
Dendritic Cell Vaccines: A Promising Approach in the Fight against Ovarian Cancer

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Immunotherapies in OC
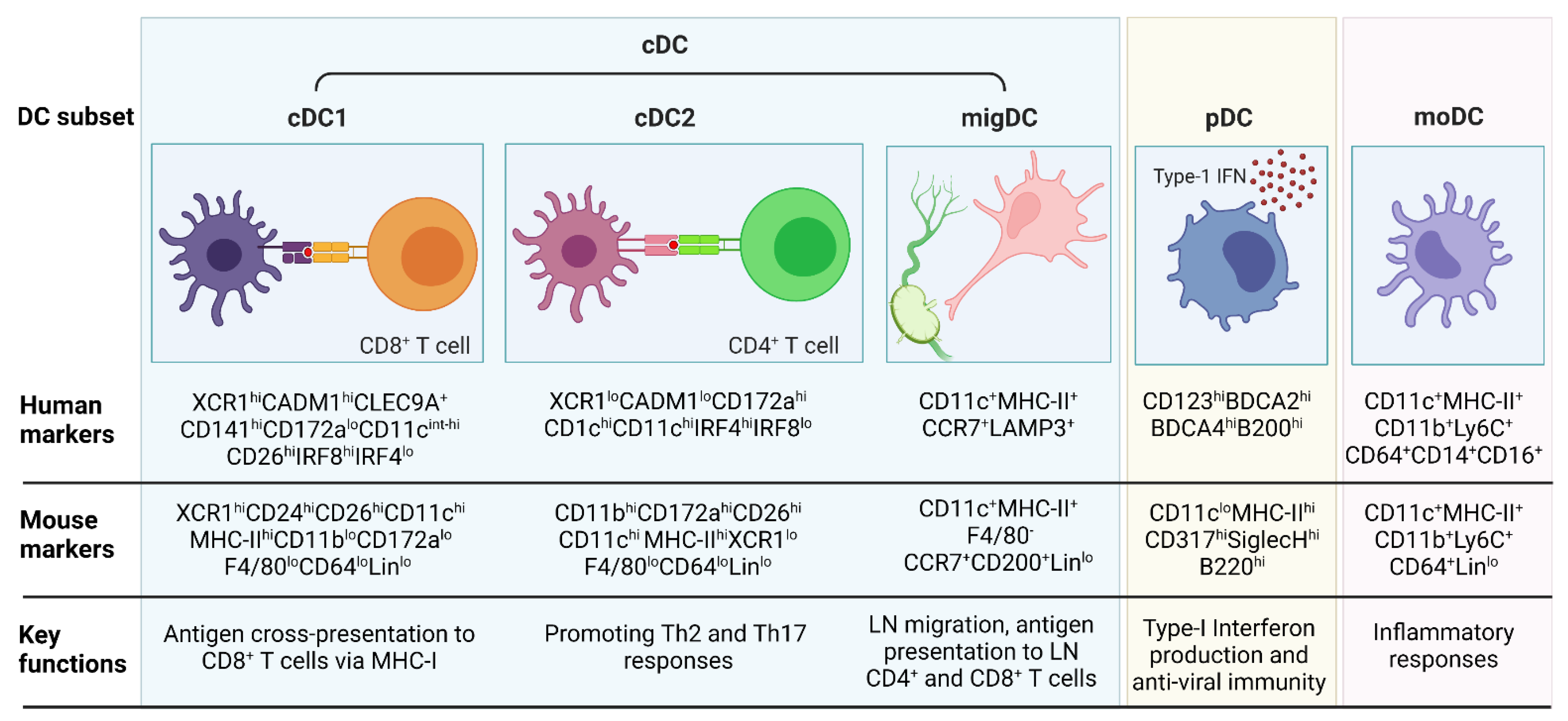
3. DC Subsets and Functions in Inflammation and Cancer
3.1. cDC1
3.2. cDC2
3.3. migDC
3.4. pDC
3.5. moDCs
4. Role of DCs in OC
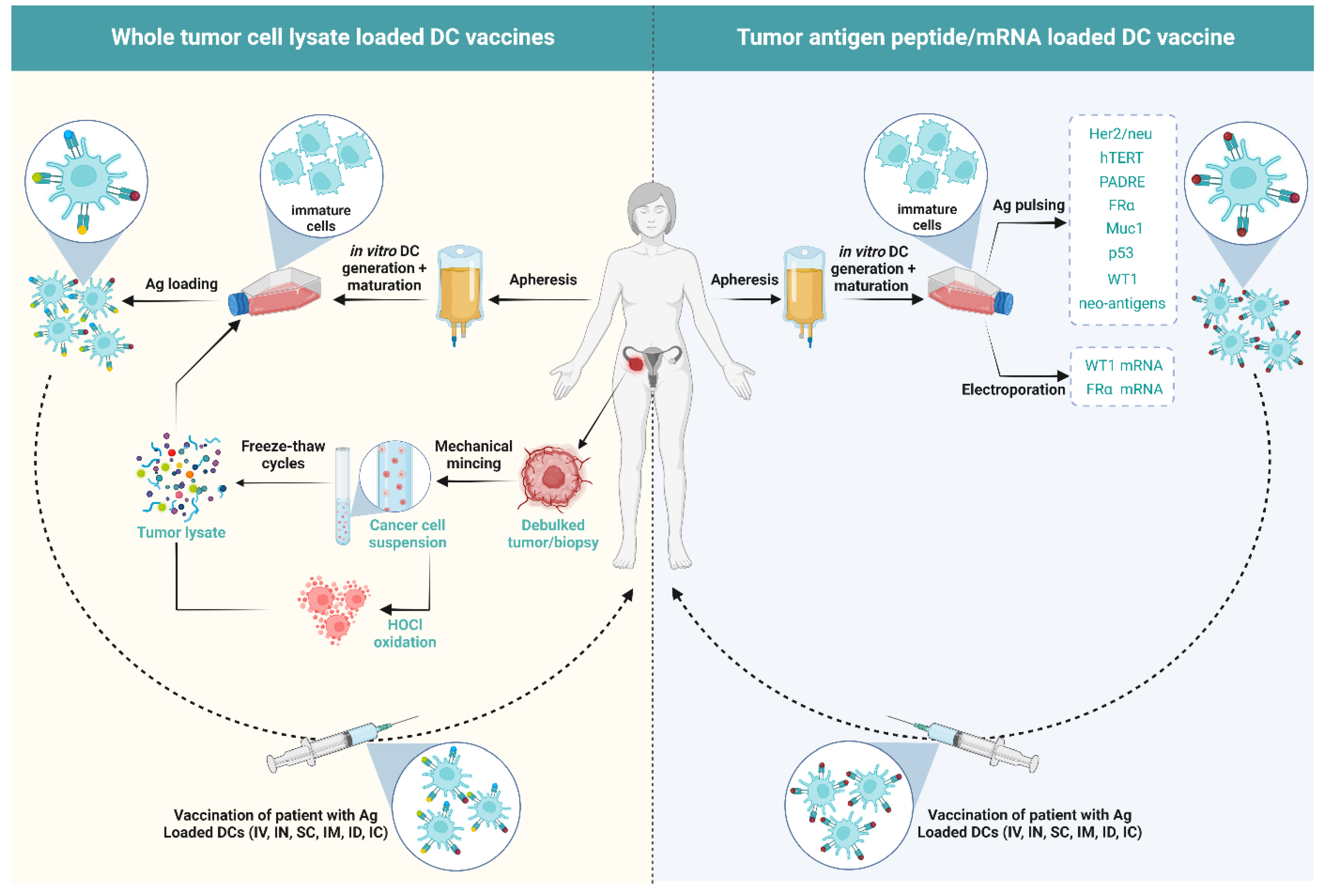
5. DCs Vaccines in OC
5.1. Influence of the Type of Antigen/Antigen Loading in DC Vaccination on the Immunological Outcome of Patients
5.2. Influence of Combination Therapies with DC Vaccine on the Immunological Outcome of Patients

{kind=link}
{kind=link}
| Phase of Study | No. of Patients | DC Generation | No. of Doses/No. of DCs per Dose | Injection Site | Combination | Response Rate | Refs. |
|---|---|---|---|---|---|---|---|
| Pilot | 3 | Monocytes from PBMCs were cultured in a medium with IL-4, GM-CSF, and TNFα followed by pulsing with Her-2/neu and mucin 1 peptides. | 3–9 doses of 2.8–8.7 million DCs | SC near inguinal LNs | - | SD | [107] |
| Phase I | 6 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4; pulsed with Keyhole Limpet Hemocyanin (KLH) and autologous tumor cell lysate in the presence of GM-CSF and TNFα. | 3–23 doses of 1–90 million DCs | IC near axillary LNs | - | 1/6-dead from disease 3/6-PD 2/6-SD | [115] |
| Phase I | 1 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4 followed by pulsing with the mannan-mucin 1 fusion protein. | Multiple doses of 40 million DCs | ID and SC | - | SD | [109] |
| Pilot | 4 | Monocytes from PBMCs were cultured in a medium with GM-CSF, IL4 and TNFα followed by pulsing with tumor cell lysate and treatment with 50% polyethylene glycol. | 6 doses of 10–26 million | SC near the neck or groin | IL12 | 1/4 patients-PD with transient reduction of CA125 | [116] |
| Pilot | 1 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4, followed by electroporation with FRα mRNA and culturing in IL-1β, IL-6, TNFα, and prostaglandin E2. | 10 doses of 2–21 million DCs | IN in the inguinal LNs | - | PR | [110] |
| Phase I | 4 | PBMCs were cultured with ionomycin and 10 µg/mL or 40 µg/mL BA7072 antigen (fusion protein containing sequences from intracellular and extracellular domains of Her-2 linked to GM-CSF. | 3 doses of 109 cells | IV | - | 2/4-SD 2/4-PD | [117] |
| Phase I/II | 11 | Monocytes from PBMCs were cultured in a medium with IL-13 and GM-CSF, followed by maturation with membrane components of Klebsiella pneumoniae and IFNγ; pulsing with hTERT, Her-2/neu, and PADRE peptides. | 4 doses of 35 million DCs | ID into the medial thigh | Arm1: DC vaccine Arm2: DC Vaccine + Cy Eligible patients were also given IM Prevnar heptavalent Pneumococcal vaccine with the first dose of DCs | Arm1-2/5 PD, 2/5 CR Arm2-2/6 PD, 4/6 CR | [108] |
