
Learn to Estimate Genetic Mutation and Microsatellite Instability with Histopathology H&E Slides in Colon Carcinoma
 ,
,
Abstract
:Simple Summary
Abstract
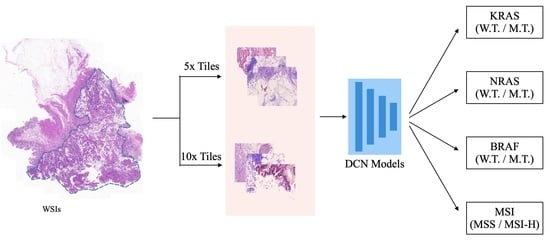
1. Introduction
- We proposed a cascaded deep convolutional framework to simultaneously generate gene mutation prediction and MSI status estimation in colorectal cancer.
- We introduced a simple yet efficient average voting ensemble strategy to produce high fidelity gene mutation prediction and MSI status estimation of the WSI.
- We further analyzed the effectiveness of the number of features selected for model ensembling to understand its effects on the performances of deep CNN models.
2. Materials and Methods
2.1. Data
2.2. Methodology
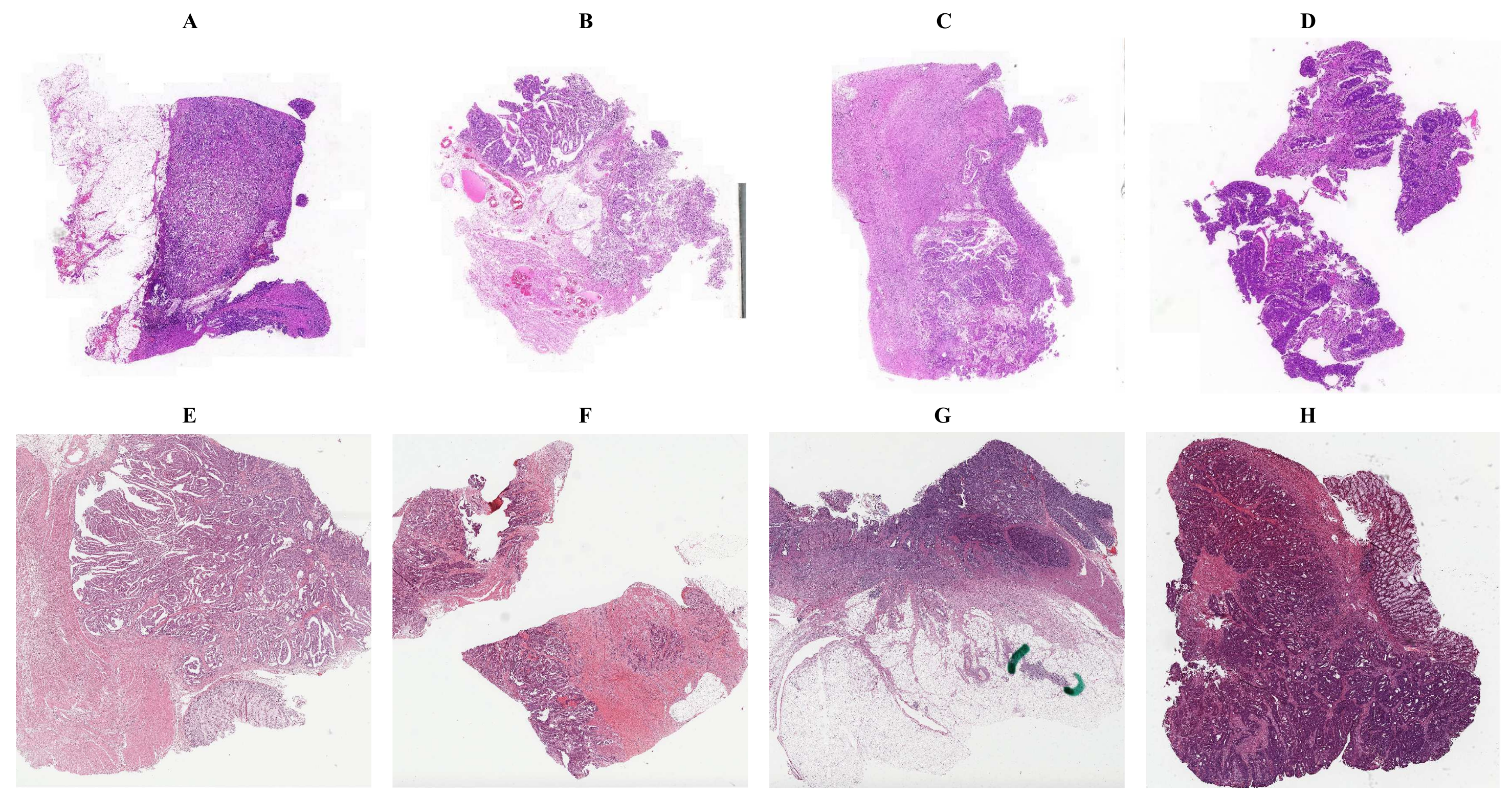
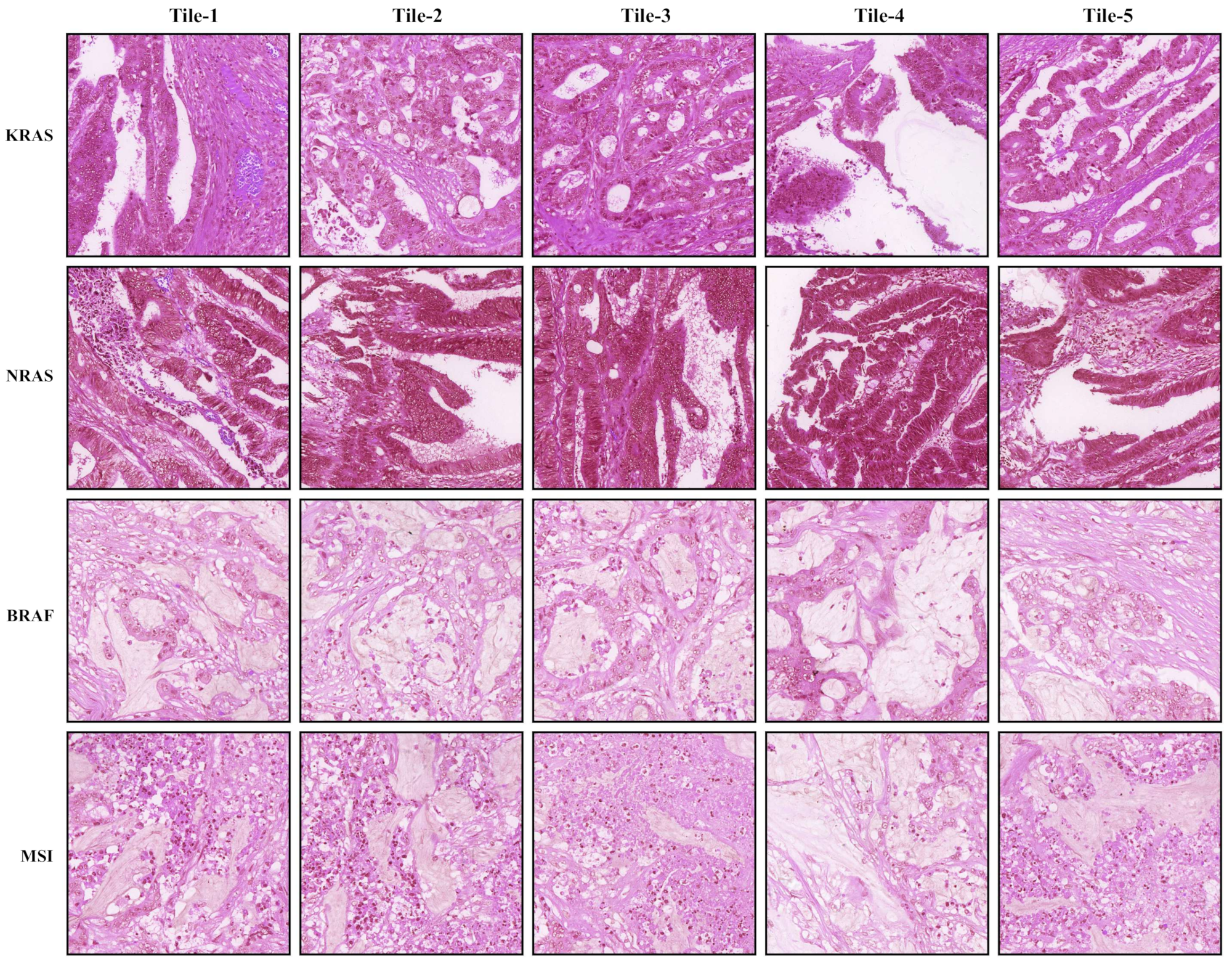
2.2.1. Data Preprocessing
2.2.2. Network Architectures
2.2.3. Model Ensemble
3. Results
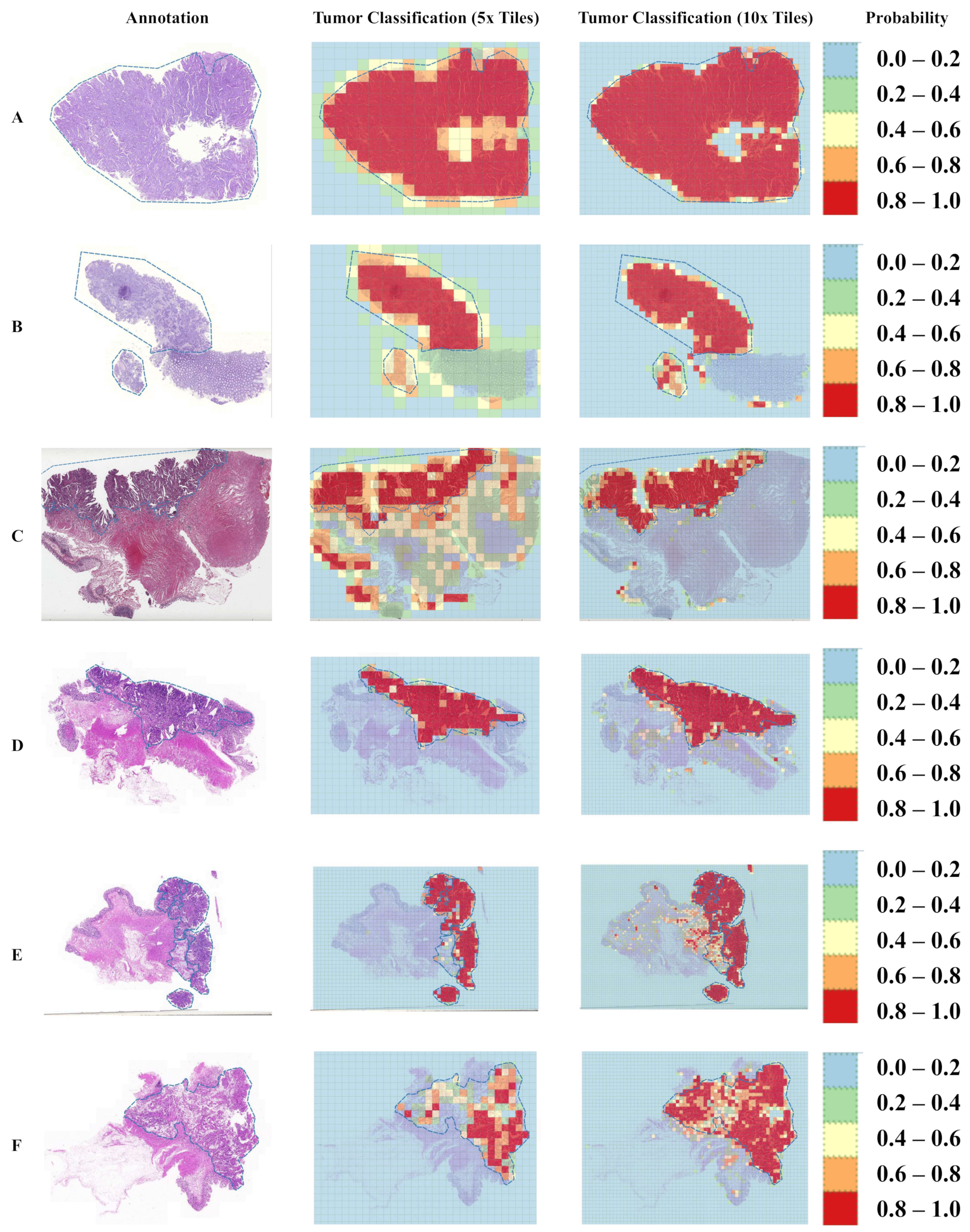
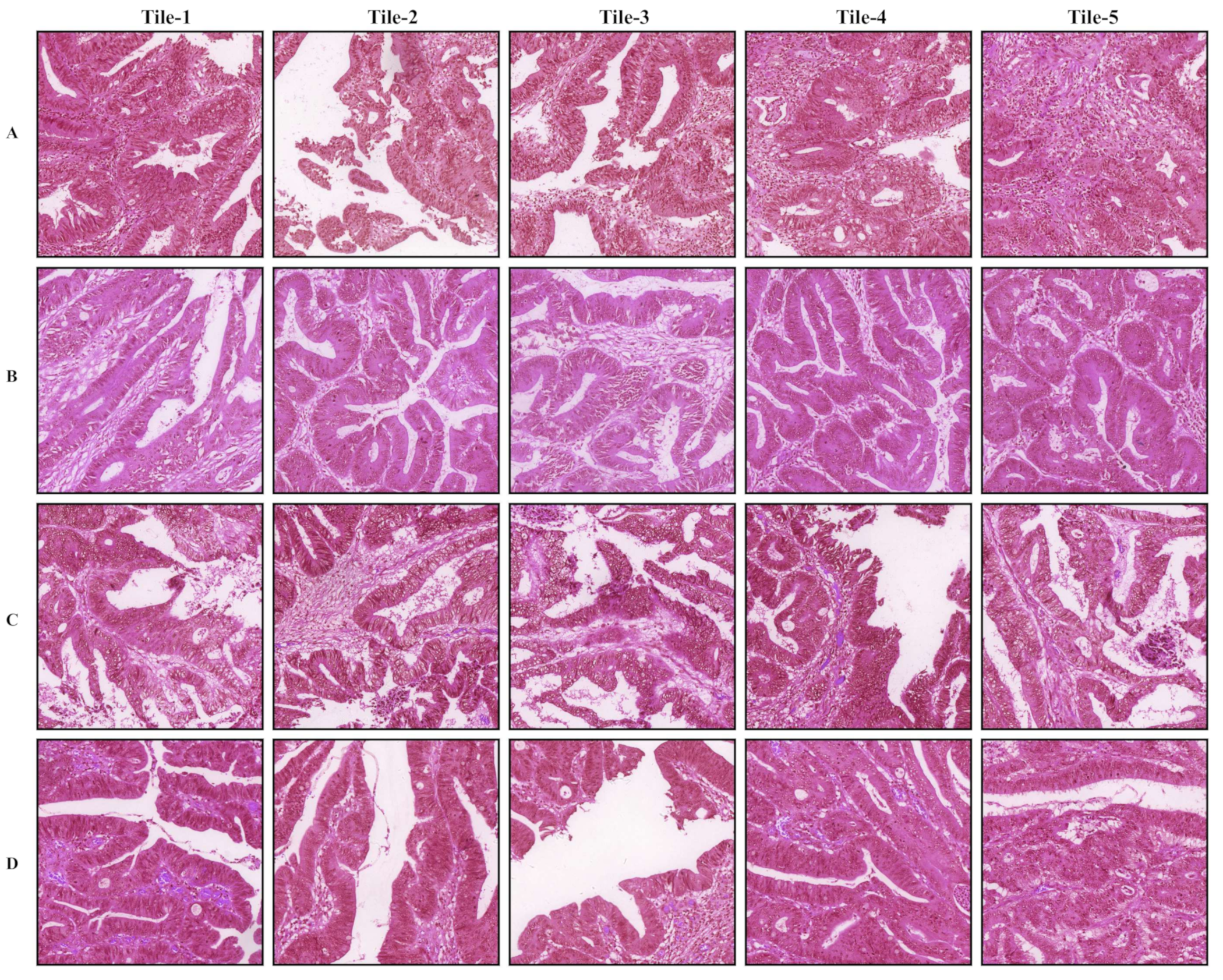
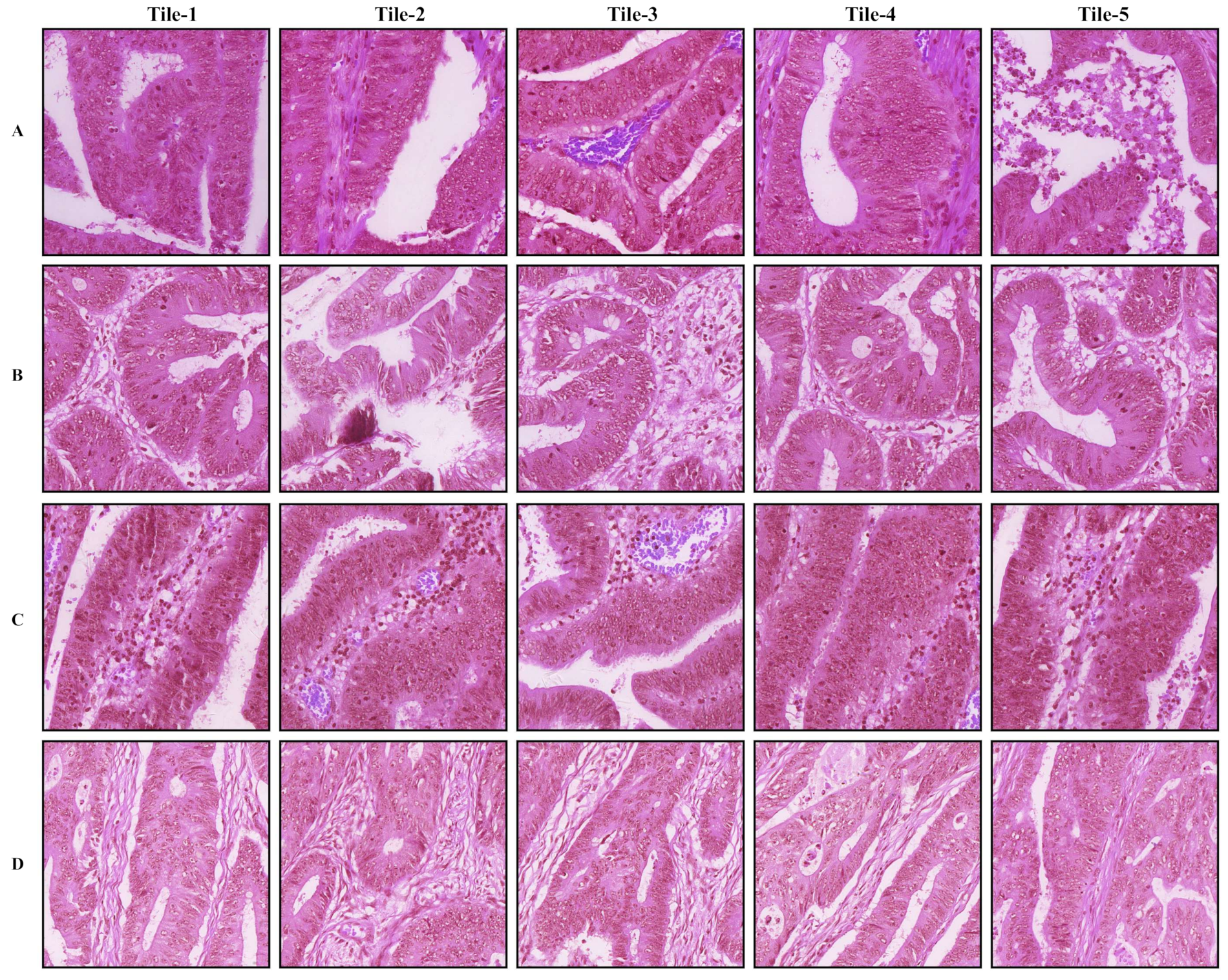
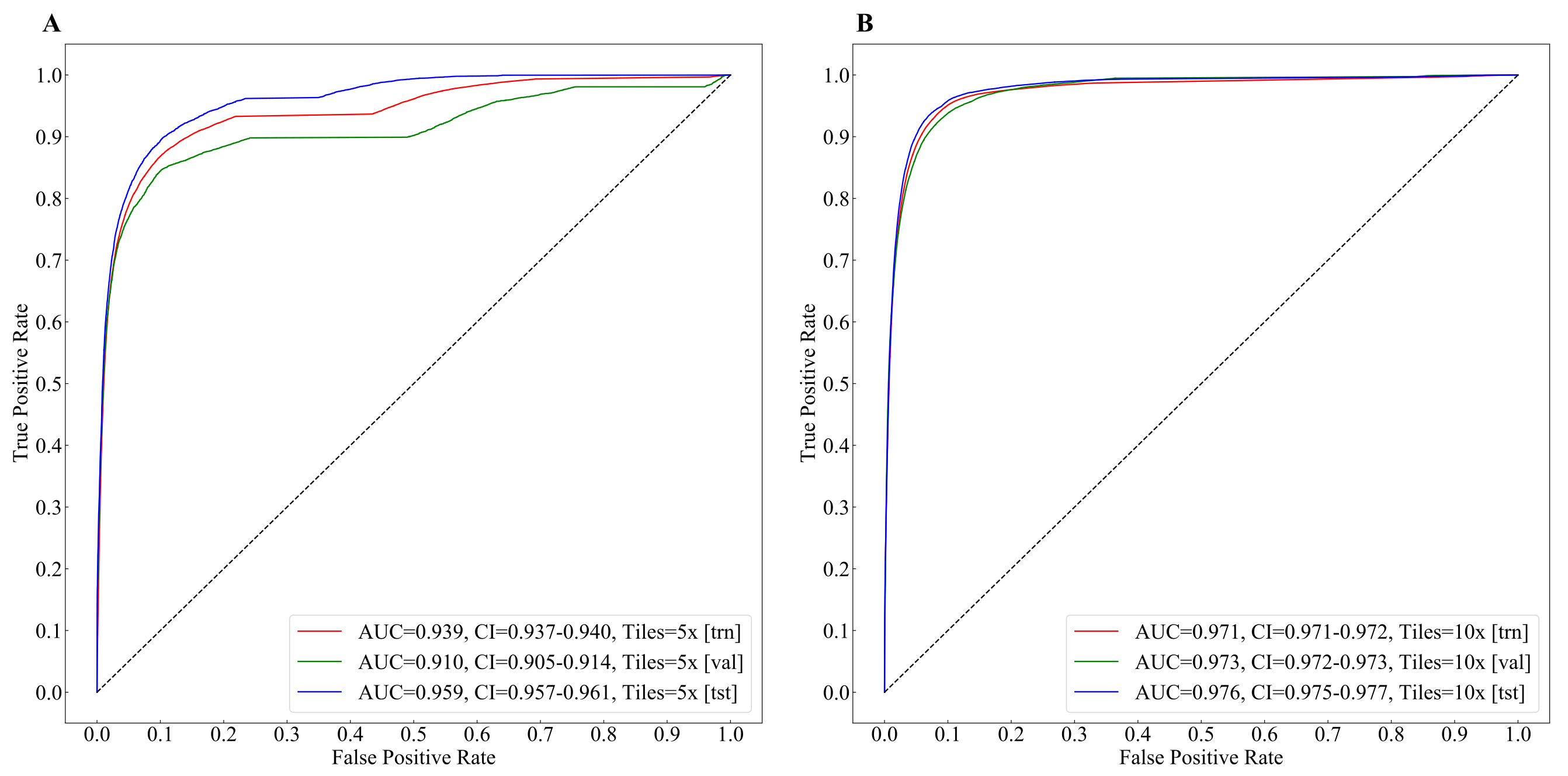
