
Interleukin 4 Controls the Pro-Tumoral Role of Macrophages in Mammary Cancer Pulmonary Metastasis in Mice
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Cell Lines and Tissue Culture
2.3. Primary Culture of Bone-Marrow-Derived Macrophages
2.4. Endothelial Cell Permeability Assay and Transendothelial Resistance
2.5. Experimental Transendothelial Migration Assay
2.6. Experimental Metastasis Assay
2.7. Adoptive Transfer Experiments
2.8. Real-Time PCR
2.9. Ingenuity Pathway Analysis
2.10. Flow Cytometry
2.11. Intravital Imaging
2.12. Statistical Analysis
3. Results
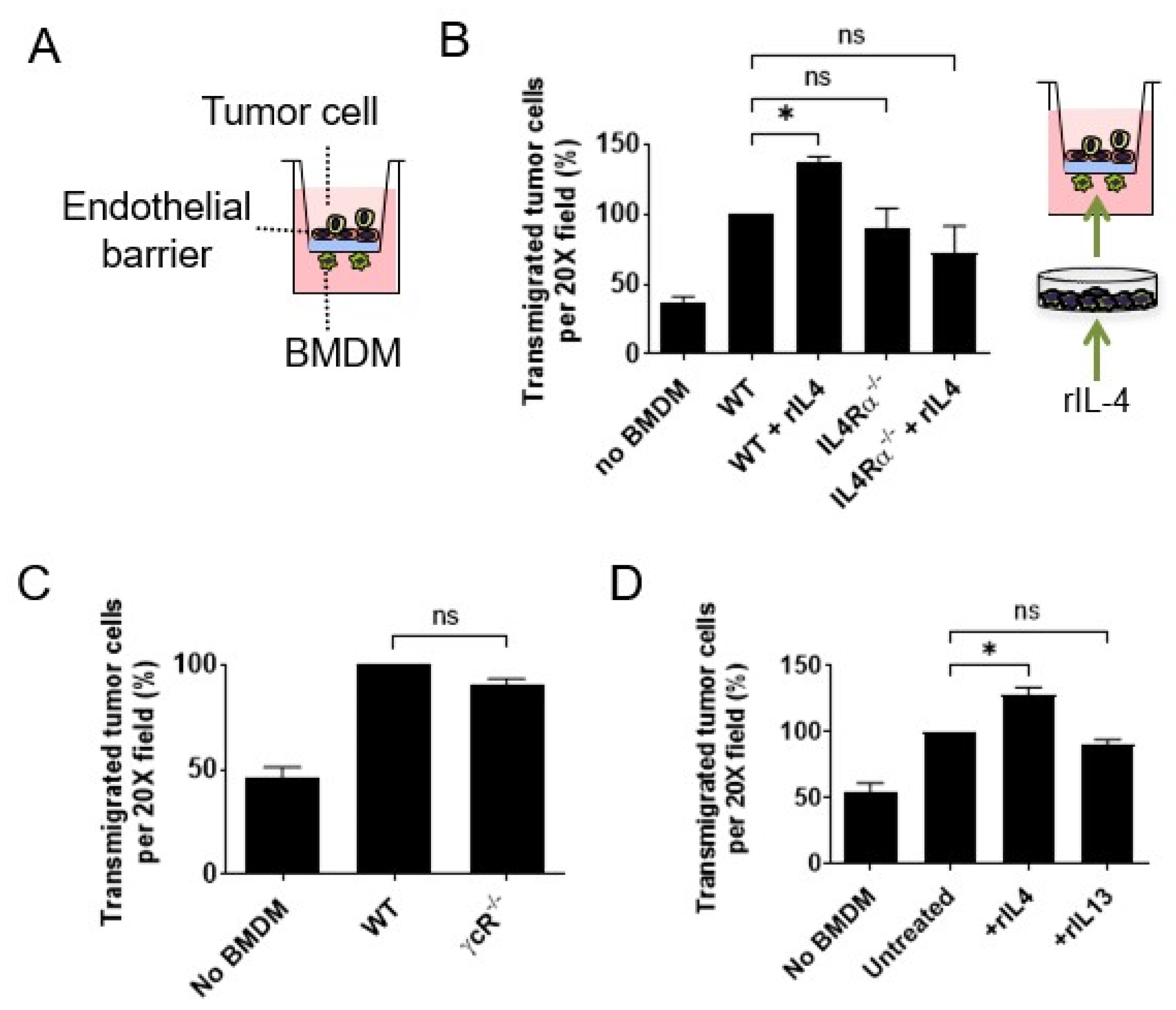
3.1. Interleukin 4 Signaling in Macrophages Enhances Cancer Cell Transendothelial Migration
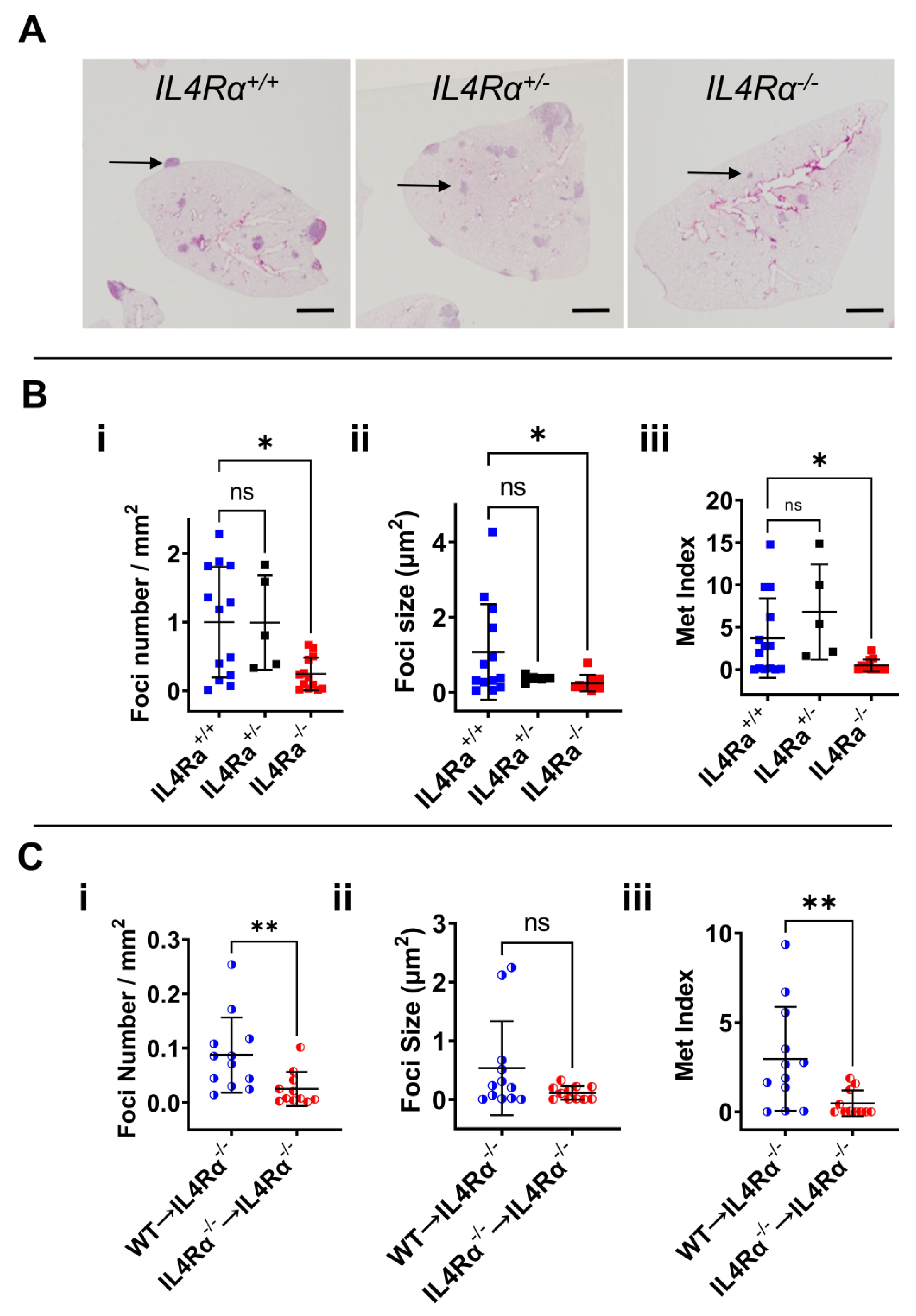
3.2. IL4Rα Is Important for Efficient Lung Metastasis Development
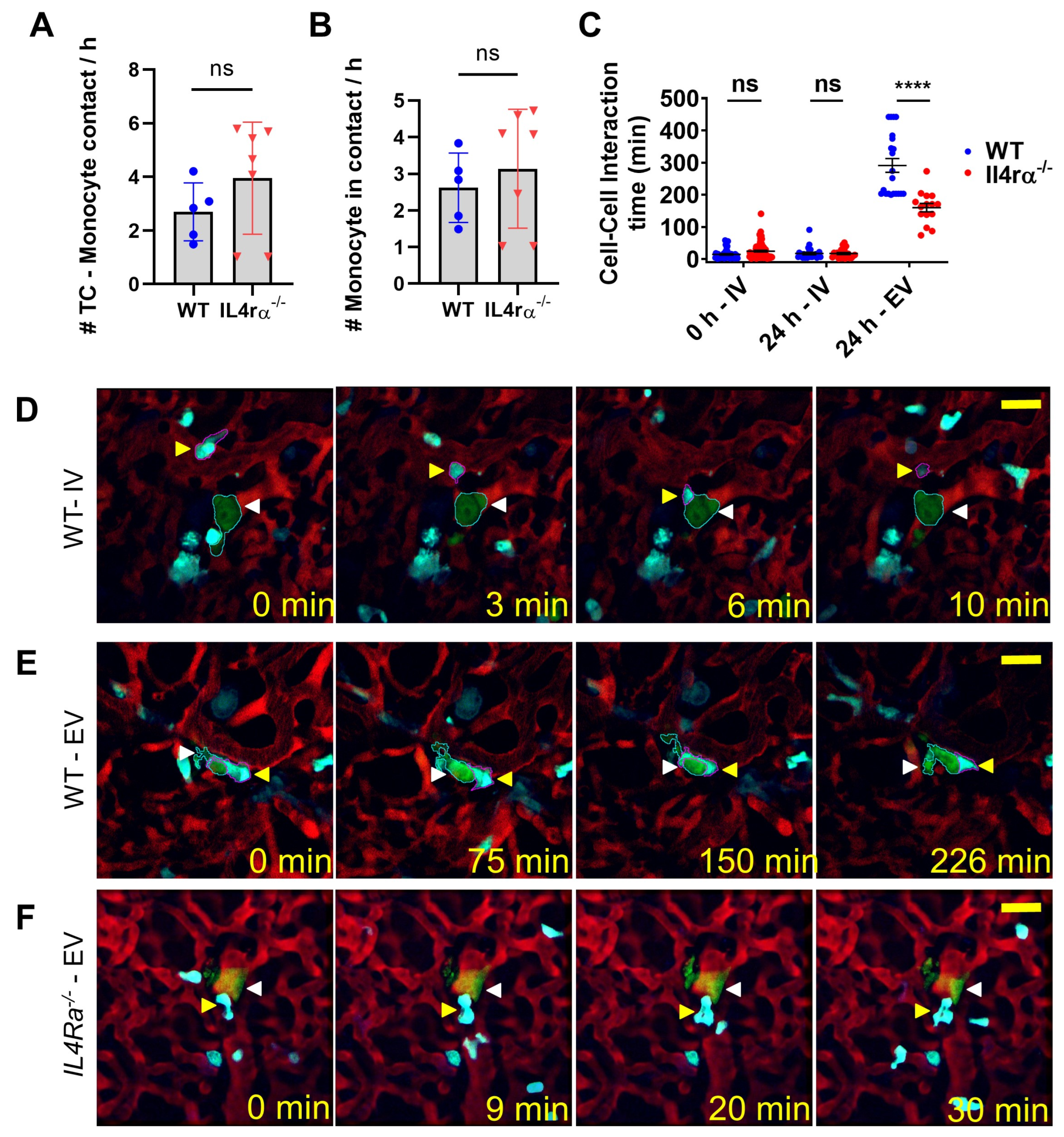
3.3. IL4 Signaling Stabilize Tumor Cell–Macrophage Contact In Vivo
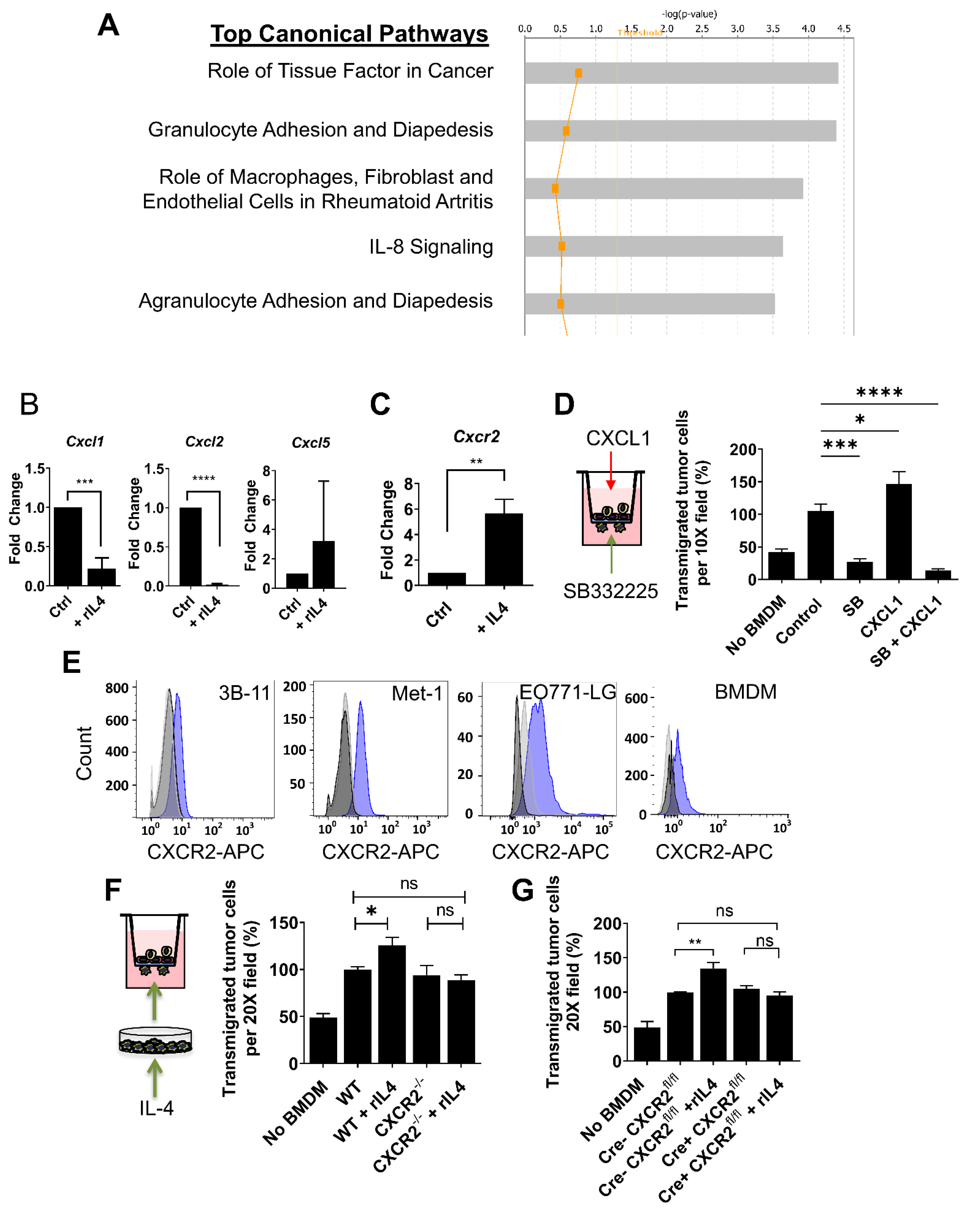
3.4. CXCR2 Is Downstream of IL4 in Macrophages and Mediates the IL4-Dependent Increase in Tumor Cell Transendothelial Migration
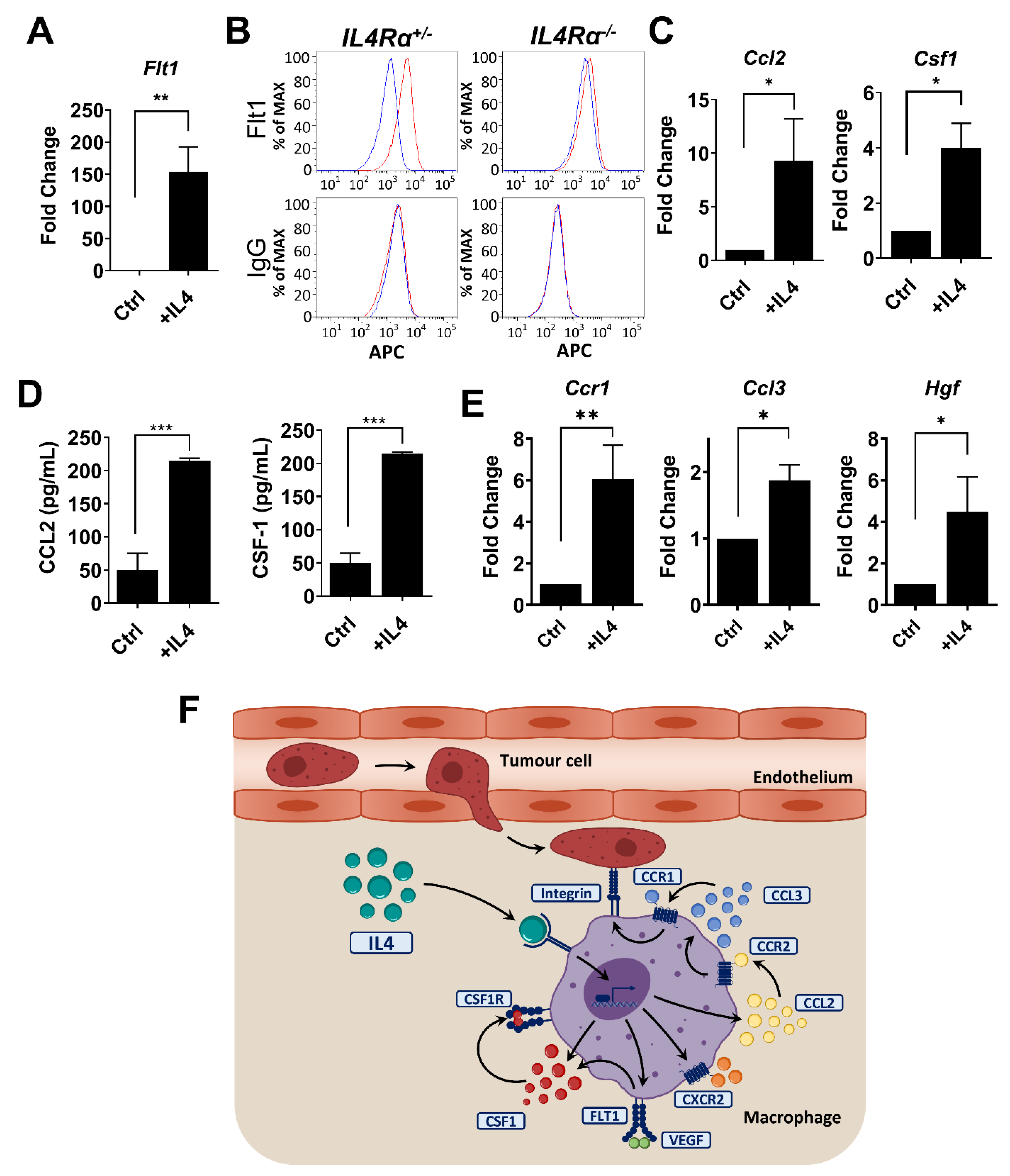
3.5. IL4 Regulates Genes in Macrophages Required for Metastatic Seeding and Expansion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Jemal, A.; Grey, N.; Ferlay, J.; Forman, D. Global cancer transitions according to the Human Development Index (2008-2030): A population-based study. Lancet Oncol. 2012, 13, 790–801. [Google Scholar] [CrossRef]