| Phase II | 21 | Monocytes from PBMCs were cultured in a medium with IL-4 and GM-CSF, followed by maturation using CD40L and pulsing with p53 peptide. | 4 doses of 20 million DCs | IV | Arm1: p53 peptide + Montanide ISA-51 + GM-CSF + IL-2 Arm2: DC vaccine + IL-2 | Arm1-2/13 CR, 11/13 PD Arm2-2/7 CR, 5/7 PD | [105] |
| Pilot | 6 | Monocytes from PBMCs were cultured in a medium with IL-4 and GM-CSF. | 3 doses of 5–10 million DCs | ID | Arm1: Bev/Cy + DC vaccine. Arm2: Bev/Cy + in vitro generated 5 × 109 T cells | Arm1-2/6 SD, 2/6 PR, 2/6 PD Arm2-1/3 CR, 1/3 SD, 1/3 PD | [112] |
| Early phase I | 5 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4, followed by pulsing with hypochlorous acid (HOCl)-oxidized whole tumor lysate and maturation with LPS and IFNγ. | 5 doses of 5–10 million DCs | IN in the inguinal LN | - | 2/5-PD 1/5-mixed response 2/5-Improved PFS | [100] |
| Pilot study | 2 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4, followed by electroporation with Wilm’s Tumor 1 (WT1) mRNA and culturing with TNFα and IL-1β. | 4 doses of 21 million DCs | ID in the groin | - | Improved OS after chemotherapy following cessation of DC vaccination | [111] |
| Phase II | 26 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4 followed by incubating with mannosylated mucin 1 protein. | 3–7 doses of 25–40 million DCs | ID in the upper arm and thighs | - | 2/26-PD 2/26-PR 1/26-CR | [118] |
| Retrospective study | 56 | PBMCs were cultured in a medium with GM-CSF and IL-4 followed by stimulation with OK-432 (streptococcal immunological adjuvant) and prostaglandin E2, and pulsed with either WT1, mucin 1, or CA125 proteins. | 5–7 doses of 10 million DCs | ID near axial or inguinal LNs | OK-432 in patients without allergies to penicillin or other drugs | 42-PD 7-SD 1-PR | [119] |
| Phase II | 7 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4, followed by pulsing with whole tumor lysate and stimulation with Poly I:C. | 6 doses of >1 million DCs | IV | - | 4-PD 2-SD 1-PR | [120] |
| Phase I/II | 10 | Monocytes from PBMCs were cultured in a medium with GM-CSF, and IL-4, and matured in TNFα, followed by pulsing with tumor lysate and Keyhole Limpet Hemocyanin (KLH). | 2 doses of 40 million DCs | SC near axillary LN | DC vaccine + IL-2 | 5/10-CR 4/10-PD 1/10-dead from disease | [101] |
| Phase II | 56 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4, followed by pulsing with oxidized mannan coupled with mucin 1-glutathione S-transferase fusion protein. | 6–10 doses of 60 million DCs | ID in upper arms and thighs | Arm1: Standard-of-care treatment Arm2: DC vaccine | No improved PFS or OS between Arm1 and Arm2 | [99] |
| Phase I | 25 | Monocyte from PBMCs were cultured in a medium with GM-CSF and IL-4, followed by pulsing with HOCl-oxidized whole tumor lysate and maturation with LPS and IFNγ. | 5 doses of 5–10 million DCs | IN in the inguinal LN | Arm1: DC vaccine Arm2: DC vaccine + Bev Arm3: DC vaccine + Bev/Cy | Arm1: 2/5 PD, 3/5 SD Arm2: 5/10 PD, 5/10 SD Arm3: 2/10 PD, 8/10 SD | [113] |