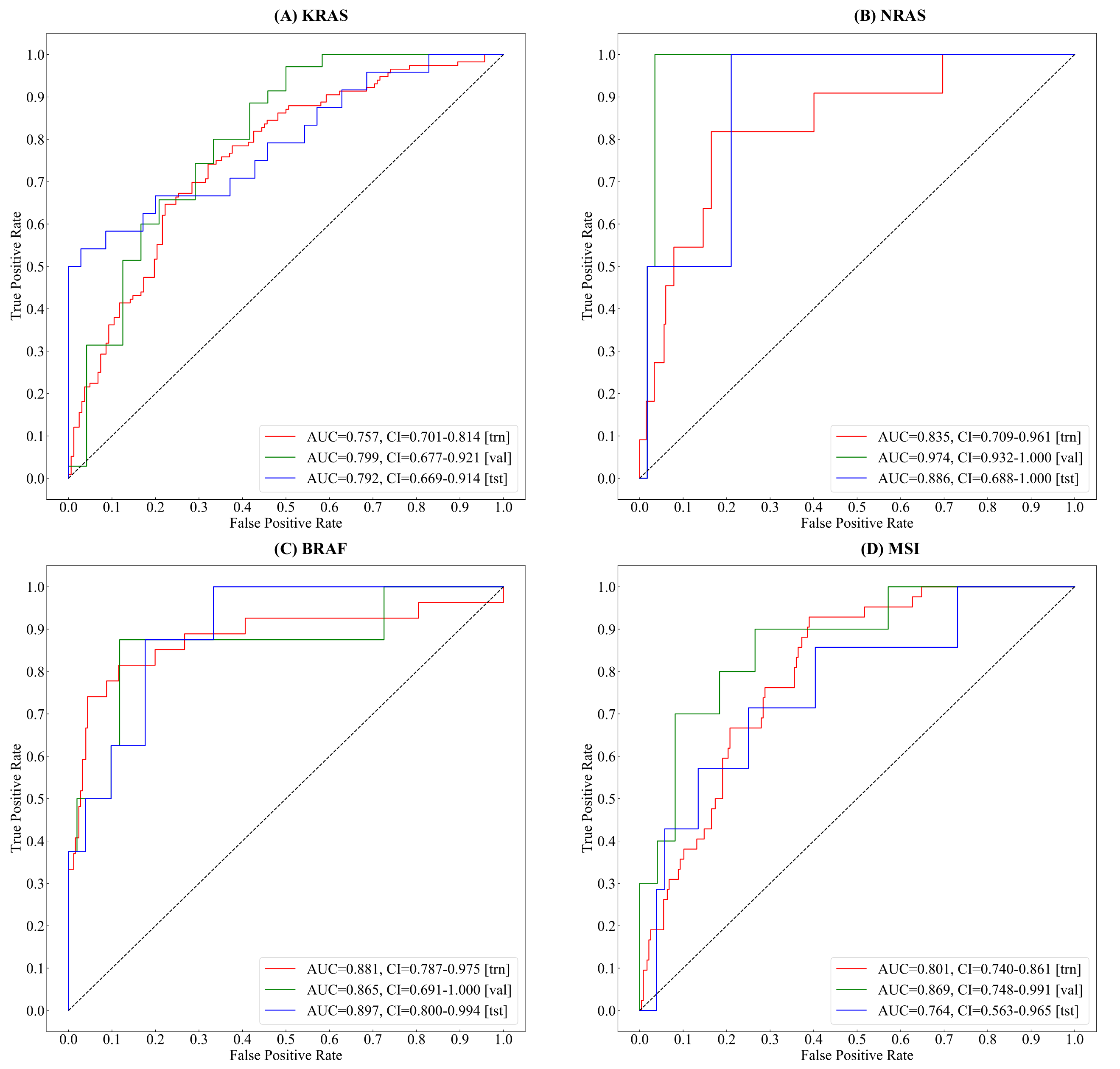
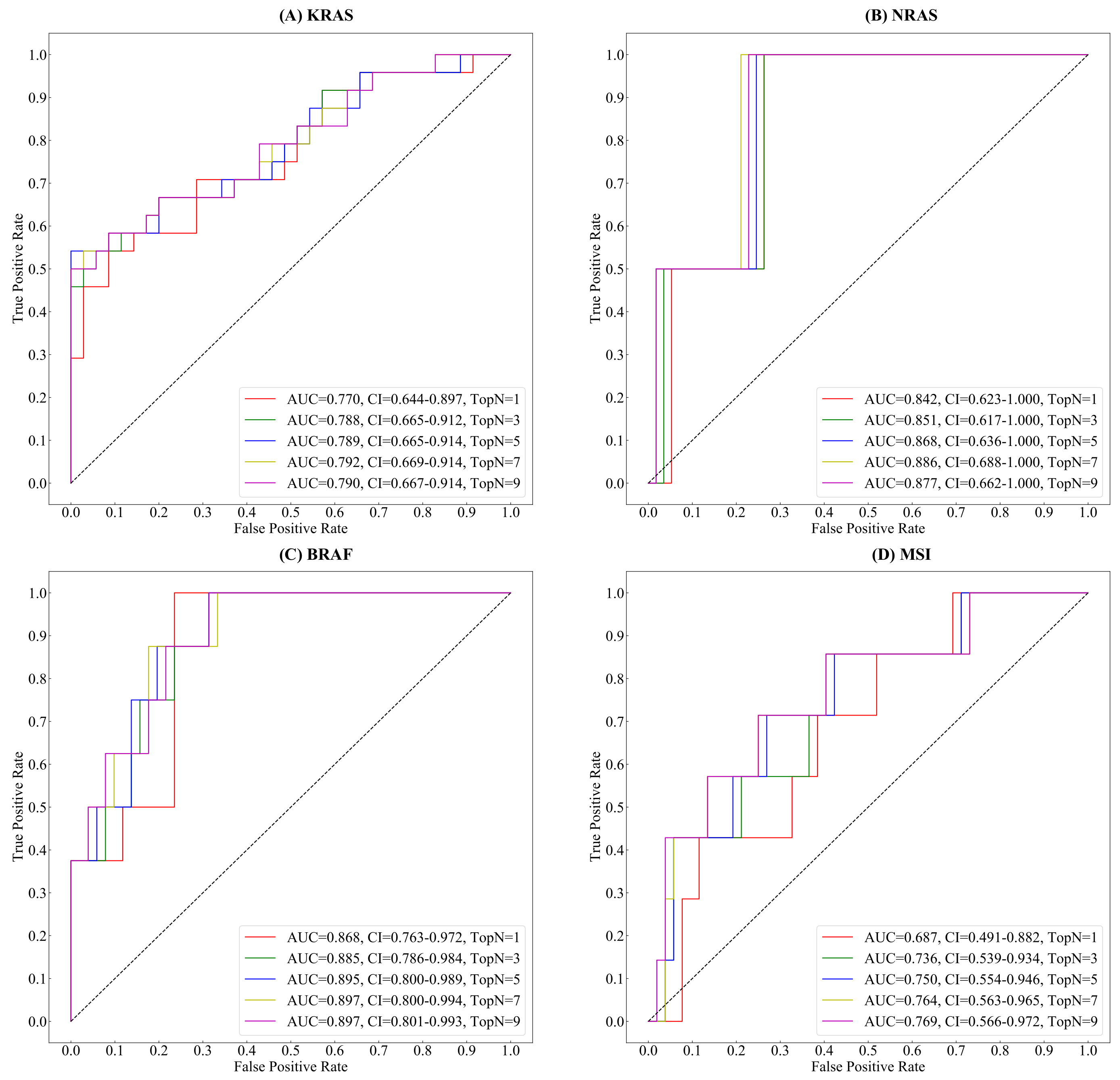
3.1. Tumor Classification
3.2. Gene&MSI Classification
4. Discussion
4.1. Regarding the Cascaded Framework
4.2. Accuracies, Uncertainties, and Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| CRC | Colorectal Cancer |
| MSI | Microsatellite Instability |
| WSI | Whole-Side Image |
| CNN | Convolutional Neural Network |
References
- Wang, Z.; Zhou, C.; Feng, X.; Mo, M.; Shen, J.; Zheng, Y. Comparison of cancer incidence and mortality between China and the United States. Precis. Cancer Med. 2021, 4. [Google Scholar] [CrossRef]
- Xia, C.; Dong, X.; Li, H.; Cao, M.; Sun, D.; He, S.; Yang, F.; Yan, X.; Zhang, S.; Li, N.; et al. Cancer statistics in China and United States, 2022: Profiles, trends, and determinants. Chin. Med. J. 2022, 135, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.M.; Cardona, K.; Russell, M.C. Historical perspective: Two decades of progress in treating metastatic colorectal cancer. J. Surg. Oncol. 