- Alwan, A. Global Status Report on Noncommunicable Diseases 2010; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-Stimulating Factor 1 Promotes Progression of Mammary Tumors to Malignancy. J. Exp. Med. 2001, 193, 727. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, M.J.; Sojka, D.K.; Kim, K.; Knolhoff, B.L.; Zho, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Gadea, B.B.; Wang, H.W.; Gocheva, V.; Hunter, K.E.; Tang, L.H.; Joyce, J.A. Deficiency of the macrophage growth factor CSF-1 disrupts pancreatic neuroendocrine tumor development. Oncogene 2011, 31, 1459–1467. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Knowles, H.J.; Harris, A.L. Macrophages and the hypoxic tumour microenvironment. Front. Biosci. A J. Virtual Libr. 2007, 12, 4298–4314. [Google Scholar] [CrossRef]
- Leek, R.D.; Lewis, C.E.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A.L. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar]
- Yang, M.; McKay, D.; Pollard, J.W.; Lewis, C.E. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res. 2018, 78, 5492–5503. [Google Scholar] [CrossRef]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.-Z.Z.; Zhang, H.; Li, J.; He, T.; Yeo, E.-J.J.; Soong, D.Y.; Carragher, N.O.; Munro, A.; Chang, A.; Bresnick, A.R.; et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J. Exp. Med. 2015, 212, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Doughty-Shenton, D.; Cassetta, L.; Fragkogianni, S.; Brownlie, D.; Kato, Y.; Carragher, N.; Pollard, J.W. Monocytes Differentiate to Immune Suppressive Precursors of Metastasis-Associated Macrophages in Mouse Models of Metastatic Breast Cancer. Front. Immunol. 2017, 8, 2004. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, X.H.; Massague, J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 2011, 20, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef]
- Shurin, M.R.; Lu, L.; Kalinski, P.; Stewart-Akers, A.M.; Lotze, M.T. Th1/Th2 balance in cancer, transplantation and pregnancy. Springer Semin. Immunopathol 1999, 21, 339–359. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, L.; Qin, Z. Paradoxical roles of IL-4 in tumor immunity. Cell. Mol. Immunol. 2009, 6, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Leland, P.; Taguchi, J.; Husain, S.R.; Kreitman, R.J.; Pastan, I.; Puri, R.K. Human breast carcinoma cells express type II IL-4 receptors and are sensitive to antitumor activity of a chimeric IL-4-Pseudomonas exotoxin fusion protein in vitro and in vivo. Mol. Med. 2000, 6, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Kawakami, M.; Husain, S.R.; Puri, R.K. Effect of interleukin (IL)-4 cytotoxin on breast tumor growth after in vivo gene transfer of IL-4 receptor alpha chain. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1826–1836. [Google Scholar]