| Phase I/II | 3 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4, followed by maturation in TNFα and pulsing with WT1 peptide. | 5 doses of 10–20 million DCs | ID into bilateral axillary parts | OK-432 lyophilized mixture of group A Streptococcus pyrogenes. | 1/3-SD 2/3-PD | [106] |
| Pilot study | 1 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4 followed by maturation with TNFα and IFNα; pulsed with autologous HLA type-1 restricted neoantigen peptides. | 4 doses of 5–12 million DCs | IN in the inguinal LNs | - | Improved symptoms | [102] |
| Early phase I | 19 | Monocytes from PBMCs were cultured in a medium with IL-4, GM-CSF, IL-15, and methylsulanylimidazole (p38 MAPK inhibitor), followed by maturation in TNFα, IL-1β, and prostaglandin E2 and then pulsed with 4 folate receptor-α (FRα) peptides. | 5 doses of 10–20 million DCs | ID into two ipsilateral areas of the body | - | 39% RFS of over 48 months from the time of enrollment into the study | [103] |
| Phase II | 71 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4, followed by pulsing with hydrostatic pressurized human OV-90 and SKOV-3 cell lysate; and maturation with poly(I:C) (TLR3 ligand). | 10 doses of 10 million DCs | SC near the inguinal LNs | Arm1: Chemotherapy Arm2: DC vaccine + chemotherapy | Improved OS of Arm2 over Arm1 | [121] |
| Phase I | 30 | Monocyte from PBMCs were cultured in a medium with GM-CSF and IL-4, followed by pulsing with hypochlorous acid (HOCl)-oxidized whole tumor cell lysate and maturation with LPS and IFNγ. | 5 doses of 5–10 million DCs | IN in the inguinal LN | Arm1: DC vaccine + Bev/Cy Arm2: DC vaccine + Bev/Cy + ASA Arm3: DC vaccine + Bev/Cy + ASA + IL-2 | Arm1: 8/10 >3 yr OS Arm2: 4/10 >3 yr OS Arm3: 4/10 >3 yr OS | [114] |
| Phase II | 136 | Monocytes from PBMCs were cultured in a medium with GM-CSF and IL-4, followed by pulsing with hydrostatic pressurized human OV-90 and SKOV-3 cell lysate; and maturation with poly(I:C) (TLR3 ligand). | 10–15 doses of 10 million DCs | - | Arm1: DC vaccine in parallel with chemotherapy Arm2: DC vaccine after chemotherapy Arm3: Chemotherapy only | Arm1: 20.3 months PFS Arm2: PFS not reached by the end of the study Arm3: 21.4 months PFS | [96,97] |
6. Potential Reasons for Poor Outcome of DC Vaccines in OC
6.1. Cancer Immunoediting, Antigen Loss, HLA Polymorphisms, and Defective Antigen-Presenting Machinery
6.2. Immunosuppressive TME in OC
6.3. Meagre Outcomes of moDC Vaccination
7. Future Perspectives
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lisio, M.-A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization—International Agency for Research on Cancer Global Cancer Observatory—Cancer Tomorrow. Available online: https://gco.iarc.fr/tomorrow/en/dataviz/bars?mode=population (accessed on 3 January 2022).