2019, 119, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Mo, S.; Dai, W.; Xiang, W.; Han, L.; Li, Q.; Wang, R.; Liu, L.; Zhang, L.; Cai, S.; et al. Prognostic nomograms for predicting cause-specific survival and overall survival of stage I–III colon cancer patients: A large population-based study. Cancer Cell Int. 2019, 19, 1–15. [Google Scholar] [CrossRef]
- Carr, P.R.; Weigl, K.; Edelmann, D.; Jansen, L.; Chang-Claude, J.; Brenner, H.; Hoffmeister, M. Estimation of absolute risk of colorectal cancer based on healthy lifestyle, genetic risk, and colonoscopy status in a population-based study. Gastroenterology 2020, 159, 129–138. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 1–30. [Google Scholar] [CrossRef]
- Mizukami, T.; Izawa, N.; Nakajima, T.E.; Sunakawa, Y. Targeting EGFR and RAS/RAF signaling in the treatment of metastatic colorectal cancer: From current treatment strategies to future perspectives. Drugs 2019, 79, 633–645. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. New Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Tol, J.; Nagtegaal, I.D.; Punt, C.J. BRAF mutation in metastatic colorectal cancer. New Engl. J. Med. 2009, 361, 98–99. [Google Scholar] [CrossRef] [Green Version]
