, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.J.; Fayomi, A.P.; Anyaeche, V.I.; Bai, S.; Buckanovich, R.J. An evolving paradigm of cancer stem cell hierarchies: Therapeutic implications. Theranostics 2020, 10, 3083–3098. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Nakamura, T.; Miyashita, K.; Fukamachi, I.; Seino, Y.; Shoji, M. Novel ELISAs to measure total and phosphorylated tau in cerebrospinal fluid. Neurosci. Lett. 2020, 722, 134826. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 TRx0237, µM |
|---|---|
| OVCAR-4 | 6.7 ± 0.2 |
| STOSE | 29.0 ± 2.1 |
| OVCAR4-PR2 | 8.6 ± 0.9 |
| STOSE-PR25 | 36.6 ± 3.2 |
| STOSE-PR50 | 60.2 ± 6.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbolina, M.V. Targeting Microtubule-Associated Protein Tau in Chemotherapy-Resistant Models of High-Grade Serous Ovarian Carcinoma. Cancers 2022, 14, 4535. https://doi.org/10.3390/cancers14184535
Barbolina MV. Targeting Microtubule-Associated Protein Tau in Chemotherapy-Resistant Models of High-Grade Serous Ovarian Carcinoma. Cancers. 2022; 14(18):4535. https://doi.org/10.3390/cancers14184535
Chicago/Turabian StyleBarbolina, Maria V. 2022. "Targeting Microtubule-Associated Protein Tau in Chemotherapy-Resistant Models of High-Grade Serous Ovarian Carcinoma" Cancers 14, no. 18: 4535. https://doi.org/10.3390/cancers14184535