- World Health Organization—International Agency for Research on Cancer Global Cancer Observatory—Cancer Today. Available online: https://gco.iarc.fr/today/online-analysis-multi-bars?v=2020&mode=cancer&mode_population=countries&population=900&populations=900&key=asr&sex=2&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&nb_items=10&group_cancer=1&include_nmsc=1&include_nmsc_other=1&type_multiple=%257B%2522inc%2522%253Afalse%252C%2522mort%2522%253Atrue%252C%2522prev%2522%253Afalse%257D&orientation=horizontal&type_sort=0&type_nb_items=%257B%2522top%2522%253Atrue%252C%2522bottom%2522%253Afalse%257D (accessed on 3 January 2022).
- Colombo, N.; Sessa, C.; du Bois, A.; Ledermann, J.; McCluggage, W.G.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO-ESGO Consensus Conference Recommendations on Ovarian Cancer: Pathology and Molecular Biology, Early and Advanced Stages, Borderline Tumours and Recurrent Disease. Int. J. Gynecol. Cancer 2019, 29, 728–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlan, A.Y.; Testa, G. Exploiting Epigenetic Dependencies in Ovarian Cancer Therapy. Int. J. Cancer 2021, 149, 1732–1743. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; Desantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian Cancer Statistics, 2018. CA A Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Redondo, A.; Guerra, E.; Manso, L.; Martin-Lorente, C.; Martinez-Garcia, J.; Perez-Fidalgo, J.A.; Varela, M.Q.; Rubio, M.J.; Barretina-Ginesta, M.P.; González-Martín, A. A SEOM Clinical Guideline in Ovarian Cancer (2020). Clin. Transl. Oncol. 2021, 23, 961–968. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Pujade-Lauraine, E. Olaparib as Maintenance Treatment for Patients with Platinum-Sensitive Relapsed Ovarian Cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919849753. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; Rubin, S.C.; et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Hwang, W.-T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic Significance of Tumor-Infiltrating T-Cells in Ovarian Cancer: A Meta-Analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8 Tumor-Infiltrating Lymphocytes and a High CD8 Regulatory T Cell Ratio Are Associated with Favorable Prognosis in Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18543. [Google Scholar] [CrossRef] [Green Version]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed Cell Death 1 Ligand 1 and Tumor-Infiltrating CD8 T Lymphocytes Are Prognostic Factors of Human Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Hasegawa, K.; Matsushita, H.; Fujieda, N.; Sato, S.; Miyagi, E.; Kakimi, K.; Fujiwara, K. Expression of Multiple Immune Checkpoint Molecules on T Cells in Malignant Ascites from Epithelial Ovarian Carcinoma. Oncol. Lett. 2018, 15, 6457–6468. [Google Scholar] [CrossRef] [PubMed]
- González-Martín, A.; Sánchez-Lorenzo, L. Immunotherapy with Checkpoint Inhibitors in Patients with Ovarian Cancer: Still Promising? Cancer 2019, 125, 4616–4622. [Google Scholar] [CrossRef] [Green Version]
- Borella, F.; Ghisoni, E.; Giannone, G.; Cosma, S.; Benedetto, C.; Valabrega, G.; Katsaros, D. Immune Checkpoint Inhibitors in Epithelial Ovarian Cancer: An Overview on Efficacy and Future Perspectives. Diagnostics 2020, 10, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynam, S.; Lugade, A.A.; Odunsi, K. Immunotherapy for Gynecologic Cancer: Current Applications and Future Directions. Clin. Obstet. Gynecol. 2020, 63, 48–63. [Google Scholar] [CrossRef]
- Zhu, J.; Yan, L.; Wang, Q. Efficacy of PD-1/PD-L1 Inhibitors in Ovarian Cancer: A Single-Arm Meta-Analysis. J. Ovarian Res. 2021, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Coosemans, A.; Orsulic, S.; Cibula, D.; Vergote, I.; Galluzzi, L.; Spisek, R. Immunological Configuration of Ovarian Carcinoma: Features and Impact on Disease Outcome. J. ImmunoTherapy Cancer 2021, 9, e002873. [Google Scholar] [CrossRef]
- Varga, A.; Piha-Paul, S.; Ott, P.A.; Mehnert, J.M.; Berton-Rigaud, D.; Morosky, A.; Yang, P.; Ruman, J.; Matei, D. Pembrolizumab in Patients with Programmed Death Ligand 1–Positive Advanced Ovarian Cancer: Analysis of KEYNOTE-028. Gynecol. Oncol. 2019, 152, 243–250. [Google Scholar] [CrossRef]
- Kandalaft, L.E.; Odunsi, K.; Coukos, G. Immunotherapy in Ovarian Cancer: Are We There Yet? J. Clin. Oncol. 2019, 37, 2460–2471. [Google Scholar] [CrossRef]
