
Multiple Myeloma Therapy: Emerging Trends and Challenges

 , , , , ,
, , , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Evolution and Molecular Basis of Multiple Myeloma
3. Factors Influencing Treatment Strategy and Current Challenges
4. Traditional Therapies
4.1. Alkylating Agents
4.2. Immunomodulatory Drugs
4.3. Proteasome Inhibitors
5. Immunotherapy
5.1. Immune System Dysreguation
5.2. Naked Monocloal Antibodies
5.2.1. Anti-CD38 mAb
5.2.2. Anti-SLAMF7 mAb
5.3. Immune Checkpoint Inhibitors
5.4. Antibody Drug Conjugates
5.4.1. B Cell Maturation Antigen (BCMA)
5.4.2. Other ADC Targets
5.5. Bispecific Antibodies
5.5.1. Bispecific T Cell Engagers
5.5.2. Bispecific NK Cell Engagers
5.6. Chimeric Antigen Receptor (CAR) T Cell Therapy
5.7. Peptite Vaccines
6. Targeted Therapies and Small Molecules
6.1. Exportin Inhibitors
6.2. Histone Deacetylase Inhibitors
6.3. BCL2 Inhibitors
6.4. Hypomethylating Agents
6.5. Proteolysis-Targeting Chimera
7. Emerging Approaches and Future Directions
7.1. Protein Disulfide Isomerase 1 Inhibitors
7.2. Peptidylprolyl Isomerase A
7.3. Sec61 Translocon
7.4. Cyclin-Dependent Kinase 6 (CKD6)
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Short, K.D.; Rajkumar, S.V.; Larson, D.; Buadi, F.; Hayman, S.; Dispenzieri, A.; Gertz, M.; Kumar, S.; Mikhael, J.; Roy, V.; et al. Incidence of extramedullary disease in patients with multiple myeloma in the era of novel therapy, and the activity of pomalidomide on extramedullary myeloma. Leukemia 2011, 25, 906–908. [Google Scholar] [CrossRef] [PubMed]
- Varettoni, M.; Corso, A.; Pica, G.; Mangiacavalli, S.; Pascutto, C.; Lazzarino, M. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: A longitudinal study on 1003 consecutive patients. Ann. Oncol. 2010, 21, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Kumar, S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020, 10, 94. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef]
- Dutta, A.K.; Fink, J.L.; Grady, J.P.; Morgan, G.J.; Mullighan, C.G.; To, L.B.; Hewett, D.R.; Zannettino, A.C.W. Subclonal evolution in disease progression from MGUS/SMM to multiple myeloma is characterised by clonal stability. Leukemia 2019, 33, 457–468. [Google Scholar] [CrossRef]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Melchor, L.; Brioli, A.; Wardell, C.P.; Murison, A.; Potter, N.E.; Kaiser, M.F.; Fryer, R.A.; Johnson, D.C.; Begum, D.B.; Hulkki Wilson, S.; et al. Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia 2014, 28, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Bahlis, N.J. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012, 120, 927–928. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Rustad, E.H.; Hultcrantz, M.; Munshi, N.; Landgren, O. Moving From Cancer Burden to Cancer Genomics for Smoldering Myeloma: A Review. JAMA Oncol. 2020, 6, 425–432. [Google Scholar] [CrossRef]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef]
- Landgren, O.; Hofmann, J.N.; McShane, C.M.; Santo, L.; Hultcrantz, M.; Korde, N.; Mailankody, S.; Kazandjian, D.; Murata, K.; Thoren, K.; et al. Association of Immune Marker Changes with Progression of Monoclonal Gammopathy of Undetermined Significance to Multiple Myeloma. JAMA Oncol. 2019, 5, 1293–1301. [Google Scholar] [CrossRef]
- Mailankody, S.; Kazandjian, D.; Korde, N.; Roschewski, M.; Manasanch, E.; Bhutani, M.; Tageja, N.; Kwok, M.; Zhang, Y.; Zingone, A.; et al. Baseline mutational patterns and sustained MRD negativity in patients with high-risk smoldering myeloma. Blood Adv. 2017, 1, 1911–1918. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed]
- Greipp, P.R.; San Miguel, J.; Durie, B.G.; Crowley, J.J.; Barlogie, B.; Bladé, J.; Boccadoro, M.; Child, J.A.; Avet-Loiseau, H.; Kyle, R.A.; et al. International staging system for multiple myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, R.; Flinchum, R.; Brown, N.; Ramsey, J.; Riccitelli, S.; Heuck, C.; Barlogie, B.; Shaughnessy, J.D., Jr. Translating a gene expression signature for multiple myeloma prognosis into a robust high-throughput assay for clinical use. BMC Med. Genom. 2014, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, R.; Broyl, A.; de Knegt, Y.; van Vliet, M.H.; van Beers, E.H.; van der Holt, B.; el Jarari, L.; Mulligan, G.; Gregory, W.; Morgan, G.; et al. A gene expression signature for high-risk multiple myeloma. Leukemia 2012, 26, 2406–2413. [Google Scholar] [CrossRef]
- Shah, V.; Sherborne, A.L.; Johnson, D.C.; Ellis, S.; Price, A.; Chowdhury, F.; Kendall, J.; Jenner, M.W.; Drayson, M.T.; Owen, R.G.; et al. Predicting ultrahigh risk multiple myeloma by molecular profiling: An analysis of newly diagnosed transplant eligible myeloma XI trial patients. Leukemia 2020, 34, 3091–3096. [Google Scholar] [CrossRef]
- Poczta, A.; Rogalska, A.; Marczak, A. Treatment of Multiple Myeloma and the Role of Melphalan in the Era of Modern Therapies-Current Research and Clinical Approaches. J. Clin. Med. 2021, 10, 1841. [Google Scholar] [CrossRef]
- Schjesvold, F.; Oriol, A. Current and Novel Alkylators in Multiple Myeloma. Cancers 2021, 13, 2465. [Google Scholar] [CrossRef]
- Mateos, M.V.; Bladé, J.; Bringhen, S.; Ocio, E.M.; Efebera, Y.; Pour, L.; Gay, F.; Sonneveld, P.; Gullbo, J.; Richardson, P.G. Melflufen: A Peptide-Drug Conjugate for the Treatment of Multiple Myeloma. J. Clin. Med. 2020, 9, 3120. [Google Scholar] [CrossRef]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2010, 24, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Davies, F.E.; Raje, N.; Hideshima, T.; Lentzsch, S.; Young, G.; Tai, Y.T.; Lin, B.; Podar, K.; Gupta, D.; Chauhan, D.; et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001, 98, 210–216. [Google Scholar] [CrossRef]
- Hayashi, T.; Hideshima, T.; Akiyama, M.; Podar, K.; Yasui, H.; Raje, N.; Kumar, S.; Chauhan, D.; Treon, S.P.; Richardson, P.; et al. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: Clinical application. Br. J. Haematol. 2005, 128, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Fedele, P.L.; Willis, S.N.; Liao, Y.; Low, M.S.; Rautela, J.; Segal, D.H.; Gong, J.N.; Huntington, N.D.; Shi, W.; Huang, D.C.S.; et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood 2018, 132, 2166–2178. [Google Scholar] [CrossRef]
- Hsu, A.K.; Quach, H.; Tai, T.; Prince, H.M.; Harrison, S.J.; Trapani, J.A.; Smyth, M.J.; Neeson, P.; Ritchie, D.S. The immunostimulatory effect of lenalidomide on NK-cell function is profoundly inhibited by concurrent dexamethasone therapy. Blood 2011, 117, 1605–1613. [Google Scholar] [CrossRef]
- Hideshima, T.; Ogiya, D.; Liu, J.; Harada, T.; Kurata, K.; Bae, J.; Massefski, W.; Anderson, K.C. Immunomodulatory drugs activate NK cells via both Zap-70 and cereblon-dependent pathways. Leukemia 2021, 35, 177–188. [Google Scholar] [CrossRef]
- Galustian, C.; Meyer, B.; Labarthe, M.C.; Dredge, K.; Klaschka, D.; Henry, J.; Todryk, S.; Chen, R.; Muller, G.; Stirling, D.; et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol. Immunother. 2009, 58, 1033–1045. [Google Scholar] [CrossRef]
- Reddy, N.; Hernandez-Ilizaliturri, F.J.; Deeb, G.; Roth, M.; Vaughn, M.; Knight, J.; Wallace, P.; Czuczman, M.S. Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br. J. Haematol. 2008, 140, 36–45. [Google Scholar] [CrossRef]
- Thakurta, A.; Pierceall, W.E.; Amatangelo, M.D.; Flynt, E.; Agarwal, A. Developing next generation immunomodulatory drugs and their combinations in multiple myeloma. Oncotarget 2021, 12, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, C.C.; Kang, J.; Amatangelo, M.; Polonskaia, A.; Katz, M.; Chiu, H.; Couto, S.; Wang, M.; Ren, Y.; Ortiz, M.; et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia 2020, 34, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Donk, N.W.C.J.v.d.; Popat, R.; Zonder, J.A.; Minnema, M.C.; Larsen, J.; Nguyen, T.V.; Chen, M.S.; Bensmaine, A.; Cota, M.; et al. First clinical (phase 1b/2a) study of iberdomide (CC-220; IBER), a CELMoD, in combination with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2019, 37, 8006. [Google Scholar] [CrossRef]