- Taieb, J.; Lapeyre-Prost, A.; Laurent Puig, P.; Zaanan, A. Exploring the best treatment options for BRAF-mutant metastatic colon cancer. Br. J. Cancer 2019, 121, 434–442. [Google Scholar] [CrossRef]
- Copija, A.; Waniczek, D.; Witkoś, A.; Walkiewicz, K.; Nowakowska-Zajdel, E. Clinical significance and prognostic relevance of microsatellite instability in sporadic colorectal cancer patients. Int. J. Mol. Sci. 2017, 18, 107. [Google Scholar] [CrossRef]
- Battaglin, F.; Naseem, M.; Lenz, H.J.; Salem, M.E. Microsatellite instability in colorectal cancer: Overview of its clinical significance and novel perspectives. Clin. Adv. Hematol. Oncol. H&O 2018, 16, 735. [Google Scholar]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Arain, M.A.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Farkas, L.; et al. Colon cancer, version 2.2021, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 329–359. [Google Scholar] [CrossRef]
- McDonough, S.J.; Bhagwate, A.; Sun, Z.; Wang, C.; Zschunke, M.; Gorman, J.A.; Kopp, K.J.; Cunningham, J.M. Use of FFPE-derived DNA in next generation sequencing: DNA extraction methods. PLoS ONE 2019, 14, e0211400. [Google Scholar] [CrossRef]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef]
- Kather, J.N.; Pearson, A.T.; Halama, N.; Jäger, D.; Krause, J.; Loosen, S.H.; Marx, A.; Boor, P.; Tacke, F.; Neumann, U.P.; et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat. Med. 2019, 25, 1054–1056. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, E.Y.; Luchini, C.; Eccher, A.; Tizaoui, K.; Shin, J.I.; Lim, B.J. Artificial Intelligence for Predicting Microsatellite Instability Based on Tumor Histomorphology: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 2462. [Google Scholar] [CrossRef]
- Skrede, O.J.; De Raedt, S.; Kleppe, A.; Hveem, T.S.; Liestøl, K.; Maddison, J.; Askautrud, H.A.; Pradhan, M.; Nesheim, J.A.; Albregtsen, F.; et al. Deep learning for prediction of colorectal cancer outcome: A discovery and validation study. Lancet 2020, 395, 350–360. [Google Scholar] [CrossRef]