- Yang, S.; Yin, X.; Yue, Y.; Wang, S. Application of Adoptive Immunotherapy in Ovarian Cancer. OncoTargets Ther. 2019, 12, 7975–7991. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Xia, B.-R.; Zhang, Z.-C.; Zhang, Y.-J.; Lou, G.; Jin, W.-L. Immunotherapy for Ovarian Cancer: Adjuvant, Combination, and Neoadjuvant. Front. Immunol. 2020, 11, 577869. [Google Scholar] [CrossRef]
- Pedersen, M.; Christine, M.; Westergaard, W.; Milne, K.; Nielsen, M.; Borch, H.; Poulsen, L.G.; Hendel, W.; Kennedy, M.; Briggs, G.; et al. Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes in Patients with Metastatic Ovarian Cancer: A Pilot Study. Oncoimmunology 2018, 7, e1502905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarivalasis, A.; Morotti, M.; Mulvey, A.; Imbimbo, M.; Coukos, G. Cell Therapies in Ovarian Cancer. Ther. Adv. Med. Oncol. 2021, 13, 17588359211008399. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar]
- Yan, W.; Hu, H.; Tang, B. Advances of Chimeric Antigen Receptor T Cell Therapy in Ovarian Cancer. OncoTargets Ther. 2019, 12, 8015–8022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correll, A.; Tü, A.; Becker, C.; Jonuleit, H.; Tüttenberg, A. Increased Regulatory T-Cell Frequencies in Patients with Advanced Melanoma Correlate with a Generally Impaired T-Cell Responsiveness and Are Restored after Dendritic Cell-Based Vaccination. Exp. Dermatol. 2010, 19, 213–221. [Google Scholar] [CrossRef]
- Hübbe, M.L.; Jæhger, D.E.; Andresen, T.L.; Andersen, M.H. Leveraging Endogenous Dendritic Cells to Enhance the Therapeutic Efficacy of Adoptive T-Cell Therapy and Checkpoint Blockade. Front. Immunol. 2020, 11, 578349. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.J.; Benike, C.; Fagnoni, F.; Liles, T.M.; Czerwinski, D.; Taidi, B.; Engleman, E.G.; Levy, R. Vaccination of Patients with B-Cell Lymphoma Using Autologous Antigen-Pulsed Dendritic Cells. Nat. Med. 1996, 2, 52–58. [Google Scholar] [CrossRef]
- Zhang, X.; He, T.; Li, Y.; Chen, L.; Liu, H.; Wu, Y.; Guo, H. Dendritic Cell Vaccines in Ovarian Cancer. Front. Immunol. 2021, 11, 613773. [Google Scholar] [CrossRef]
- Sprooten, J.; Ceusters, J.; Coosemans, A.; Agostinis, P.; de Vleeschouwer, S.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; Garg, A.D. Trial Watch: Dendritic Cell Vaccination for Cancer Immunotherapy. OncoImmunology 2019, 8, e1638212. [Google Scholar]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Immunogenic Cell Death Induction by Anticancer Chemotherapeutics. OncoImmunology 2017, 6, e1386829. [Google Scholar] [CrossRef] [Green Version]
- Laureano, R.S.; Sprooten, J.; Vanmeerbeerk, I.; Borras, D.M.; Govaerts, J.; Naulaerts, S.; Berneman, Z.N.; Beuselinck, B.; Bol, K.F.; Borst, J.; et al. Trial Watch: Dendritic Cell (DC)-Based Immunotherapy for Cancer. OncoImmunology 2022, 11, 2096363. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinman, R.M.; Cohn, Z.A. Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Mempel, T.R.; Henrickson, S.E.; von Andrian, U.H. T-Cell Priming by Dendritic Cells in Lymph Nodes Occurs in Three Distinct Phases. Nature 2004, 427, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Gerhard, G.M.; Bill, R.; Messemaker, M.; Klein, A.M.; Pittet, M.J. Tumor-Infiltrating Dendritic Cell States Are Conserved across Solid Human Cancers. J. Exp. Med. 2021, 218, e20200264. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.A.; Dutertre, C.-A.; Ginhoux, F.; Murphy, K.M. Genetic Models of Human and Mouse Dendritic Cell Development and Function. Nat. Rev. Immunol. 2021, 21, 101–115. [Google Scholar] [CrossRef]
- Murphy, T.L.; Murphy, K.M. Dendritic Cells in Cancer Immunology. Cell. Mol. Immunol. 2022, 19, 3–13. [Google Scholar] [CrossRef]
- Guilliams, M.; Dutertre, C.A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; van Gassen, S.; Chen, J.; Poidinger, M.; de Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef] [Green Version]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.-J.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ Dendritic Cells (DCs) Represent a Unique Myeloid DC Subset That Cross-Presents Necrotic Cell Antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [Green Version]
- Kretzer, N.M.; Theisen, D.J.; Tussiwand, R.; Briseño, C.G.; Grajales-Reyes, G.E.; Wu, X.; Durai, V.; Albring, J.; Bagadia, P.; Murphy, T.L.; et al. RAB43 Facilitates Cross-Presentation of Cell-Associated Antigens by CD8α+ Dendritic Cells. J. Exp. Med. 2016, 213, 2871–2883. [Google Scholar] [CrossRef]