- Van de Donk, N.W.C.J.; Popat, R.; Larsen, J.; Minnema, M.C.; Jagannath, S.; Oriol, A.; Zonder, J.; Richardson, P.G.; Rodriguez-Otero, P.; Badros, A.Z.; et al. First Results of Iberdomide (IBER.; CC-220) in Combination with Dexamethasone (DEX) and Daratumumab (DARA) or Bortezomib (BORT) in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 16–17. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Havens, C.G.; Lu, G.; Rychak, E.; Mendy, D.; Gaffney, B.; Surka, C.; Lu, C.-C.; Matyskiela, M.; Khambatta, G.; et al. CC-92480 Is a Novel Cereblon E3 Ligase Modulator with Enhanced Tumoricidal and Immunomodulatory Activity Against Sensitive and Resistant Multiple Myeloma Cells. Blood 2019, 134, 1812. [Google Scholar] [CrossRef]
- Richardson, P.G.; Vangsted, A.J.; Ramasamy, K.; Trudel, S.; Martínez, J.; Mateos, M.-V.; Otero, P.R.; Lonial, S.; Popat, R.; Oriol, A.; et al. First-in-human phase I study of the novel CELMoD agent CC-92480 combined with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2020, 38, 8500. [Google Scholar] [CrossRef]
- Rasco, D.W.; Papadopoulos, K.P.; Pourdehnad, M.; Gandhi, A.K.; Hagner, P.R.; Li, Y.; Wei, X.; Chopra, R.; Hege, K.; DiMartino, J.; et al. A First-in-Human Study of Novel Cereblon Modulator Avadomide (CC-122) in Advanced Malignancies. Clin. Cancer Res. 2019, 25, 90–98. [Google Scholar] [CrossRef]
- Berdeja, J.; Ailawadhi, S.; Horwitz, S.M.; Matous, J.V.; Mehta-Shah, N.; Martin, T.; Muchtar, E.; Richardson, P.G.; Richard, S.; Bhutani, M.; et al. A Phase 1 Study of CFT7455, a Novel Degrader of IKZF1/3, in Multiple Myeloma and Non-Hodgkin Lymphoma. Blood 2021, 138, 1675. [Google Scholar] [CrossRef]
- McConkey, D.J.; Zhu, K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist. Updat. 2008, 11, 164–179. [Google Scholar] [CrossRef]
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2011, 10, 2034–2042. [Google Scholar] [CrossRef]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Gulla, A.; Morelli, E.; Samur, M.K.; Botta, C.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Malvestiti, S.; Prabhala, R.H.; Talluri, S.; et al. Bortezomib induces anti-multiple myeloma immune response mediated by cGAS/STING pathway activation. Blood Cancer Discov. 2021, 2, 468–483. [Google Scholar] [CrossRef] [PubMed]
- Spisek, R.; Charalambous, A.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: Therapeutic implications. Blood 2007, 109, 4839–4845. [Google Scholar] [CrossRef]
- Shi, J.; Tricot, G.J.; Garg, T.K.; Malaviarachchi, P.A.; Szmania, S.M.; Kellum, R.E.; Storrie, B.; Mulder, A.; Shaughnessy, J.D., Jr.; Barlogie, B.; et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood 2008, 111, 1309–1317. [Google Scholar] [CrossRef]
- Ito, S. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef]
- Fostier, K.; De Becker, A.; Schots, R. Carfilzomib: A novel treatment in relapsed and refractory multiple myeloma. OncoTargets Ther. 2012, 5, 237–244. [Google Scholar] [CrossRef]
- Gupta, N.; Hanley, M.J.; Xia, C.; Labotka, R.; Harvey, R.D.; Venkatakrishnan, K. Clinical Pharmacology of Ixazomib: The First Oral Proteasome Inhibitor. Clin. Pharmacokinet. 2019, 58, 431–449. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors—Molecular basis and current perspectives in multiple myeloma. J. Cell Mol. Med. 2014, 18, 947–961. [Google Scholar] [CrossRef]
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C., Jr.; Fenical, W.; et al. Marizomib, a proteasome inhibitor for all seasons: Preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 2011, 11, 254–284. [Google Scholar] [CrossRef]
- Hari, P.; Matous, J.V.; Voorhees, P.M.; Shain, K.H.; Obreja, M.; Frye, J.; Fujii, H.; Jakubowiak, A.J.; Rossi, D.; Sonneveld, P. Oprozomib in patients with newly diagnosed multiple myeloma. Blood Cancer J. 2019, 9, 66. [Google Scholar] [CrossRef]
- Hari, P.; Paba-Prada, C.E.; Voorhees, P.M.; Frye, J.; Chang, Y.L.; Moreau, P.; Zonder, J.; Boccia, R.; Shain, K.H. Efficacy and safety results from a phase 1b/2, multicenter, open-label study of oprozomib and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Leuk Res. 2019, 83, 106172. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, E.; Jung, C.K.; Kang, S.W.; Kim, B.G.; Jung, Y.; Kim, T.H.; Lim, J.Y.; Lee, S.E.; Min, C.K.; et al. Oral proteasome inhibitor with strong preclinical efficacy in myeloma models. BMC Cancer 2016, 16, 247. [Google Scholar] [CrossRef] [PubMed]
- Swamydas, M.; Murphy, E.V.; Ignatz-Hoover, J.J.; Malek, E.; Driscoll, J.J. Deciphering mechanisms of immune escape to inform immunotherapeutic strategies in multiple myeloma. J. Hematol. Oncol. 2022, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Mutis, T.; Lokhorst, H.M.; van de Donk, N. Immunotherapy in myeloma: How far have we come? Ther. Adv. Hematol. 2019, 10, 2040620718822660. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Vyas, M.; Müller, R.; Pogge von Strandmann, E. Antigen Loss Variants: Catching Hold of Escaping Foes. Front. Immunol. 2017, 8, 175. [Google Scholar] [CrossRef]
- Lozano, E.; Díaz, T.; Mena, M.P.; Suñe, G.; Calvo, X.; Calderón, M.; Pérez-Amill, L.; Rodríguez, V.; Pérez-Galán, P.; Roué, G.; et al. Loss of the Immune Checkpoint CD85j/LILRB1 on Malignant Plasma Cells Contributes to Immune Escape in Multiple Myeloma. J. Immunol. 2018, 200, 2581–2591. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef]
- Gulla, A.; Morelli, E.; Samur, M.K.; Botta, C.; Johnstone, M.; Bianchi, G.; Fulciniti, M.; Yamamoto, L.; Prabhala, R.; Wen, K.; et al. Gabarap Loss Mediates Immune Escape in High Risk Multiple Myeloma. Blood 2021, 138, 891. [Google Scholar] [CrossRef]
- Racanelli, V.; Leone, P.; Frassanito, M.A.; Brunetti, C.; Perosa, F.; Ferrone, S.; Dammacco, F. Alterations in the antigen processing-presenting machinery of transformed plasma cells are associated with reduced recognition by CD8+ T cells and characterize the progression of MGUS to multiple myeloma. Blood 2010, 115, 1185–1193. [Google Scholar] [CrossRef]
- Kumar, S.; Kimlinger, T.; Morice, W. Immunophenotyping in multiple myeloma and related plasma cell disorders. Best Pract. Res. Clin. Haematol. 2010, 23, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.; Rendón-Huerta, E.P.; Ortiz-Navarrete, V.; Montaño, L.F. CD38 and Regulation of the Immune Response Cells in Cancer. J. Oncol. 2021, 2021, 6630295. [Google Scholar] [CrossRef] [PubMed]
- Maecker, B.; Anderson, K.S.; von Bergwelt-Baildon, M.S.; Weller, E.; Vonderheide, R.H.; Richardson, P.G.; Schlossman, R.L.; Menezes, I.A.; Xia, Z.; Munshi, N.C.; et al. Viral antigen-specific CD8+ T-cell responses are impaired in multiple myeloma. Br. J. Haematol. 2003, 121, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Krasovsky, J.; Osman, K.; Geller, M.D. Vigorous premalignancy-specific effector T cell response in the bone marrow of patients with monoclonal gammopathy. J. Exp. Med. 2003, 198, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Suen, H.; Brown, R.; Yang, S.; Weatherburn, C.; Ho, P.J.; Woodland, N.; Nassif, N.; Barbaro, P.; Bryant, C.; Hart, D.; et al. Multiple myeloma causes clonal T-cell immunosenescence: Identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 2016, 30, 1716–1724. [Google Scholar] [CrossRef]
- Sharma, A.; Khan, R.; Joshi, S.; Kumar, L.; Sharma, M. Dysregulation in T helper 1/T helper 2 cytokine ratios in patients with multiple myeloma. Leuk Lymphoma 2010, 51, 920–927. [Google Scholar] [CrossRef]
- Bernal, M.; Garrido, P.; Jiménez, P.; Carretero, R.; Almagro, M.; López, P.; Navarro, P.; Garrido, F.; Ruiz-Cabello, F. Changes in activatory and inhibitory natural killer (NK) receptors may induce progression to multiple myeloma: Implications for tumor evasion of T and NK cells. Hum. Immunol. 2009, 70, 854–857. [Google Scholar] [CrossRef]
- Jinushi, M.; Vanneman, M.; Munshi, N.C.; Tai, Y.T.; Prabhala, R.H.; Ritz, J.; Neuberg, D.; Anderson, K.C.; Carrasco, D.R.; Dranoff, G. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc. Natl. Acad. Sci. USA 2008, 105, 1285–1290. [Google Scholar] [CrossRef]