- Kawakami, M.; Kawakami, K.; Stepensky, V.A.; Maki, R.A.; Robin, H.; Muller, W.; Husain, S.R.; Puri, R.K. Interleukin 4 receptor on human lung cancer: A molecular target for cytotoxin therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 3503–3511. [Google Scholar]
- Ma, R.Y.; Zhang, H.; Li, X.F.; Zhang, C.B.; Selli, C.; Tagliavini, G.; Lam, A.D.; Prost, S.; Sims, A.H.; Hu, H.Y.; et al. Monocyte-derived macrophages promote breast cancer bone metastasis outgrowth. J. Exp. Med. 2020, 217, e20191820. [Google Scholar] [CrossRef]
- Deng, L.; Zhou, J.F.; Sellers, R.S.; Li, J.F.; Nguyen, A.V.; Wang, Y.; Orlofsky, A.; Liu, Q.; Hume, D.A.; Pollard, J.W.; et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am. J. Pathol. 2010, 176, 952–967. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, D.A.; van Zuylen, W.J.; DeBats, C.E.E.; Alexander, K.A.; Kellie, S.; Hume, D.A. Expression of Gal4-dependent transgenes in cells of the mononuclear phagocyte system labeled with enhanced cyan fluorescent protein using Csf1r-Gal4VP16/UAS-ECFP double-transgenic mice. J. Leukoc. Biol. 2008, 83, 430–433. [Google Scholar] [CrossRef]
- Dunham, L.J.; Stewart, H.L. A survey of transplantable and transmissible animal tumors. J. Natl. Cancer Inst. 1953, 13, 1299–1377. [Google Scholar]