- Noubade, R.; Majri-Morrison, S.; Tarbell, K.V. Beyond CDC1: Emerging Roles of DC Crosstalk in Cancer Immunity. Front. Immunol. 2019, 10, 1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, S.T.; Durai, V.; Wu, R.; Theisen, D.J.; Ward, J.P.; Bern, M.D.; Davidson, J.T., 4th; Bagadia, P.; Liu, T.; Briseño, C.G.; et al. CDC1 Prime and Are Licensed by CD4 + T Cells to Induce Anti-Tumour Immunity. Nature 2020, 584, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Paulete, A.R.; Cueto, F.J.; Martínez-López, M.; Labiano, S.; Morales-Kastresana, A.; Rodríguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.-J.; et al. Checkpoint Blockade Cancer Immunotherapy Targets Tumour-Specific Mutant Antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- del Prete, A.; Sozio, F.; Barbazza, I.; Salvi, V.; Tiberio, L.; Laffranchi, M.; Gismondi, A.; Bosisio, D.; Schioppa, T.; Sozzani, S. Functional Role of Dendritic Cell Subsets in Cancer Progression and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 3930. [Google Scholar] [CrossRef]
- Eickhoff, S.; Brewitz, A.; Gerner, M.Y.; Klauschen, F.; Komander, K.; Hemmi, H.; Garbi, N.; Kaisho, T.; Germain, R.N.; Kastenmüller, W. Robust Anti-Viral Immunity Requires Multiple Distinct T Cell-Dendritic Cell Interactions. Cell 2015, 162, 1322–1337. [Google Scholar] [CrossRef] [Green Version]
- Plantinga, M.; Guilliams, M.; Vanheerswynghels, M.; Deswarte, K.; Branco-Madeira, F.; Toussaint, W.; Vanhoutte, L.; Neyt, K.; Killeen, N.; Malissen, B.; et al. Conventional and Monocyte-Derived CD11b(+) Dendritic Cells Initiate and Maintain T Helper 2 Cell-Mediated Immunity to House Dust Mite Allergen. Immunity 2013, 38, 322–335. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Nish, S.A.; Jiang, R.; Hou, L.; Licona-Limón, P.; Weinstein, J.S.; Zhao, H.; Medzhitov, R. Control of T Helper 2 Responses by Transcription Factor IRF4-Dependent Dendritic Cells. Immunity 2013, 39, 722–732. [Google Scholar] [CrossRef] [Green Version]
- Laoui, D.; Keirsse, J.; Morias, Y.; van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The Tumour Microenvironment Harbours Ontogenically Distinct Dendritic Cell Populations with Opposing Effects on Tumour Immunity. Nat. Commun. 2016, 7, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlitzer, A.; McGovern, N.; Teo, P.; Zelante, T.; Atarashi, K.; Low, D.; Ho, A.W.S.; See, P.; Shin, A.; Wasan, P.S.; et al. IRF4 Transcription Factor-Dependent CD11b+ Dendritic Cells in Human and Mouse Control Mucosal IL-17 Cytokine Responses. Immunity 2013, 38, 970–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, E.K.; Uronen-Hansson, H.; Semmrich, M.; Rivollier, A.; Hägerbrand, K.; Marsal, J.; Gudjonsson, S.; Håkansson, U.; Reizis, B.; Kotarsky, K.; et al. IRF4 Transcription-Factor-Dependent CD103(+)CD11b(+) Dendritic Cells Drive Mucosal T Helper 17 Cell Differentiation. Immunity 2013, 38, 958–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubert, K.; Lehmann, C.H.K.; Heger, L.; Baranska, A.; Staedtler, A.M.; Buchholz, V.R.; Yamazaki, S.; Heidkamp, G.F.; Eissing, N.; Zebroski, H.; et al. Antigen Delivery to CD11c+CD8− Dendritic Cells Induces Protective Immune Responses against Experimental Melanoma in Mice In Vivo. J. Immunol. 2014, 192, 5830–5838. [Google Scholar] [CrossRef] [Green Version]
- Murgaski, A.; Kiss, M.; van Damme, H.; Kancheva, D.; Vanmeerbeek, I.; Keirsse, J.; Hadadi, E.; Brughmans, J.; Arnouk, S.M.; Hamouda, A.E.I.; et al. Efficacy of CD40 Agonists Is Mediated by Distinct CDC Subsets and Subverted by Suppressive Macrophages. Cancer Res. 2022. [CrossRef]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic Cells, Monocytes and Macrophages: A Unified Nomenclature Based on Ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human Dendritic Cell Subsets: An Update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 2019, 177, 556–571. [Google Scholar] [CrossRef]
- Kumamoto, Y.; Linehan, M.; Weinstein, J.S.; Laidlaw, B.J.; Craft, J.E.; Iwasaki, A. CD301b+ Dermal Dendritic Cells Drive T Helper 2 Cell-Mediated Immunity. Immunity 2013, 39, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K.L.; Caton, M.L.; Bogunovic, M.; Greter, M.; Grajkowska, L.T.; Ng, D.; Klinakis, A.; Charo, I.F.; Jung, S.; Gommerman, J.L.; et al. Notch2 Receptor Signaling Controls Functional Differentiation of Dendritic Cells in the Spleen and Intestine. Immunity 2011, 35, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.C.; Gudjonson, H.; Pritykin, Y.; Deep, D.; Lavallée, V.-P.; Mendoza, A.; Fromme, R.; Mazutis, L.; Ariyan, C.; Leslie, C.; et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity in Brief Single-Cell Analyses of Dendritic Cells Reveals New Subsets with Distinct pro-and Anti-Inflammatory Potential. Cell 2019, 179, 846–863. [Google Scholar] [CrossRef] [Green Version]