- De Jong, M.M.E.; Kellermayer, Z.; Papazian, N.; Tahri, S.; Hofste Op Bruinink, D.; Hoogenboezem, R.; Sanders, M.A.; van de Woestijne, P.C.; Bos, P.K.; Khandanpour, C.; et al. The multiple myeloma microenvironment is defined by an inflammatory stromal cell landscape. Nat. Immunol. 2021, 22, 769–780. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Neri, P.; Bae, J.E.; Tassone, P.; Shammas, M.A.; Allam, C.K.; Daley, J.F.; Chauhan, D.; Blanchard, E.; Thatte, H.S.; et al. Dysfunctional T regulatory cells in multiple myeloma. Blood 2006, 107, 301–304. [Google Scholar] [CrossRef]
- Leone, P.; Berardi, S.; Frassanito, M.A.; Ria, R.; De Re, V.; Cicco, S.; Battaglia, S.; Ditonno, P.; Dammacco, F.; Vacca, A.; et al. Dendritic cells accumulate in the bone marrow of myeloma patients where they protect tumor plasma cells from CD8+ T-cell killing. Blood 2015, 126, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.K.; Dhodapkar, M.V.; Matayeva, E.; Steinman, R.M.; Dhodapkar, K.M. Expansion of FOXP3high regulatory T cells by human dendritic cells (DCs) in vitro and after injection of cytokine-matured DCs in myeloma patients. Blood 2006, 108, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone marrow microenvironment in multiple myeloma progression. J. Biomed. Biotechnol. 2012, 2012, 157496. [Google Scholar] [CrossRef] [PubMed]
- Dima, D.; Dower, J.; Comenzo, R.L.; Varga, C. Evaluating Daratumumab in the Treatment of Multiple Myeloma: Safety, Efficacy and Place in Therapy. Cancer Manag. Res. 2020, 12, 7891–7903. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Structure and enzymatic functions of human CD38. Mol. Med. 2006, 12, 317–323. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Quarona, V.; Toscani, D.; Costa, F.; Chillemi, A.; Pistoia, V.; Giuliani, N.; Malavasi, F. Adenosine Generated in the Bone Marrow Niche Through a CD38-Mediated Pathway Correlates with Progression of Human Myeloma. Mol. Med. 2016, 22, 694–704. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- De Weers, M.; Tai, Y.T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Jansen, J.H.; Nederend, M.; Lammerts van Bueren, J.J.; Groen, R.W.; Parren, P.W.; Leusen, J.H.; Boross, P. The Therapeutic CD38 Monoclonal Antibody Daratumumab Induces Programmed Cell Death via Fcγ Receptor-Mediated Cross-Linking. J. Immunol. 2016, 197, 807–813. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Bögels, M.; van Egmond, M.; Lammerts van Bueren, J.J.; Mutis, T.; Groen, R.W.; Breij, E.; Martens, A.C.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs 2015, 7, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Weiss, B.M.; Plesner, T.; Bahlis, N.J.; Belch, A.; Lonial, S.; Lokhorst, H.M.; Voorhees, P.M.; Richardson, P.G.; Chari, A.; et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood 2016, 128, 37–44. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Van der Veer, M.S.; de Weers, M.; van Kessel, B.; Bakker, J.M.; Wittebol, S.; Parren, P.W.; Lokhorst, H.M.; Mutis, T. Towards effective immunotherapy of myeloma: Enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica 2011, 96, 284–290. [Google Scholar] [CrossRef]
- Deckert, J.; Wetzel, M.C.; Bartle, L.M.; Skaletskaya, A.; Goldmacher, V.S.; Vallée, F.; Zhou-Liu, Q.; Ferrari, P.; Pouzieux, S.; Lahoute, C.; et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin. Cancer Res. 2014, 20, 4574–4583. [Google Scholar] [CrossRef]
- Van de Donk, N.W.; Moreau, P.; Plesner, T.; Palumbo, A.; Gay, F.; Laubach, J.P.; Malavasi, F.; Avet-Loiseau, H.; Mateos, M.V.; Sonneveld, P.; et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood 2016, 127, 681–695. [Google Scholar] [CrossRef]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): A randomised, multicentre, open-label, phase 3 study. Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef]
- Moreau, P.; Dimopoulos, M.A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Špička, I.; Baker, R.; Kim, K.; et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): A multicentre, open-label, randomised phase 3 trial. Lancet 2021, 397, 2361–2371. [Google Scholar] [CrossRef]
- Veillette, A.; Guo, H. CS1, a SLAM family receptor involved in immune regulation, is a therapeutic target in multiple myeloma. Crit. Rev. Oncol. Hematol. 2013, 88, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Lonial, S.; Jakubowiak, A.J.; Harousseau, J.L.; Anderson, K.C. Monoclonal antibodies in the treatment of multiple myeloma. Br. J. Haematol. 2011, 154, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, C.C.; Lonial, S. How to Integrate Elotuzumab and Daratumumab Into Therapy for Multiple Myeloma. J. Clin. Oncol. 2016, 34, 4421–4430. [Google Scholar] [CrossRef] [PubMed]
- Kumaresan, P.R.; Lai, W.C.; Chuang, S.S.; Bennett, M.; Mathew, P.A. CS1, a novel member of the CD2 family, is homophilic and regulates NK cell function. Mol. Immunol. 2002, 39, 1–8. [Google Scholar] [CrossRef]
- Tai, Y.T.; Dillon, M.; Song, W.; Leiba, M.; Li, X.F.; Burger, P.; Lee, A.I.; Podar, K.; Hideshima, T.; Rice, A.G.; et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 2008, 112, 1329–1337. [Google Scholar] [CrossRef]
- Wang, Y.; Sanchez, L.; Siegel, D.S.; Wang, M.L. Elotuzumab for the treatment of multiple myeloma. J. Hematol. Oncol. 2016, 9, 55. [Google Scholar] [CrossRef]
- Pazina, T.; James, A.M.; MacFarlane, A.W.t.; Bezman, N.A.; Henning, K.A.; Bee, C.; Graziano, R.F.; Robbins, M.D.; Cohen, A.D.; Campbell, K.S. The anti-SLAMF7 antibody elotuzumab mediates NK cell activation through both CD16-dependent and -independent mechanisms. Oncoimmunology 2017, 6, e1339853. [Google Scholar] [CrossRef]
- Hsi, E.D.; Steinle, R.; Balasa, B.; Szmania, S.; Draksharapu, A.; Shum, B.P.; Huseni, M.; Powers, D.; Nanisetti, A.; Zhang, Y.; et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin. Cancer Res. 2008, 14, 2775–2784. [Google Scholar] [CrossRef]
- Sola, C.; Blery, M.; Bonnafous, C.; Bonnet, E.; Fuseri, N.; Graziano, R.F.; Morel, Y.; André, P. Lirilumab Enhances Anti-Tumor Efficacy of Elotuzumab. Blood 2014, 124, 4711. [Google Scholar] [CrossRef]
- Robbins, M.; Jure-Kunkel, M.; Dito, G.; Andre, P.; Zhang, H.-f.; Bezman, N.; Graziano, R.F. Effects of IL-21, KIR Blockade, and CD137 Agonism on the Non-Clinical Activity of Elotuzumab. Blood 2014, 124, 4717. [Google Scholar] [CrossRef]
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849. [Google Scholar] [CrossRef]
- Van Rhee, F.; Szmania, S.M.; Dillon, M.; van Abbema, A.M.; Li, X.; Stone, M.K.; Garg, T.K.; Shi, J.; Moreno-Bost, A.M.; Yun, R.; et al. Combinatorial efficacy of anti-CS1 monoclonal antibody elotuzumab (HuLuc63) and bortezomib against multiple myeloma. Mol. Cancer Ther. 2009, 8, 2616–2624. [Google Scholar] [CrossRef]
- Balasa, B.; Yun, R.; Belmar, N.A.; Fox, M.; Chao, D.T.; Robbins, M.D.; Starling, G.C.; Rice, A.G. Elotuzumab enhances natural killer cell activation and myeloma cell killing through interleukin-2 and TNF-α pathways. Cancer Immunol. Immunother. 2015, 64, 61–73. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Lonial, S.; Betts, K.A.; Chen, C.; Zichlin, M.L.; Brun, A.; Signorovitch, J.E.; Makenbaeva, D.; Mekan, S.; Sy, O.; et al. Elotuzumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: Extended 4-year follow-up and analysis of relative progression-free survival from the randomized ELOQUENT-2 trial. Cancer 2018, 124, 4032–4043. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Kwon, M.; Kim, C.G.; Lee, H.; Cho, H.; Kim, Y.; Lee, E.C.; Choi, S.J.; Park, J.; Seo, I.H.; Bogen, B.; et al. PD-1 Blockade Reinvigorates Bone Marrow CD8(+) T Cells from Patients with Multiple Myeloma in the Presence of TGFβ Inhibitors. Clin. Cancer Res. 2020, 26, 1644–1655. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef]
- Tamura, H.; Ishibashi, M.; Yamashita, T.; Tanosaki, S.; Okuyama, N.; Kondo, A.; Hyodo, H.; Shinya, E.; Takahashi, H.; Dong, H.; et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 2013, 27, 464–472. [Google Scholar] [CrossRef]