- Jia, P.; Yang, X.; Guo, L.; Liu, B.; Lin, J.; Liang, H.; Sun, J.; Zhang, C.; Ye, K. MSIsensor-pro: Fast, accurate, and Matched-normal-sample-free detection of microsatellite instability. Genom. Proteom. Bioinform. 2020, 18, 65–71. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Bradley, A.P. The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognit. 1997, 30, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Xu, W. Fast implementation of DeLong’s algorithm for comparing the areas under correlated receiver operating characteristic curves. IEEE Signal Process. Lett. 2014, 21, 1389–1393. [Google Scholar] [CrossRef]
- Tan, M.; Le, Q. Efficientnet: Rethinking model scaling for convolutional neural networks. In Proceedings of the International Conference on Machine Learning, PMLR, Long Beach, CA, USA, 9–15 June 2019; pp. 6105–6114. [Google Scholar]
- LeCun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef]
- Simonyan, K.; Zisserman, A. Very deep convolutional networks for large-scale image recognition. arXiv 2014, arXiv:1409.1556. [Google Scholar]
- Lin, M.; Chen, Q.; Yan, S. Network in network. arXiv 2013, arXiv:1312.4400. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Szegedy, C.; Vanhoucke, V.; Ioffe, S.; Shlens, J.; Wojna, Z. Rethinking the inception architecture for computer vision. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 2818–2826. [Google Scholar]
- Huang, G.; Liu, Z.; Van Der Maaten, L.; Weinberger, K.Q. Densely connected convolutional networks. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017; pp. 4700–4708. [Google Scholar]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012, 25. [Google Scholar] [CrossRef]
- Baldi, P.; Sadowski, P.J. Understanding dropout. Adv. Neural Inf. Process. Syst. 2013, 26. [Google Scholar]
- Shore, J.; Johnson, R. Properties of cross-entropy minimization. IEEE Trans. Inf. Theory 1981, 27, 472–482. [Google Scholar] [CrossRef]
- Lin, T.Y.; Goyal, P.; Girshick, R.; He, K.; Dollár, P. Focal loss for dense object detection. Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017, 2980–2988.