- Bosteels, C.; Neyt, K.; Vanheerswynghels, M.; van Helden, M.J.; Sichien, D.; Debeuf, N.; de Prijck, S.; Bosteels, V.; Vandamme, N.; Martens, L.; et al. Inflammatory Type 2 CDCs Acquire Features of CDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 2020, 52, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes and Progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leylek, R.; Alcántara-Hernández, M.; Lanzar, Z.; Lüdtke, A.; Perez, O.A.; Reizis, B.; Idoyaga, J. Integrated Cross-Species Analysis Identifies a Conserved Transitional Dendritic Cell Population. Cell Rep. 2019, 29, 3736–3750. [Google Scholar] [CrossRef] [Green Version]
- Tussiwand, R.; Everts, B.; Grajales-Reyes, G.E.; Kretzer, N.M.; Iwata, A.; Bagaitkar, J.; Wu, X.; Wong, R.; Anderson, D.A.; Murphy, T.L.; et al. Klf4 Expression in Conventional Dendritic Cells Is Required for T Helper 2 Cell Responses. Immunity 2015, 42, 916–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334. [Google Scholar] [CrossRef]
- Maier, B.; Leader, A.M.; Chen, S.T.; Tung, N.; Chang, C.; LeBerichel, J.; Chudnovskiy, A.; Maskey, S.; Walker, L.; Finnigan, J.P.; et al. A Conserved Dendritic-Cell Regulatory Program Limits Antitumour Immunity. Nature 2020, 580, 257–262. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M.; Merad, M. Expanding Dendritic Cell Nomenclature in the Single-Cell Era. Nat. Rev. Immunol. 2022, 22, 67–68. [Google Scholar] [CrossRef]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845. [Google Scholar] [CrossRef]
- Randolph, G.J.; Angeli, V.; Swartz, M.A. Dendritic-Cell Trafficking to Lymph Nodes through Lymphatic Vessels. Nat. Rev. Immunol. 2005, 5, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N. Spreading the Load: Antigen Transfer between Migratory and Lymph Node-Resident Dendritic Cells Promotes T-Cell Priming. Eur. J. Immunol. 2017, 47, 1798–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reizis, B.; Bunin, A.; Ghosh, H.S.; Lewis, K.L.; Sisirak, V. Plasmacytoid Dendritic Cells: Recent Progress and Open Questions. Annu. Rev. Immunol. 2011, 29, 163–183. [Google Scholar] [CrossRef] [Green Version]
- Poropatich, K.; Dominguez, D.; Chan, W.C.; Andrade, J.; Zha, Y.; Wray, B.; Miska, J.; Qin, L.; Cole, L.; Coates, S.; et al. OX40+ Plasmacytoid Dendritic Cells in the Tumor Microenvironment Promote Antitumor Immunity. J. Clin. Investig. 2020, 130, 3528–3542. [Google Scholar] [CrossRef]
- van Beek, J.J.P.; Flórez-Grau, G.; Gorris, M.A.J.; Bol, K.F.; Textor, J.; Jolanda, I.; de Vries Correspondence, M. Human PDCs Are Superior to CDC2s in Attracting Cytolytic Lymphocytes in Melanoma Patients Receiving DC Vaccination. Cell Rep. 2020, 30, 1027–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghirelli, C.; Reyal, F.; Jeanmougin, M.; Zollinger, R.; Sirven, P.; Michea, P.; Caux, C.; Bendriss-Vermare, N.; Donnadieu, M.H.; Caly, M.; et al. Breast Cancer Cell-Derived GM-CSF Licenses Regulatory Th2 Induction by Plasmacytoid Predendritic Cells in Aggressive Disease Subtypes. Cancer Res. 2015, 75, 2775–2787. [Google Scholar] [CrossRef] [Green Version]
- Kvedaraite, E.; Ginhoux, F. Human Dendritic Cells in Cancer. Sci. Immunol. 2022, 7, eabm9409. [Google Scholar] [CrossRef]
- Helft, J.; Böttcher, J.; Chakravarty, P.; Zelenay, S.; Huotari, J.; Schraml, B.U.; Goubau, D.; Reis e Sousa, C. GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c+MHCII+ Macrophages and Dendritic Cells. Immunity 2015, 42, 1197–1211. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer Chemotherapy-Induced Intratumoral Recruitment and Differentiation of Antigen-Presenting Cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [Green Version]
- Mastelic-Gavillet, B.; Sarivalasis, A.; Lozano, L.E.; Wyss, T.; Inoges, S.; de Vries, I.J.M.; Dartiguenave, F.; Jichlinski, P.; Derrè, L.; Coukos, G.; et al. Quantitative and Qualitative Impairments in Dendritic Cell Subsets of Patients with Ovarian or Prostate Cancer. Eur. J. Cancer 2020, 135, 173–182. [Google Scholar] [CrossRef]