- Mussetti, A.; Pellegrinelli, A.; Cieri, N.; Garzone, G.; Dominoni, F.; Cabras, A.; Montefusco, V. PD-L1, LAG3, and HLA-DR are increasingly expressed during smoldering myeloma progression. Ann. Hematol. 2019, 98, 1713–1720. [Google Scholar] [CrossRef]
- Lucas, F.; Pennell, M.; Huang, Y.; Benson, D.M.; Efebera, Y.A.; Chaudhry, M.; Hughes, T.; Woyach, J.A.; Byrd, J.C.; Zhang, S.; et al. T Cell Transcriptional Profiling and Immunophenotyping Uncover LAG3 as a Potential Significant Target of Immune Modulation in Multiple Myeloma. Biol. Blood Marrow Transplant. 2020, 26, 7–15. [Google Scholar] [CrossRef]
- Guillerey, C.; Harjunpää, H.; Carrié, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT immune checkpoint blockade restores CD8(+) T-cell immunity against multiple myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef]
- Asimakopoulos, F. TIGIT checkpoint inhibition for myeloma. Blood 2018, 132, 1629–1630. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef]
- Ansell, S.; Gutierrez, M.E.; Shipp, M.A.; Gladstone, D.; Moskowitz, A.; Borello, I.; Popa-Mckiver, M.; Farsaci, B.; Zhu, L.; Lesokhin, A.M.; et al. A Phase 1 Study of Nivolumab in Combination with Ipilimumab for Relapsed or Refractory Hematologic Malignancies (CheckMate 039). Blood 2016, 128, 183. [Google Scholar] [CrossRef]
- Görgün, G.; Samur, M.K.; Cowens, K.B.; Paula, S.; Bianchi, G.; Anderson, J.E.; White, R.E.; Singh, A.; Ohguchi, H.; Suzuki, R.; et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4607–4618. [Google Scholar] [CrossRef]
- Verkleij, C.P.M.; Jhatakia, A.; Broekmans, M.E.C.; Frerichs, K.A.; Zweegman, S.; Mutis, T.; Bezman, N.A.; van de Donk, N. Preclinical Rationale for Targeting the PD-1/PD-L1 Axis in Combination with a CD38 Antibody in Multiple Myeloma and Other CD38-Positive Malignancies. Cancers 2020, 12, 3713. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Orlowski, R.Z.; Siegel, D.S.D.; Reece, D.E.; Moreau, P.; Ocio, E.M.; Shah, J.J.; Rodríguez-Otero, P.; Munshi, N.C.; Avigan, D.; et al. Pembrolizumab in combination with lenalidomide and low-dose dexamethasone for relapsed/refractory multiple myeloma (RRMM): Final efficacy and safety analysis. J. Clin. Oncol. 2016, 34, 8010. [Google Scholar] [CrossRef]
- Badros, A.; Hyjek, E.; Ma, N.; Lesokhin, A.; Dogan, A.; Rapoport, A.P.; Kocoglu, M.; Lederer, E.; Philip, S.; Milliron, T.; et al. Pembrolizumab, pomalidomide, and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood 2017, 130, 1189–1197. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Yimer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Cho, H.J.; Costa, L.J.; Davies, F.E.; Neparidze, N.; Vij, R.; Feng, Y.; Teterina, A.; Wassner Fritsch, E.; Wenger, M.; Kaufman, J.L. Atezolizumab in Combination with Daratumumab with or without Lenalidomide or Pomalidomide: A Phase Ib Study in Patients with Multiple Myeloma. Blood 2018, 132, 597. [Google Scholar] [CrossRef]
- Verkleij, C.P.M.; Minnema, M.C.; de Weerdt, O.; Bosman, P.W.C.; Frerichs, K.A.; Croockewit, A.J.; Klein, S.K.; Bos, G.; Mutis, T.; Plattel, W.J.; et al. Efficacy and Safety of Nivolumab Combined with Daratumumab with or without Low-Dose Cyclophosphamide in Relapsed/Refractory Multiple Myeloma; Interim Analysis of the Phase 2 Nivo-Dara Study. Blood 2019, 134, 1879. [Google Scholar] [CrossRef]
- Cohen, Y.C.; Oriol, A.; Wu, K.L.; Lavi, N.; Vlummens, P.; Jackson, C.; Garvin, W.; Carson, R.; Crist, W.; Fu, J.; et al. Daratumumab With Cetrelimab, an Anti-PD-1 Monoclonal Antibody, in Relapsed/Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk 2021, 21, 46–54.e44. [Google Scholar] [CrossRef]
- Frerichs, K.A.; Verkleij, C.P.M.; Dimopoulos, M.A.; Marin Soto, J.A.; Zweegman, S.; Young, M.H.; Newhall, K.J.; Mutis, T.; van de Donk, N. Efficacy and Safety of Durvalumab Combined with Daratumumab in Daratumumab-Refractory Multiple Myeloma Patients. Cancers 2021, 13, 2452. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef]
- Yu, B.; Liu, D. Antibody-drug conjugates in clinical trials for lymphoid malignancies and multiple myeloma. J. Hematol. Oncol. 2019, 12, 94. [Google Scholar] [CrossRef]
- Herrera, A.F.; Molina, A. Investigational Antibody-Drug Conjugates for Treatment of B-lineage Malignancies. Clin. Lymphoma Myeloma Leuk 2018, 18, 452–468.e454. [Google Scholar] [CrossRef]
- Skaletskaya, A.; Setiady, Y.Y.; Park, P.U.; Lutz, R.J. Abstract 770: Lorvotuzumab mertansine (IMGN901) immune effector activity and its effect on human NK cells. Cancer Res. 2011, 71, 770. [Google Scholar] [CrossRef]
- Tai, Y.T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Yu, B.; Jiang, T.; Liu, D. BCMA-targeted immunotherapy for multiple myeloma. J. Hematol. Oncol. 2020, 13, 125. [Google Scholar] [CrossRef]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef]
- Tai, Y.T.; Anderson, K.C. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015, 7, 1187–1199. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Anderson, L.D., Jr.; Sutherland, H.J.; Yong, K.; et al. Targeting B-cell maturation antigen with GSK2857916 antibody-drug conjugate in relapsed or refractory multiple myeloma (BMA117159): A dose escalation and expansion phase 1 trial. Lancet Oncol. 2018, 19, 1641–1653. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody-drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9, 37. [Google Scholar] [CrossRef]
- Richardson, P.G.; Lee, H.C.; Abdallah, A.O.; Cohen, A.D.; Kapoor, P.; Voorhees, P.M.; Hoos, A.; Wang, K.; Baron, J.; Piontek, T.; et al. Single-agent belantamab mafodotin for relapsed/refractory multiple myeloma: Analysis of the lyophilised presentation cohort from the pivotal DREAMM-2 study. Blood Cancer J. 2020, 10, 106. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Sborov, D.; Suvannasankha, A.; et al. Longer term outcomes with single-agent belantamab mafodotin in patients with relapsed or refractory multiple myeloma: 13-month follow-up from the pivotal DREAMM-2 study. Cancer 2021, 127, 4198–4212. [Google Scholar] [CrossRef]
- Tai, Y.T.; Anderson, K.C. B cell maturation antigen (BCMA)-based immunotherapy for multiple myeloma. Expert Opin. Biol. Ther. 2019, 19, 1143–1156. [Google Scholar] [CrossRef]
- Lee, H.C.; Raje, N.S.; Landgren, O.; Upreti, V.V.; Wang, J.; Avilion, A.A.; Hu, X.; Rasmussen, E.; Ngarmchamnanrith, G.; Fujii, H.; et al. Phase 1 study of the anti-BCMA antibody-drug conjugate AMG 224 in patients with relapsed/refractory multiple myeloma. Leukemia 2021, 35, 255–258. [Google Scholar] [CrossRef]
- Kinneer, K.; Flynn, M.; Thomas, S.B.; Meekin, J.; Varkey, R.; Xiao, X.; Zhong, H.; Breen, S.; Hynes, P.G.; Fleming, R.; et al. Preclinical assessment of an antibody-PBD conjugate that targets BCMA on multiple myeloma and myeloma progenitor cells. Leukemia 2019, 33, 766–771. [Google Scholar] [CrossRef]
- Xing, L.; Lin, L.; Yu, T.; Li, Y.; Wen, K.; Cho, S.-F.; Hsieh, P.A.; Kinneer, K.; Munshi, N.C.; Anderson, K.C.; et al. Anti-Bcma PBD MEDI2228 Combats Drug Resistance and Synergizes with Bortezomib and Inhibitors to DNA Damage Response in Multiple Myeloma. Blood 2019, 134, 1817. [Google Scholar] [CrossRef]
- Xing, L.; Lin, L.; Yu, T.; Li, Y.; Cho, S.F.; Liu, J.; Wen, K.; Hsieh, P.A.; Kinneer, K.; Munshi, N.; et al. A novel BCMA PBD-ADC with ATM/ATR/WEE1 inhibitors or bortezomib induce synergistic lethality in multiple myeloma. Leukemia 2020, 34, 2150–2162. [Google Scholar] [CrossRef]
- Kumar, S.K.; Migkou, M.; Bhutani, M.; Spencer, A.; Ailawadhi, S.; Kalff, A.; Walcott, F.; Pore, N.; Gibson, D.; Wang, F.; et al. Phase 1, First-in-Human Study of MEDI2228, a BCMA-Targeted ADC in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 26–27. [Google Scholar] [CrossRef]
- Willert, E.K.; Robinson, G.L.; Higgins, J.P.; Liu, J.; Lee, J.; Syed, S.; Zhang, Y.; Tavares, D.; Lublinsky, A.; Chattopadhyay, N.; et al. Abstract 2384: TAK-169, an exceptionally potent CD38 targeted engineered toxin body, as a novel direct cell kill approach for the treatment of multiple myeloma. Cancer Res. 2019, 79, 2384. [Google Scholar] [CrossRef]