- Hinton, G.; Srivastava, N.; Swersky, K. Overview of mini-batch gradient descent. Neural Netw. Mach. Learn. 2012, 575. [Google Scholar]
- Jin, Q.; Meng, Z.; Sun, C.; Cui, H.; Su, R. RA-UNet: A hybrid deep attention-aware network to extract liver and tumor in CT scans. Front. Bioeng. Biotechnol. 2020, 1471. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, C.; Huang, J.; Liu, S.; Zhuo, Y.; Lu, X. Deep learning framework based on integration of S-Mask R-CNN and Inception-v3 for ultrasound image-aided diagnosis of prostate cancer. Future Gener. Comput. Syst. 2021, 114, 358–367. [Google Scholar] [CrossRef]
- Ismael, S.A.A.; Mohammed, A.; Hefny, H. An enhanced deep learning approach for brain cancer MRI images classification using residual networks. Artif. Intell. Med. 2020, 102, 101779. [Google Scholar] [CrossRef]
- Hildebrand, L.A.; Pierce, C.J.; Dennis, M.; Paracha, M.; Maoz, A. Artificial intelligence for histology-based detection of microsatellite instability and prediction of response to immunotherapy in colorectal cancer. Cancers 2021, 13, 391. [Google Scholar] [CrossRef]
- Echle, A.; Laleh, N.G.; Quirke, P.; Grabsch, H.; Muti, H.; Saldanha, O.; Brockmoeller, S.; van den Brandt, P.; Hutchins, G.; Richman, S.; et al. Artificial intelligence for detection of microsatellite instability in colorectal cancer—A multicentric analysis of a pre-screening tool for clinical application. ESMO Open 2022, 7, 100400. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, B.; Topatana, W.; Cao, J.; Zhu, H.; Juengpanich, S.; Mao, Q.; Yu, H.; Cai, X. Classification and mutation prediction based on histopathology H&E images in liver cancer using deep learning. NPJ Precis. Oncol. 2020, 4, 1–7. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WSIs | |||||
|---|---|---|---|---|---|
| Train (n = 278) | Val (n = 59) | Test (n = 59) | Overall (n = 396) | ||
| Age(year) | Min. | 22 | 29 | 36 | 22 |
| Max. | 90 | 90 | 90 | 90 | |
| Median | 65.5 | 67 | 68 | 66 | |
| Gender | Male | 138 | 30 | 33 | 201 |
| Female | 140 | 29 | 26 | 195 | |
| KRAS | W.T. | 162 | 24 | 35 | 221 |
| M.T | 116 | 35 | 24 | 175 | |
| NRAS | W.T. | 267 | 57 | 57 | 381 |
| M.T | 11 | 2 | 2 | 15 | |
| BRAF | W.T. | 251 | 51 | 51 | 353 |
| M.T. | 27 | 8 | 8 | 43 | |
| MSI | MSI-H | 236 | 49 | 52 | 337 |
| MSS/MSI-L | 42 | 10 | 7 | 59 | |
| Tiles | 5× Mag. | 283,126 | 49,988 | 55,787 | 388,901 |
| 10× Mag. | 1,152,481 | 203,183 | 227,595 | 1,583,259 | |
| Stage | Operator | Resolution | Channels | Layers |
|---|---|---|---|---|
| 0 | 512 × 512 | 3 | 0 | |
| 1 | Conv3 × 3 | 512 × 512 | 32 | 1 |
| 2 | MBConv1, k3 × 3 | 256 × 256 | 16 | 1 |
| 3 | MBConv6, k3 × 3 | 256 × 256 | 24 | 2 |
| 4 | MBConv6, k5 × 5 | 128 × 128 | 40 | 2 |
| 5 | MBConv6, k3 × 3 | 64 × 64 | 80 | 3 |
| 6 | MBConv6, k5 × 5 | 32 × 32 | 112 | 3 |
| 7 | MBConv6, k5 × 5 | 32 × 32 | 192 | 4 |
| 8 | MBConv6, k3 × 3 | 16 × 16 | 320 | 1 |
| 9 | Conv1 × 1&Pooling | 16 × 16 | 1280 | 1 |
| 10 | Dropout&FC | 1280 × 1 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Lyu, T.; Liu, S.; Zhang, W.; Zhou, Y.; Zeng, C.; Wu, G. Learn to Estimate Genetic Mutation and Microsatellite Instability with Histopathology H&E Slides in Colon Carcinoma. Cancers 2022, 14, 4144. https://doi.org/10.3390/cancers14174144
Guo Y, Lyu T, Liu S, Zhang W, Zhou Y, Zeng C, Wu G. Learn to Estimate Genetic Mutation and Microsatellite Instability with Histopathology H&E Slides in Colon Carcinoma. Cancers. 2022; 14(17):4144. https://doi.org/10.3390/cancers14174144
Chicago/Turabian StyleGuo, Yimin, Ting Lyu, Shuguang Liu, Wei Zhang, Youjian Zhou, Chao Zeng, and Guangming Wu. 2022. "Learn to Estimate Genetic Mutation and Microsatellite Instability with Histopathology H&E Slides in Colon Carcinoma" Cancers 14, no. 17: 4144. https://doi.org/10.3390/cancers14174144