- Borowsky, A.D.; Namba, R.; Young, L.; Hunter, K.W.; Hodgson, G.J.; Tepper, C.G.; McGoldrick, E.T.; Muller, W.J.; Cardiff, R.D.; Gregg, J.P. Syngeneic mouse mammary carcinoma cell lines: Two closely related cell lines with divergent metastatic behavior. Clin. Exp. Metastasis 2005, 22, 47–59. [Google Scholar] [CrossRef]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, B.S.; Lund, L.R.; Christensen, I.J.; Johnsen, M.; Usher, P.A.; Wulf-Andersen, L.; Frandsen, T.L.; Dano, K.; Gundersen, H.J. A precise and efficient stereological method for determining murine lung metastasis volumes. Am. J. Pathol. 2001, 158, 1997–2003. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Milde, R.; Varin, A.; Melgert, B.N.; Draijer, C.; Thomas, B.; Fabbri, M.; Crawshaw, A.; Ho, L.P.; et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: Similarities and differences. Blood 2013, 121, e57–e69. [Google Scholar] [CrossRef]
- Entenberg, D.; Rodriguez-Tirado, C.; Kato, Y.; Kitamura, T.; Pollard, J.W.; Condeelis, J. In vivo subcellular resolution optical imaging in the lung reveals early metastatic proliferation and motility. Intravital 2015, 4, 1–11. [Google Scholar] [CrossRef]
- Rodriguez-Tirado, C.; Kitamura, T.; Kato, Y.; Pollard, J.W.; Condeelis, J.S.; Entenberg, D. Long-term High-Resolution Intravital Microscopy in the Lung with a Vacuum Stabilized Imaging Window. J. Vis. Exp. 2016, 6, 54603. [Google Scholar] [CrossRef]