- Vogl, D.T.; Kaufman, J.L.; Holstein, S.A.; Nadeem, O.; O’Donnell, E.; Suryanarayan, K.; Collins, S.; Parot, X.; Chaudhry, M. TAK-573, an Anti-CD38/Attenuated Ifnα Fusion Protein, Has Clinical Activity and Modulates the Ifnα Receptor (IFNAR) Pathway in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 37–38. [Google Scholar] [CrossRef]
- Sherbenou, D.W.; Aftab, B.T.; Su, Y.; Behrens, C.R.; Wiita, A.; Logan, A.C.; Acosta-Alvear, D.; Hann, B.C.; Walter, P.; Shuman, M.A.; et al. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J. Clin. Investig. 2016, 126, 4640–4653. [Google Scholar] [CrossRef]
- Burton, J.D.; Ely, S.; Reddy, P.K.; Stein, R.; Gold, D.V.; Cardillo, T.M.; Goldenberg, D.M. CD74 is expressed by multiple myeloma and is a promising target for therapy. Clin. Cancer Res. 2004, 10, 6606–6611. [Google Scholar] [CrossRef]
- Stein, R.; Mattes, M.J.; Cardillo, T.M.; Hansen, H.J.; Chang, C.H.; Burton, J.; Govindan, S.; Goldenberg, D.M. CD74: A new candidate target for the immunotherapy of B-cell neoplasms. Clin. Cancer Res. 2007, 13, 5556s–5563s. [Google Scholar] [CrossRef]
- Abrahams, C.L.; Li, X.; Embry, M.; Yu, A.; Krimm, S.; Krueger, S.; Greenland, N.Y.; Wen, K.W.; Jones, C.; DeAlmeida, V.; et al. Targeting CD74 in multiple myeloma with the novel, site-specific antibody-drug conjugate STRO-001. Oncotarget 2018, 9, 37700–37714. [Google Scholar] [CrossRef]
- Shah, N.N.; Krishnan, A.Y.; Shah, N.D.; Burke, J.M.; Melear, J.M.; Spira, A.I.; Popplewell, L.L.; Andreadis, C.B.; Chhabra, S.; Sharman, J.P.; et al. Preliminary Results of a Phase 1 Dose Escalation Study of the First-in-Class Anti-CD74 Antibody Drug Conjugate (ADC), STRO-001, in Patients with Advanced B-Cell Malignancies. Blood 2019, 134, 5329. [Google Scholar] [CrossRef]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef]
- Lancman, G.; Sastow, D.L.; Cho, H.J.; Jagannath, S.; Madduri, D.; Parekh, S.S.; Richard, S.; Richter, J.; Sanchez, L.; Chari, A. Bispecific Antibodies in Multiple Myeloma: Present and Future. Blood Cancer Discov. 2021, 2, 423–433. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef]
- Elkins, K.; Zheng, B.; Go, M.; Slaga, D.; Du, C.; Scales, S.J.; Yu, S.F.; McBride, J.; de Tute, R.; Rawstron, A.; et al. FcRL5 as a target of antibody-drug conjugates for the treatment of multiple myeloma. Mol. Cancer Ther. 2012, 11, 2222–2232. [Google Scholar] [CrossRef]
- Dement-Brown, J.; Newton, C.S.; Ise, T.; Damdinsuren, B.; Nagata, S.; Tolnay, M. Fc receptor-like 5 promotes B cell proliferation and drives the development of cells displaying switched isotypes. J. Leukoc. Biol. 2012, 91, 59–67. [Google Scholar] [CrossRef]
- Ross, T.; Reusch, U.; Wingert, S.; Haneke, T.; Klausz, K.; Otte, A.-K.; Schub, N.; Knackmuss, S.; Müller, T.; Ellwanger, K.; et al. Preclinical Characterization of AFM26, a Novel B Cell Maturation Antigen (BCMA)-Directed Tetravalent Bispecific Antibody for High Affinity Retargeting of NK Cells Against Myeloma. Blood 2018, 132, 1927. [Google Scholar] [CrossRef]
- Draghi, M.; Schafer, J.L.; Nelson, A.; Frye, Z.; Oliphant, A.; Haserlat, S.; Lajoie, J.; Rogers, K.; Villinger, F.; Schmidt, M.; et al. Abstract 4972: Preclinical development of a first-in-class NKp30xBCMA NK cell engager for the treatment of multiple myeloma. Cancer Res. 2019, 79, 4972. [Google Scholar] [CrossRef]
- Watkins-Yoon, J.; Guzman, W.; Oliphant, A.; Haserlat, S.; Leung, A.; Chottin, C.; Ophir, M.; Vekeria, J.; Nelson, A.P.; Frye, Z.; et al. CTX-8573, an Innate-Cell Engager Targeting BCMA, is a Highly Potent Multispecific Antibody for the Treatment of Multiple Myeloma. Blood 2019, 134, 3182. [Google Scholar] [CrossRef]
- Shah, Z.; Malik, M.N.; Batool, S.S.; Kotapati, S.; Akhtar, A.; Rehman, O.u.; Ghani, M.; Sadiq, M.; Akbar, A.; Ashraf, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Based Immunotherapy for Treatment of Relapsed Refractory Multiple Myeloma (RRMM): A Systematic Review of Preclinical and Clinical Trials. Blood 2019, 134, 5567. [Google Scholar] [CrossRef]
- Harrison, S.J.; Minnema, M.C.; Lee, H.C.; Spencer, A.; Kapoor, P.; Madduri, D.; Larsen, J.; Ailawadhi, S.; Kaufman, J.L.; Raab, M.S.; et al. A Phase 1 First in Human (FIH) Study of AMG 701, an Anti-B-Cell Maturation Antigen (BCMA) Half-Life Extended (HLE) BiTE® (bispecific T-cell engager) Molecule, in Relapsed/Refractory (RR) Multiple Myeloma (MM). Blood 2020, 136, 28–29. [Google Scholar] [CrossRef]
- Cho, S.F.; Lin, L.; Xing, L.; Li, Y.; Wen, K.; Yu, T.; Hsieh, P.A.; Munshi, N.; Wahl, J.; Matthes, K.; et al. The immunomodulatory drugs lenalidomide and pomalidomide enhance the potency of AMG 701 in multiple myeloma preclinical models. Blood Adv. 2020, 4, 4195–4207. [Google Scholar] [CrossRef]
- Hipp, S.; Tai, Y.T.; Blanset, D.; Deegen, P.; Wahl, J.; Thomas, O.; Rattel, B.; Adam, P.J.; Anderson, K.C.; Friedrich, M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia 2017, 31, 1743–1751. [Google Scholar] [CrossRef]
- Costa, L.J.; Wong, S.W.; Bermúdez, A.; de la Rubia, J.; Mateos, M.-V.; Ocio, E.M.; Rodríguez-Otero, P.; San-Miguel, J.; Li, S.; Sarmiento, R.; et al. First Clinical Study of the B-Cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Interim Results of a Phase 1 Multicenter Trial. Blood 2019, 134, 143. [Google Scholar] [CrossRef]
- Chari, A.; Berdeja, J.G.; Oriol, A.; van de Donk, N.W.C.J.; Rodriguez, P.; Askari, E.; Mateos, M.-V.; Minnema, M.C.; Verona, R.; Girgis, S.; et al. A Phase 1, First-in-Human Study of Talquetamab, a G Protein-Coupled Receptor Family C Group 5 Member D (GPRC5D) × CD3 Bispecific Antibody, in Patients with Relapsed and/or Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 40–41. [Google Scholar] [CrossRef]
- Girgis, S.; Lin, S.X.W.; Pillarisetti, K.; Verona, R.; Vieyra, D.; Casneuf, T.; Fink, D.; Miao, X.; Chen, Y.; Stephenson, T.; et al. Teclistamab and talquetamab modulate levels of soluble B-cell maturation antigen in patients with relapsed and/or refractory multiple myeloma. J. Clin. Oncol. 2021, 39, 8047. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Krishnan, A.Y.; Oriol, A.; Donk, N.W.C.J.v.d.; Rodríguez-Otero, P.; Askari, E.; Mateos, M.-V.; Minnema, M.C.; Costa, L.J.; Verona, R.; et al. Updated results of a phase 1, first-in-human study of talquetamab, a G protein-coupled receptor family C group 5 member D (GPRC5D) × CD3 bispecific antibody, in relapsed/refractory multiple myeloma (MM). J. Clin. Oncol. 2021, 39, 8008. [Google Scholar] [CrossRef]
- Rodriguez, C.; D’Souza, A.; Shah, N.; Voorhees, P.M.; Buelow, B.; Vij, R.; Kumar, S.K. Initial Results of a Phase I Study of TNB-383B, a BCMA × CD3 Bispecific T-Cell Redirecting Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 43–44. [Google Scholar] [CrossRef]
- Cohen, A.D.; Harrison, S.J.; Krishnan, A.; Fonseca, R.; Forsberg, P.A.; Spencer, A.; Berdeja, J.G.; Laubach, J.P.; Li, M.; Choeurng, V.; et al. Initial Clinical Activity and Safety of BFCR4350A, a FcRH5/CD3 T-Cell-Engaging Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 42–43. [Google Scholar] [CrossRef]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Lancman, G.; Richter, J.; Chari, A. Bispecifics, trispecifics, and other novel immune treatments in myeloma. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 264–271. [Google Scholar] [CrossRef]
- Plesner, T.; Harrison, S.J.; Quach, H.; Lee, C.H.; Bryant, A.; Vangsted, A.J.; Estell, J.; Delforge, M.; Offner, F.; Twomey, P.; et al. A Phase I Study of RO7297089, a B-Cell Maturation Antigen (BCMA)-CD16a Bispecific Antibody in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2021, 138, 2755. [Google Scholar] [CrossRef]
- Chan, W.K.; Kang, S.; Youssef, Y.; Glankler, E.N.; Barrett, E.R.; Carter, A.M.; Ahmed, E.H.; Prasad, A.; Chen, L.; Zhang, J.; et al. A CS1-NKG2D Bispecific Antibody Collectively Activates Cytolytic Immune Cells against Multiple Myeloma. Cancer Immunol. Res. 2018, 6, 776–787. [Google Scholar] [CrossRef]
- Gantke, T.; Weichel, M.; Herbrecht, C.; Reusch, U.; Ellwanger, K.; Fucek, I.; Eser, M.; Müller, T.; Griep, R.; Molkenthin, V.; et al. Trispecific antibodies for CD16A-directed NK cell engagement and dual-targeting of tumor cells. Protein Eng. Des. Sel. 2017, 30, 673–684. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hudecek, M.; Jensen, M.C.; Riddell, S.R. Engineered T cells for anti-cancer therapy. Curr. Opin Immunol. 2012, 24, 633–639. [Google Scholar] [CrossRef]
- Sadelain, M.; Rivière, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Till, B.G.; Jensen, M.C.; Wang, J.; Chen, E.Y.; Wood, B.L.; Greisman, H.A.; Qian, X.; James, S.E.; Raubitschek, A.; Forman, S.J.; et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008, 112, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Mikkilineni, L.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood 2017, 130, 2594–2602. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef]
- Wang, X.; Rivière, I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther. Oncolytics 2016, 3, 16015. [Google Scholar] [CrossRef]
- Gattinoni, L.; Finkelstein, S.E.; Klebanoff, C.A.; Antony, P.A.; Palmer, D.C.; Spiess, P.J.; Hwang, L.N.; Yu, Z.; Wrzesinski, C.; Heimann, D.M.; et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005, 202, 907–912. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Zhao, W.H.; Liu, J.; Wang, B.Y.; Chen, Y.X.; Cao, X.M.; Yang, Y.; Zhang, Y.L.; Wang, F.X.; Zhang, P.Y.; Lei, B.; et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef]
- Xu, J.; Chen, L.J.; Yang, S.S.; Sun, Y.; Wu, W.; Liu, Y.F.; Xu, J.; Zhuang, Y.; Zhang, W.; Weng, X.Q.; et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 9543–9551. [Google Scholar] [CrossRef]
- Van de Donk, N.; Themeli, M.; Usmani, S.Z. Determinants of response and mechanisms of resistance of CAR T-cell therapy in multiple myeloma. Blood Cancer Discov. 2021, 2, 302–318. [Google Scholar] [CrossRef]
- Li, C.; Wang, Q.; Zhu, H.; Mao, X.; Wang, Y.; Zhang, Y.; Zhou, J. T Cells Expressing Anti B-Cell Maturation Antigen Chimeric Antigen Receptors for Plasma Cell Malignancies. Blood 2018, 132, 1013. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef]
- Green, D.J.; Pont, M.; Sather, B.D.; Cowan, A.J.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, E.N., III; Becker, P.S.; Coffey, D.G.; et al. Fully Human Bcma Targeted Chimeric Antigen Receptor T Cells Administered in a Defined Composition Demonstrate Potency at Low Doses in Advanced Stage High Risk Multiple Myeloma. Blood 2018, 132, 1011. [Google Scholar] [CrossRef]
- Samur, M.K.; Fulciniti, M.; Aktas Samur, A.; Bazarbachi, A.H.; Tai, Y.T.; Prabhala, R.; Alonso, A.; Sperling, A.S.; Campbell, T.; Petrocca, F.; et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 2021, 12, 868. [Google Scholar] [CrossRef] [PubMed]
- Da Vià, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Heidemeier, A.; Zhou, X.; Danhof, S.; Kraus, S.; Chatterjee, M.; et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat. Med. 2021, 27, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Zah, E.; Nam, E.; Bhuvan, V.; Tran, U.; Ji, B.Y.; Gosliner, S.B.; Wang, X.; Brown, C.E.; Chen, Y.Y. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 2020, 11, 2283. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Medema, J.P.; de Jong, J.; van Hall, T.; Melief, C.J.; Offringa, R. Immune escape of tumors in vivo by expression of cellular FLICE-inhibitory protein. J. Exp. Med. 1999, 190, 1033–1038. [Google Scholar] [CrossRef]
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520. [Google Scholar] [CrossRef]
- Wang, M.; Pruteanu, I.; Cohen, A.D.; Garfall, A.L.; Milone, M.C.; Tian, L.; Gonzalez, V.E.; Gill, S.; Frey, N.V.; Barrett, D.M.; et al. Identification and Validation of Predictive Biomarkers to CD19- and BCMA-Specific CAR T-Cell Responses in CAR T-Cell Precursors. Blood 2019, 134, 622. [Google Scholar] [CrossRef]
- Finney, O.C.; Yeri, A.; Mao, P.; Pandya, C.; Alonzo, E.; Hopkins, G.; Hymson, S.; Hu, T.; Foos, M.; Bhadoriya, S.; et al. Molecular and Phenotypic Profiling of Drug Product and Post-Infusion Samples from CRB-402, an Ongoing: Phase I Clinical Study of bb21217 a BCMA-Directed CAR T Cell Therapy. Blood 2020, 136, 3–4. [Google Scholar] [CrossRef]
- Leblay, N.; Maity, R.; Barakat, E.; McCulloch, S.; Duggan, P.; Jimenez-Zepeda, V.; Bahlis, N.J.; Neri, P. Cite-Seq Profiling of T Cells in Multiple Myeloma Patients Undergoing BCMA Targeting CAR-T or Bites Immunotherapy. Blood 2020, 136, 11–12. [Google Scholar] [CrossRef]
- Garfall, A.L.; Dancy, E.K.; Cohen, A.D.; Hwang, W.T.; Fraietta, J.A.; Davis, M.M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 2019, 3, 2812–2815. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Cherkassky, L.; Villena-Vargas, J.; Colovos, C.; Servais, E.; Plotkin, J.; Jones, D.R.; Sadelain, M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014, 6, 261ra151. [Google Scholar] [CrossRef]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef]
- Chen, K.H.; Wada, M.; Pinz, K.G.; Liu, H.; Shuai, X.; Chen, X.; Yan, L.E.; Petrov, J.C.; Salman, H.; Senzel, L.; et al. A compound chimeric antigen receptor strategy for targeting multiple myeloma. Leukemia 2018, 32, 402–412. [Google Scholar] [CrossRef]
- Fernández de Larrea, C.; Staehr, M.; Lopez, A.V.; Ng, K.Y.; Chen, Y.; Godfrey, W.D.; Purdon, T.J.; Ponomarev, V.; Wendel, H.G.; Brentjens, R.J.; et al. Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape-Driven Relapse in Multiple Myeloma. Blood Cancer Discov. 2020, 1, 146–154. [Google Scholar] [CrossRef]
- Suarez, E.R.; de Chang, K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef]
- Drent, E.; Poels, R.; Ruiter, R.; van de Donk, N.; Zweegman, S.; Yuan, H.; de Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin. Cancer Res. 2019, 25, 4014–4025. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Mailankody, S.; Matous, J.V.; Liedtke, M.; Sidana, S.; Malik, S.; Nath, R.; Oluwole, O.O.; Karski, E.E.; Lovelace, W.; Zhou, X.; et al. Universal: An Allogeneic First-in-Human Study of the Anti-Bcma ALLO-715 and the Anti-CD52 ALLO-647 in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 24–25. [Google Scholar] [CrossRef]
- Carmon, L.; Avivi, I.; Kovjazin, R.; Zuckerman, T.; Dray, L.; Gatt, M.E.; Or, R.; Shapira, M.Y. Phase I/II study exploring ImMucin, a pan-major histocompatibility complex, anti-MUC1 signal peptide vaccine, in multiple myeloma patients. Br. J. Haematol. 2015, 169, 44–56. [Google Scholar] [CrossRef]
- Kovjazin, R.; Volovitz, I.; Kundel, Y.; Rosenbaum, E.; Medalia, G.; Horn, G.; Smorodinsky, N.I.; Brenner, B.; Carmon, L. ImMucin: A novel therapeutic vaccine with promiscuous MHC binding for the treatment of MUC1-expressing tumors. Vaccine 2011, 29, 4676–4686. [Google Scholar] [CrossRef] [PubMed]
- Kovjazin, R.; Horn, G.; Smorodinsky, N.I.; Shapira, M.Y.; Carmon, L. Cell surface-associated anti-MUC1-derived signal peptide antibodies: Implications for cancer diagnostics and therapy. PLoS ONE 2014, 9, e85400. [Google Scholar] [CrossRef]
- Choi, C.; Witzens, M.; Bucur, M.; Feuerer, M.; Sommerfeldt, N.; Trojan, A.; Ho, A.; Schirrmacher, V.; Goldschmidt, H.; Beckhove, P. Enrichment of functional CD8 memory T cells specific for MUC1 in bone marrow of patients with multiple myeloma. Blood 2005, 105, 2132–2134. [Google Scholar] [CrossRef]
- Bae, J.; Samur, M.; Munshi, A.; Hideshima, T.; Keskin, D.; Kimmelman, A.; Lee, A.H.; Dranoff, G.; Anderson, K.C.; Munshi, N.C. Heteroclitic XBP1 peptides evoke tumor-specific memory cytotoxic T lymphocytes against breast cancer, colon cancer, and pancreatic cancer cells. Oncoimmunology 2014, 3, e970914. [Google Scholar] [CrossRef]
- Bae, J.; Prabhala, R.; Voskertchian, A.; Brown, A.; Maguire, C.; Richardson, P.; Dranoff, G.; Anderson, K.C.; Munshi, N.C. A multiepitope of XBP1, CD138 and CS1 peptides induces myeloma-specific cytotoxic T lymphocytes in T cells of smoldering myeloma patients. Leukemia 2015, 29, 218–229. [Google Scholar] [CrossRef]
- Nooka, A.K.; Wang, M.L.; Yee, A.J.; Kaufman, J.L.; Bae, J.; Peterkin, D.; Richardson, P.G.; Raje, N.S. Assessment of Safety and Immunogenicity of PVX-410 Vaccine With or Without Lenalidomide in Patients With Smoldering Multiple Myeloma: A Nonrandomized Clinical Trial. JAMA Oncol. 2018, 4, e183267. [Google Scholar] [CrossRef]