- Entenberg, D.; Wyckoff, J.; Gligorijevic, B.; Roussos, E.T.; Verkhusha, V.V.; Pollard, J.W.; Condeelis, J. Setup and use of a two-laser multiphoton microscope for multichannel intravital fluorescence imaging. Nat. Protoc. 2011, 6, 1500–1520. [Google Scholar] [CrossRef]
- Güç, E.; Brownlie, D.; Rodriguez-Tirado, C.; Kitamura, T.; Pollard, J.W. Generation of mouse bone marrow-derived macrophages using tumor coculture assays to mimic the tumor microenvironment. Methods Enzym. 2020, 632, 91–111. [Google Scholar] [CrossRef]
- Stevenson, C.S.; Coote, K.; Webster, R.; Johnston, H.; Atherton, H.C.; Nicholls, A.; Giddings, J.; Sugar, R.; Jackson, A.; Press, N.J.; et al. Characterization of cigarette smoke-induced inflammatory and mucus hypersecretory changes in rat lung and the role of CXCR2 ligands in mediating this effect. Am. J. Physiology. Lung Cell. Mol. Physiol. 2005, 288, 22. [Google Scholar] [CrossRef]
- Kitamura, T.; Kato, Y.; Brownlie, D.; Soong, D.Y.H.; Sugano, G.; Kippen, N.; Li, J.; Doughty-Shenton, D.; Carragher, N.; Pollard, J.W. Mammary Tumor Cells with High Metastatic Potential Are Hypersensitive to Macrophage-Derived HGF. Cancer Immunol. Res. 2019, 7, 2052–2064. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.N.; Rafii, S.; Lyden, D. Preparing the “soil”: The premetastatic niche. Cancer Res. 2006, 66, 11089–11093. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Taftaf, R.; Kawaguchi, M.; Chang, Y.F.; Chen, W.; Entenberg, D.; Zhang, Y.; Gerratana, L.; Huang, S.; Patel, D.B.; et al. Homophilic CD44 Interactions Mediate Tumor Cell Aggregation and Polyclonal Metastasis in Patient-Derived Breast Cancer Models. Cancer Discov. 2019, 9, 96–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Mills, C.D.; Shearer, J.; Evans, R.; Caldwell, M.D. Macrophage arginine metabolism and the inhibition or stimulation of cancer. J. Immunol. 1992, 149, 2709–2714. [Google Scholar]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez-Tirado, C.; Entenberg, D.; Li, J.; Qian, B.-Z.; Condeelis, J.S.; Pollard, J.W. Interleukin 4 Controls the Pro-Tumoral Role of Macrophages in Mammary Cancer Pulmonary Metastasis in Mice. Cancers 2022, 14, 4336. https://doi.org/10.3390/cancers14174336
Rodriguez-Tirado C, Entenberg D, Li J, Qian B-Z, Condeelis JS, Pollard JW. Interleukin 4 Controls the Pro-Tumoral Role of Macrophages in Mammary Cancer Pulmonary Metastasis in Mice. Cancers. 2022; 14(17):4336. https://doi.org/10.3390/cancers14174336
Chicago/Turabian StyleRodriguez-Tirado, Carolina, David Entenberg, Jiufeng Li, Bin-Zhi Qian, John S. Condeelis, and Jeffrey W. Pollard. 2022. "Interleukin 4 Controls the Pro-Tumoral Role of Macrophages in Mammary Cancer Pulmonary Metastasis in Mice" Cancers 14, no. 17: 4336. https://doi.org/10.3390/cancers14174336