- Jørgensen, N.G.; Klausen, U.; Grauslund, J.H.; Helleberg, C.; Aagaard, T.G.; Do, T.H.; Ahmad, S.M.; Olsen, L.R.; Klausen, T.W.; Breinholt, M.F.; et al. Peptide Vaccination Against PD-L1 With IO103 a Novel Immune Modulatory Vaccine in Multiple Myeloma: A Phase I First-in-Human Trial. Front. Immunol. 2020, 11, 595035. [Google Scholar] [CrossRef] [PubMed]
- Hallett, W.H.; Jing, W.; Drobyski, W.R.; Johnson, B.D. Immunosuppressive effects of multiple myeloma are overcome by PD-L1 blockade. Biol. Blood Marrow Transplant. 2011, 17, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Avivi, I.; Vasir, B.; Uhl, L.; Munshi, N.C.; Katz, T.; Dey, B.R.; Somaiya, P.; Mills, H.; Campigotto, F.; et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin. Cancer Res. 2013, 19, 3640–3648. [Google Scholar] [CrossRef] [PubMed]
- Billel, G.; Smith, E.L.; Dogan, A.; Hsu, M.; Devlin, S.; Pichardo, J.D.; Chung, D.J.; Koehne, G.; Korde, N.S.; Landau, H.J.; et al. Presence of PD-1 Expressing T Cells Predicts for Inferior Overall Survival in Newly Diagnosed Multiple Myeloma. Blood 2015, 126, 1785. [Google Scholar] [CrossRef]
- Qian, J.; Xie, J.; Hong, S.; Yang, J.; Zhang, L.; Han, X.; Wang, M.; Zhan, F.; Shaughnessy, J.D., Jr.; Epstein, J.; et al. Dickkopf-1 (DKK1) is a widely expressed and potent tumor-associated antigen in multiple myeloma. Blood 2007, 110, 1587–1594. [Google Scholar] [CrossRef]
- Qian, J.; Zheng, Y.; Zheng, C.; Wang, L.; Qin, H.; Hong, S.; Li, H.; Lu, Y.; He, J.; Yang, J.; et al. Active vaccination with Dickkopf-1 induces protective and therapeutic antitumor immunity in murine multiple myeloma. Blood 2012, 119, 161–169. [Google Scholar] [CrossRef]
- Strambio-De-Castillia, C.; Niepel, M.; Rout, M.P. The nuclear pore complex: Bridging nuclear transport and gene regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 490–501. [Google Scholar] [CrossRef]
- Theodoropoulos, N.; Lancman, G.; Chari, A. Targeting Nuclear Export Proteins in Multiple Myeloma Therapy. Target. Oncol. 2020, 15, 697–708. [Google Scholar] [CrossRef]
- Azizian, N.G.; Li, Y. XPO1-dependent nuclear export as a target for cancer therapy. J. Hematol. Oncol. 2020, 13, 61. [Google Scholar] [CrossRef]
- Turner, J.G.; Dawson, J.; Sullivan, D.M. Nuclear export of proteins and drug resistance in cancer. Biochem. Pharmacol. 2012, 83, 1021–1032. [Google Scholar] [CrossRef]
- Schmidt, J.; Braggio, E.; Kortuem, K.M.; Egan, J.B.; Zhu, Y.X.; Xin, C.S.; Tiedemann, R.E.; Palmer, S.E.; Garbitt, V.M.; McCauley, D.; et al. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia 2013, 27, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, T.; Argueta, C.; Aboukameel, A.; Unger, T.J.; Klebanov, B.; Mohammad, R.M.; Muqbil, I.; Azmi, A.S.; Drolen, C.; Senapedis, W.; et al. Selinexor, a Selective Inhibitor of Nuclear Export (SINE) compound, acts through NF-κB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death. Oncotarget 2016, 7, 78883–78895. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.G.; Kashyap, T.; Dawson, J.L.; Gomez, J.; Bauer, A.A.; Grant, S.; Dai, Y.; Shain, K.H.; Meads, M.; Landesman, Y.; et al. XPO1 inhibitor combination therapy with bortezomib or carfilzomib induces nuclear localization of IκBα and overcomes acquired proteasome inhibitor resistance in human multiple myeloma. Oncotarget 2016, 7, 78896–78909. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, U.H.; Senapedis, W.; Baloglu, E.; Unger, T.J.; Chari, A.; Vogl, D.; Cornell, R.F. Clinical Implications of Targeting XPO1-mediated Nuclear Export in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk 2018, 18, 335–345. [Google Scholar] [CrossRef]
- Kanai, M.; Hanashiro, K.; Kim, S.H.; Hanai, S.; Boulares, A.H.; Miwa, M.; Fukasawa, K. Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat. Cell Biol. 2007, 9, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.K.; Jiang, H.; Aoki, M. Triple layer control: Phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle 2005, 4, 908–913. [Google Scholar] [CrossRef]
- Tai, Y.T.; Landesman, Y.; Acharya, C.; Calle, Y.; Zhong, M.Y.; Cea, M.; Tannenbaum, D.; Cagnetta, A.; Reagan, M.; Munshi, A.A.; et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: Molecular mechanisms and therapeutic implications. Leukemia 2014, 28, 155–165. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Sutherland, H.; White, D.; Sebag, M.; Lentzsch, S.; Kotb, R.; Venner, C.P.; Gasparetto, C.; Del Col, A.; Neri, P.; et al. Selinexor plus low-dose bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma. Blood 2018, 132, 2546–2554. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef]
- Tandon, N.; Ramakrishnan, V.; Kumar, S.K. Clinical use and applications of histone deacetylase inhibitors in multiple myeloma. Clin. Pharmacol. 2016, 8, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Maru, Y.; Tanaka, J. Action mechanisms of histone deacetylase inhibitors in the treatment of hematological malignancies. Cancer Sci. 2016, 107, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Gandhi, V.; Konopleva, M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma 2017, 58, 2026–2039. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef]
- Paner, A.; Patel, P.; Dhakal, B. The evolving role of translocation t(11;14) in the biology, prognosis, and management of multiple myeloma. Blood Rev. 2020, 41, 100643. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Gasparetto, C.; Schjesvold, F.H.; Moreau, P.; Touzeau, C.; Facon, T.; Boise, L.H.; Jiang, Y.; Yang, X.; Dunbar, F.; et al. Targeting BCL-2 with venetoclax and dexamethasone in patients with relapsed/refractory t(11;14) multiple myeloma. Am. J. Hematol. 2021, 96, 418–427. [Google Scholar] [CrossRef]
- Lasica, M.; Anderson, M.A. Review of Venetoclax in CLL, AML and Multiple Myeloma. J. Pers. Med. 2021, 11, 463. [Google Scholar] [CrossRef]
- Khouri, J.; Faiman, B.M.; Grabowski, D.; Mahfouz, R.Z.; Khan, S.N.; Wei, W.; Valent, J.; Dean, R.; Samaras, C.; Jha, B.K.; et al. DNA methylation inhibition in myeloma: Experience from a phase 1b study of low-dose continuous azacitidine in combination with lenalidomide and low-dose dexamethasone in relapsed or refractory multiple myeloma. Semin. Hematol. 2021, 58, 45–55. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef]
- An, S.; Fu, L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 2018, 36, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lee, H.C.; Shirazi, F.; Baladandayuthapani, V.; Lin, H.; Kuiatse, I.; Wang, H.; Jones, R.J.; Berkova, Z.; Singh, R.K.; et al. Protein targeting chimeric molecules specific for bromodomain and extra-terminal motif family proteins are active against pre-clinical models of multiple myeloma. Leukemia 2018, 32, 2224–2239. [Google Scholar] [CrossRef]
- Lim, S.L.; Damnernsawad, A.; Shyamsunder, P.; Chng, W.J.; Han, B.C.; Xu, L.; Pan, J.; Pravin, D.P.; Alkan, S.; Tyner, J.W.; et al. Proteolysis targeting chimeric molecules as therapy for multiple myeloma: Efficacy, biomarker and drug combinations. Haematologica 2019, 104, 1209–1220. [Google Scholar] [CrossRef]
- Lu, X.; Sabbasani, V.R.; Osei-Amponsa, V.; Evans, C.N.; King, J.C.; Tarasov, S.G.; Dyba, M.; Das, S.; Chan, K.C.; Schwieters, C.D.; et al. Structure-guided bifunctional molecules hit a DEUBAD-lacking hRpn13 species upregulated in multiple myeloma. Nat. Commun. 2021, 12, 7318. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef]
- Freedman, R.B.; Hirst, T.R.; Tuite, M.F. Protein disulphide isomerase: Building bridges in protein folding. Trends Biochem. Sci. 1994, 19, 331–336. [Google Scholar] [CrossRef]
- Laurindo, F.R.; Pescatore, L.A.; Fernandes Dde, C. Protein disulfide isomerase in redox cell signaling and homeostasis. Free Radic. Biol. Med. 2012, 52, 1954–1969. [Google Scholar] [CrossRef]
- Turano, C.; Coppari, S.; Altieri, F.; Ferraro, A. Proteins of the PDI family: Unpredicted non-ER locations and functions. J. Cell. Physiol. 2002, 193, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Beer, D.G.; Kardia, S.L.; Huang, C.C.; Giordano, T.J.; Levin, A.M.; Misek, D.E.; Lin, L.; Chen, G.; Gharib, T.G.; Thomas, D.G.; et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 2002, 8, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.M.; Jeon, Y.J. Proteostasis In The Endoplasmic Reticulum: Road to Cure. Cancers 2019, 11, 1739. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar]
- Hasipek, M.; Grabowski, D.; Guan, Y.; Alugubelli, R.R.; Tiwari, A.D.; Gu, X.; DeAvila, G.A.; Silva, A.S.; Meads, M.B.; Parker, Y.; et al. Therapeutic Targeting of Protein Disulfide Isomerase PDIA1 in Multiple Myeloma. Cancers 2021, 13, 2649. [Google Scholar] [CrossRef]
- Vatolin, S.; Phillips, J.G.; Jha, B.K.; Govindgari, S.; Hu, J.; Grabowski, D.; Parker, Y.; Lindner, D.J.; Zhong, F.; Distelhorst, C.W.; et al. Novel Protein Disulfide Isomerase Inhibitor with Anticancer Activity in Multiple Myeloma. Cancer Res. 2016, 76, 3340–3350. [Google Scholar] [CrossRef]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Reitz, A.B.; Manevich, Y.; McClure, J.J.; Champion, M.M.; Chou, C.J.; Sharik, M.E.; et al. Inhibitors of the protein disulfide isomerase family for the treatment of multiple myeloma. Leukemia 2019, 33, 1011–1022. [Google Scholar] [CrossRef]
- Cohen, Y.C.; Zada, M.; Wang, S.Y.; Bornstein, C.; David, E.; Moshe, A.; Li, B.; Shlomi-Loubaton, S.; Gatt, M.E.; Gur, C.; et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nat. Med. 2021, 27, 491–503. [Google Scholar] [CrossRef]
- Lang, S.; Pfeffer, S.; Lee, P.H.; Cavalié, A.; Helms, V.; Förster, F.; Zimmermann, R. An Update on Sec61 Channel Functions, Mechanisms, and Related Diseases. Front. Physiol. 2017, 8, 887. [Google Scholar] [CrossRef]
- Domenger, A.; Choisy, C.; Baron, L.; Mayau, V.; Perthame, E.; Deriano, L.; Arnulf, B.; Bories, J.C.; Dadaglio, G.; Demangel, C. The Sec61 translocon is a therapeutic vulnerability in multiple myeloma. EMBO Mol. Med. 2022, 14, e14740. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.L.D.; Ramberger, E.; Bohl, S.R.; Dolnik, A.; Steinebach, C.; Conrad, T.; Müller, S.; Popp, O.; Kull, M.; Haji, M.; et al. Proteomic profiling reveals CDK6 upregulation as a targetable resistance mechanism for lenalidomide in multiple myeloma. Nat. Commun. 2022, 13, 1009. [Google Scholar] [CrossRef] [PubMed]






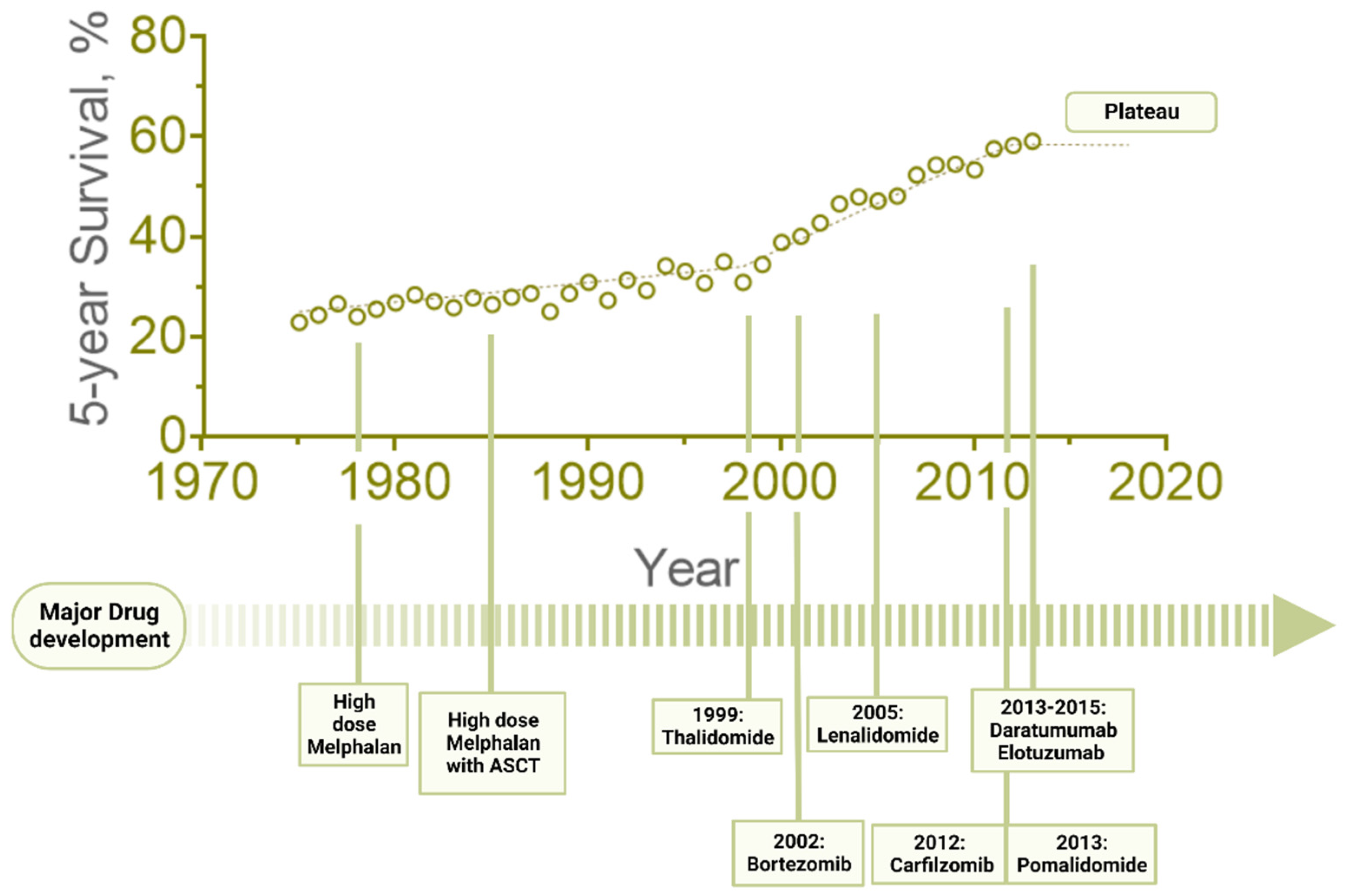{kind=link}
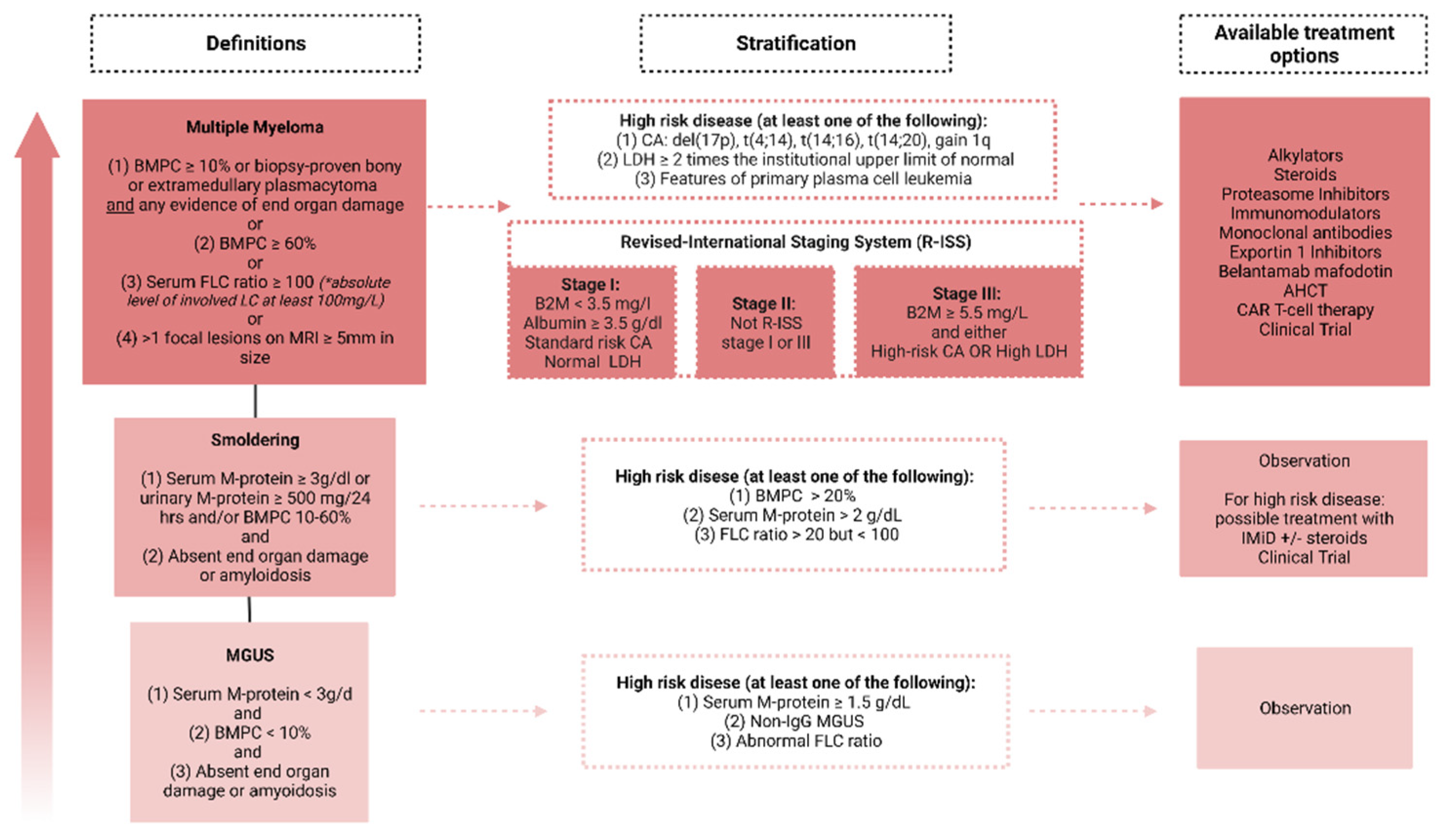{kind=link}
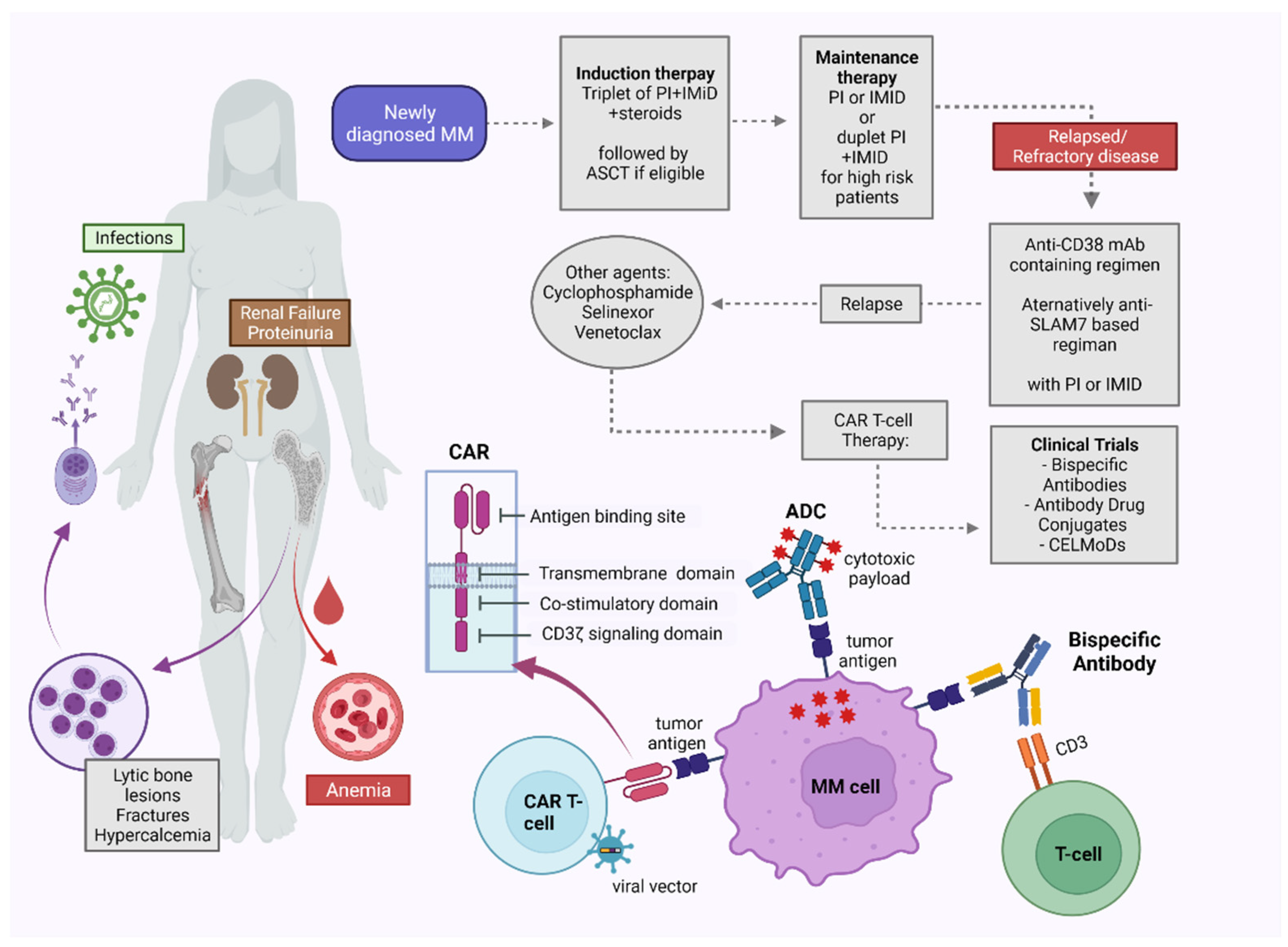{kind=link}
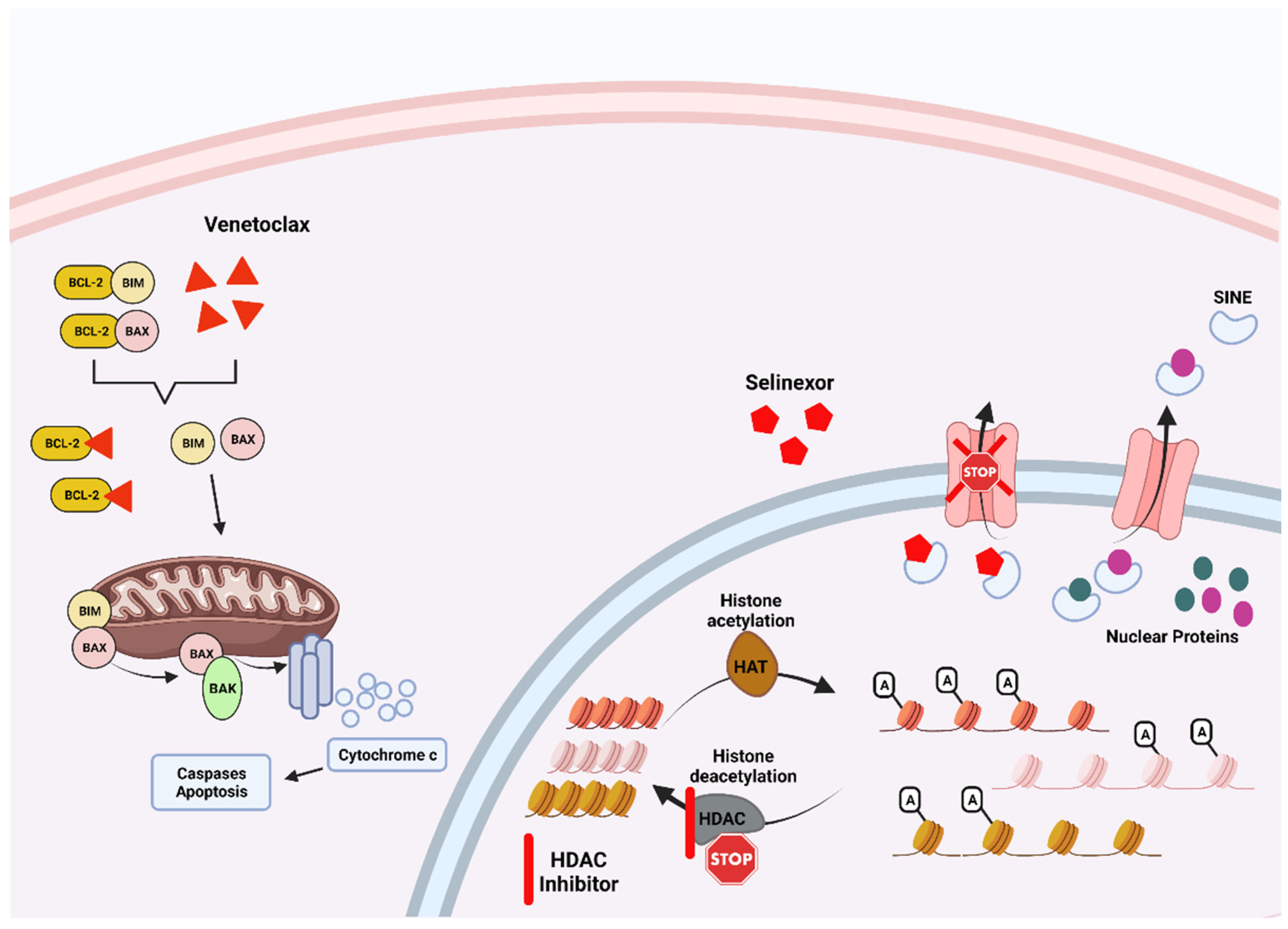{kind=link}
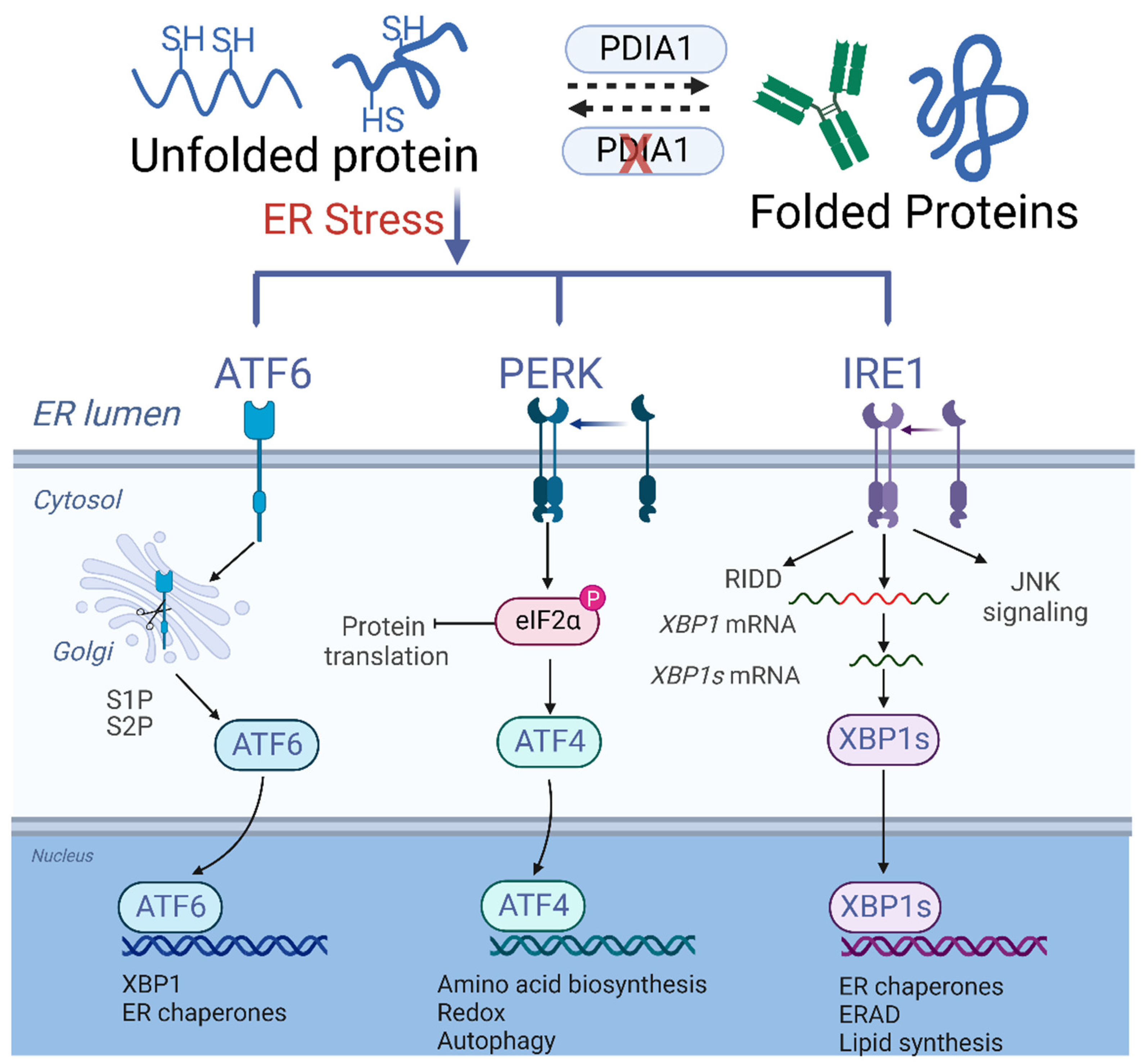{kind=link}
| Regimen | Trial Name NCT Number | Phase | N | Disease Status | Outcomes |
|---|---|---|---|---|---|
| Dara-Vd vs. Vd | CASTOR NCT02136134 | 3 | 498 | RRMM | ORR: 83.8% vs. 63.2% (p < 0.0001) ≥CR: 28.8% vs. 9.8% (p < 0.0001) m-PFS: 16.7 vs. 7.1 months (p < 0.0001) |
| Dara-Rd vs. Rd | POLLUX NCT02076009 | 3 | 569 | RRMM | ORR: 92.9 vs. 76.4% (p < 0.0001) ≥CR: 56.6 vs. 23.2% (p < 0.0001 m-PFS: 44.5 vs. 17.5 months (p < 0.0001) |
| Dara-Kd vs. Kd | CANDOR NCT03158688 | 3 | 446 | RRMM | ORR: 84% vs. 75% (p = 0.0080) ≥CR: 33% vs. 13% m-PFS: 28.6 vs. 15.2 months (p < 0.0001) |
| Dara-Pd vs. Pd | APOLLO NCT03180736 | 3 | 304 | RRMM | ORR: 69% vs. 46% (p < 0.0001) ≥CR: 25 vs. 4%, (p < 0.0001) m-PFS: 12.4 vs. 6.9 months (p = 0.0018) |
| Dara-Rd vs. RD | MAIA NCT02252172 | 3 | 737 | NDMM | ORR: 92.9% vs. 81.6 (p < 0.0001) ≥CR: 51% vs. 30% (p < 0.0001) m-PFS: NR vs. 34.4 months (p < 0.0001) |
| Dara-VMP vs. VMP | ALCYONE (NCT02195479) | 3 | 706 | NDMM | ORR: 90.9% vs. 73.9% (p < 0.0001) ≥CR: 46% vs. 25% (p < 0.0001) m-PFS: 36.4 vs. 19.3 months (p < 0.0001) |
| Dara-VTd vs. VTd | CASSIOPEIA NCT02541383 | 3 | 1085 | NDMM | At day 100 post AHCT: CR: 29% vs. 20% (p = 0.001) ≥CR: 39% vs. 26% (p < 0.0001) m-PFS: NR for both groups (p < 0.0001) |
| Dara-VRd vs. VRd | GRIFFIN NCT03710603 | 2 | 207 | NDMM | At the end of post-AHCT consolidation: ORR: 99% vs. 91.8 (p = 0.016) sCR: 42.4% vs. 32% (p = 0.068) m-PFS: NR for both groups |
| Isa-Pd vs. Pd | ICARIA NCT02990338 | 3 | 307 | RRMM | ORR: 60% vs. 35% (p < 0.0001) m-PFS: 11.5 vs. 6.5 months (p = 0.001) m-OS: 24.6 vs. 17.7 months (p = 0.028) |
| Isa-Kd vs. Kd | IKEMA NCT03275285 | 3 | 302 | RRMM | CR: 40% vs. 28% ≥VGPR: 73% vs. 56% (p = 0.0011) m-PFS: 19.15 months vs. NR (p = 0.0007) |
| Elo-Rd vs. Rd | ELOQUENT-2 NCT01239797 | 3 | 321 | RRMM | ORR: 79%, vs. 66% (p < 0.001) m-PFS: 19.4 vs. 14.9 months (p < 0.001) m-OS: 48.3 vs. 39.6 months (p = 0.0408) |
| Elo-Pd vs. Pd | ELOQUENT-3 NCT02654132 | 2 | 117 | RRMM | ORR: 53% vs. 26% m-PFS: 10.3 vs. 4.7 months (p = 0.008) m-OS: 29.8 vs. 17.4 months (p = 0.0217) |
| Agent Name | Target Antigen | NCT Number (Trial Name) | Phase | N | Disease Status | Outcomes | CRS ICANS |
|---|---|---|---|---|---|---|---|
| CAR -T Cell Therapy Products | |||||||
| bb21217 | BCMA | NCT03274219 (CRB-402) | 1 | 46 | RRMM | ▪ ORR: 55%, ≥CR: 18%, VGPR: 30% ▪ mPFS: 7.2 months | ▪ CRS: 67% ▪ ICANS: 22% |
| Idecabtagene vicleucel | BCMA | NCT02658929 (CRB-401) | 1 | 62 | RRMM | ▪ ORR: 76%, ≥CR: 39%, ≥VGPR: 65% ▪ mPFS/mOS: 8.8/34.2 months | ▪ CRS: 76% ▪ ICANS: 44% |
| NCT03361748 (KarMMa) | 2 | 128 | RRMM | ▪ ORR: 73%, ≥CR: 33%, ≥VGPR: 52% ▪ mPFS/mOS: 8.8/19.4 months | ▪ CRS: 84% ▪ ICANS: 18% | ||
| Ciltacabtagene autoleucel | BCMA | NCT03548207 (CARTITUDE-1) | 1/2 | 113 | RRMM | ▪ ORR: 98%, CR: 82.5%, VGPR: 95% ▪ mPFS and mOS: NR | ▪ CRS: 95% ▪ ICANS: 21% |
| NCT04133636 (CARTITUDE-2) | 2 | 20 | RRMM | ▪ ORR: 95%, CR: 75%, ≥VGPR: 85% ▪ mPFS and mOS: NR | ▪ CRS: 85% ▪ ICANS: 20% | ||
| Zevorcabtagene autoleucel | BCMA | NCT03716856 NCT03302403 NCT03380039 | 1 | 24 | RRMM | ▪ ORR: 87.5%, ≥CR: 80% ▪ mPFS 9.2 months | ▪ CRS: 62.5% ▪ ICANS: 12.5% |
| NCT03975907 (LUMMICAR-1) | 1/2 | 38 | RRMM | ▪ ORR: 92%, CR: 79% ▪ mPFS: 22.7 months | ▪ CRS: 73.7% | ||
| NCT03915184 (LUMMICAR-2) | 1/2 | 34 | RRMM | ▪ ORR: 100% ▪ mPFS and mOS: NR | ▪ CRS: 86% | ||
| Orvacabtagene autoleucel (JCARH125) | BCMA | NCT03090659 (LEGEND-2) | 1 | 74 | RRMM | ▪ ORR 88%, CR 73% ▪ mPFS: 18 months, mOS: NR | ▪ CRS: 92% ▪ ICANS: 21% |
| NCT03430011 (EVOLVE) | 1/2 | 115 | RRMM | ▪ ORR: 82% ▪ mPFS: NR | ▪ CRS: 75% | ||
| CT103A | BCMA | NCT05066646 (FUMANBA-1) | 1/2 | 79 | RRMM | ▪ ORR: 94.9% ▪ CR/sCR: 69.6% | ▪ CRS 94.9% ▪ ICANS: 2.5% |
| CART-ddBCMA | BCMA | NCT04155749 | 1 | 25 | RRMM | ▪ ORR: 100%, ≥CR: 75% ▪ mPFS and mOS: NR | ▪ CRS: 100% ▪ ICANS: 16% |
| GC012F | BCMA/CD19 | NCT04236011 NCT04182581 | 1 | 28 | RRMM | ▪ ORR: 80–100% | ▪ CRS: 100% |
| OriCAR-017 | GPRC5D | NCT05016778 | 1 | 11 | RRMM | ▪ ORR: 100% ▪ MRD (10−5) negativity: 100% | ▪ CRS: 100% |
| Bispecific Antibodies | |||||||
| Teclistamab | BCMAxCD3 | NCT03145181 NCT04557098 (MajesTEC-1) | 1/2 | 165 | RRMM | ▪ ORR: 63%, ≥CR 39.4% ▪ MRD (10−5) negativity: 26.7% ▪ mPFS: 11.3 months | ▪ CRS: 72% ▪ ICANS: 3% |
| Teclistamab + daratumumab | NCT04108195 (TRIMM-2) | 1b | 46 | RRMM | ▪ ORR: 78% ▪ ≥VGPR: 73% | ▪ CRS: 61% ▪ ICANS: 2.1% | |
| Elranatamab | BCMAxCD3 | NCT03269136 (MagnetisMM-1) | 1 | 55 | RRMM | ▪ ORR: 64% | ▪ CRS: 67% |
| NCT04649359 (MagnetisMM-3) | 2 | 60 | RRMM | ▪ ORR: NR | ▪ CRS 58.9% ▪ ICANS: 3.6% | ||
| Talquetamab | GPRC5DxCD3 | NCT03399799 (MonumenTAL-1) | 1 | 174 | RRMM | ▪ ORR 63% ▪ ≥VGPR 50% | ▪ CRS 79% ▪ ICANS: 7% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dima, D.; Jiang, D.; Singh, D.J.; Hasipek, M.; Shah, H.S.; Ullah, F.; Khouri, J.; Maciejewski, J.P.; Jha, B.K. Multiple Myeloma Therapy: Emerging Trends and Challenges. Cancers 2022, 14, 4082. https://doi.org/10.3390/cancers14174082
Dima D, Jiang D, Singh DJ, Hasipek M, Shah HS, Ullah F, Khouri J, Maciejewski JP, Jha BK. Multiple Myeloma Therapy: Emerging Trends and Challenges. Cancers. 2022; 14(17):4082. https://doi.org/10.3390/cancers14174082
Chicago/Turabian StyleDima, Danai, Dongxu Jiang, Divya Jyoti Singh, Metis Hasipek, Haikoo S. Shah, Fauzia Ullah, Jack Khouri, Jaroslaw P. Maciejewski, and Babal K. Jha. 2022. "Multiple Myeloma Therapy: Emerging Trends and Challenges" Cancers 14, no. 17: 4082. https://doi.org/10.3390/cancers14174082