- Truxova, I.; Kasikova, L.; Hensler, M.; Skapa, P.; Laco, J.; Pecen, L.; Belicova, L.; Praznovec, I.; Halaska, M.J.; Brtnicky, T.; et al. Mature Dendritic Cells Correlate with Favorable Immune Infiltrate and Improved Prognosis in Ovarian Carcinoma Patients. J. ImmunoTherapy Cancer 2018, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, U.K.; Rutkowski, M.R.; Rauwerdink, A.M.; Fields, J.; Escovar-Fadul, X.; Baird, J.; Cubillos-Ruiz, J.R.; Jacobs, A.C.; Gonzalez, J.L.; Weaver, J.; et al. Ovarian Cancer Progression Is Controlled by Phenotypic Changes in Dendritic Cells. J. Exp. Med. 2012, 209, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Conejo-Garcia, J.R.; Benencia, F.; Courreges, M.C.; Kang, E.; Mohamed-Hadley, A.; Buckanovich, R.J.; Holtz, D.O.; Jenkins, A.; Na, H.; Zhang, L.; et al. Tumor-Infiltrating Dendritic Cell Precursors Recruited by a β-Defensin Contribute to Vasculogenesis under the Influence of Vegf-A. Nat. Med. 2004, 10, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Huarte, E.; Cubillos-Ruiz, J.R.; Nesbeth, Y.C.; Scarlett, U.K.; Martinez, D.G.; Buckanovich, R.J.; Benencia, F.; Stan, R.V.; Keler, T.; Sarobe, P.; et al. Depletion of Dendritic Cells Delays Ovarian Cancer Progression by Boosting Antitumor Immunity. Cancer Res. 2008, 68, 7684–7691. [Google Scholar] [CrossRef] [Green Version]
- Labidi-Galy, S.I.; Treilleux, I.; Goddard-Leon, S.; Combes, J.D.; Blay, J.Y.; Ray-Coquard, I.; Caux, C.; Bendriss-Vermare, N. Plasmacytoid Dendritic Cells Infiltrating Ovarian Cancer Are Associated with Poor Prognosis. Oncoimmunology 2012, 1, 380–382. [Google Scholar] [CrossRef] [Green Version]
- Curiel, T.J.; Cheng, P.; Mottram, P.; Alvarez, X.; Moons, L.; Evdemon-Hogan, M.; Wei, S.; Zou, L.; Kryczek, I.; Hoyle, G.; et al. Dendritic Cell Subsets Differentially Regulate Angiogenesis in Human Ovarian Cancer. Cancer Res. 2004, 64, 5535–5538. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Kryczek, I.; Zou, L.; Daniel, B.; Cheng, P.; Mottram, P.; Curiel, T.; Lange, A.; Zou, W. Plasmacytoid Dendritic Cells Induce CD8+ Regulatory T Cells in Human Ovarian Carcinoma. Cancer Res. 2005, 65, 5020–5026. [Google Scholar] [CrossRef] [Green Version]
- Conrad, C.; Gregorio, J.; Wang, Y.H.; Ito, T.; Meller, S.; Hanabuchi, S.; Anderson, S.; Atkinson, N.; Ramirez, P.T.; Liu, Y.J.; et al. Plasmacytoid Dendritic Cells Promote Immunosuppression in Ovarian Cancer via ICOS Costimulation of Foxp3+ T-Regulatory Cells. Cancer Res. 2012, 72, 5240–5249. [Google Scholar] [CrossRef] [Green Version]
- Tang-Huau, T.L.; Gueguen, P.; Goudot, C.; Durand, M.; Bohec, M.; Baulande, S.; Pasquier, B.; Amigorena, S.; Segura, E. Human in Vivo-Generated Monocyte-Derived Dendritic Cells and Macrophages Cross-Present Antigens through a Vacuolar Pathway. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human Inflammatory Dendritic Cells Induce Th17 Cell Differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Bol, K.F.; Aarntzen, E.H.; Hout, F.E.I.T.; Schreibelt, G.; Creemers, J.H.A.; Lesterhuis, W.J.; Gerritsen, W.R.; Grunhagen, D.J.; Verhoef, C.; Punt, C.J.A.; et al. Favorable Overall Survival in Stage III Melanoma Patients after Adjuvant Dendritic Cell Vaccination. Oncoimmunology 2016, 5, e1057673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; de Vries, I.J.M.; Bol, K.F. Dendritic Cell Cancer Therapy: Vaccinating the Right Patient at the Right Time. Front. Immunol. 2018, 9, 2265. [Google Scholar] [CrossRef] [PubMed]
- Murgaski, A.; Bardet, P.M.R.; Arnouk, S.M.; Clappaert, E.J.; Laoui, D. Unleashing Tumour-Dendritic Cells to Fight Cancer by Tackling Their Three A’s: Abundance, Activation and Antigen-Delivery. Cancers 2019, 11, 670. [Google Scholar] [CrossRef] [Green Version]
- Rob, L.; Cibula, D.; Knapp, P.; Mallmann, P.; Klat, J.; Minar, L.; Bartos, P.; Chovanec, J.; Valha, P.; Pluta, M.; et al. Safety and Efficacy of Dendritic Cell-Based Immunotherapy DCVAC/OvCa Added to First-Line Chemotherapy (Carboplatin plus Paclitaxel) for Epithelial Ovarian Cancer: A Phase 2, Open-Label, Multicenter, Randomized Trial. J. ImmunoTherapy Cancer 2022, 10, e003190. [Google Scholar] [CrossRef]
- Fucikova, J.; Hensler, M.; Kasikova, L.; Lanickova, T.; Pasulka, J.; Rakova, J.; Drozenova, J.; Fredriksen, T.; Hraska, M.; Hrnciarova, T.; et al. An Autologous Dendritic Cell Vaccine Promotes Anticancer Immunity in Ovarian Cancer Patients with Low Mutational Burden and Cold Tumors. Clin. Cancer Res. 2022, 28, 3053–3065. [Google Scholar] [CrossRef] [PubMed]
- Hensler, M.; Rakova, J.; Kasikova, L.; Lanickova, T.; Pasulka, J.; Holicek, P.; Hraska, M.; Hrnciarova, T.; Kadlecova, P.; Schoenenberger, A.; et al. Peripheral Gene Signatures Reveal Distinct Cancer Patient Immunotypes with Therapeutic Implications for Autologous DC-Based Vaccines. OncoImmunology 2022, 11, 2101596. [Google Scholar] [CrossRef] [PubMed]
- Gray, H.J.; Benigno, B.; Berek, J.; Chang, J.; Mason, J.; Mileshkin, L.; Mitchell, P.; Moradi, M.; Recio, F.O.; Michener, C.M.; et al. Progression-Free and Overall Survival in Ovarian Cancer Patients Treated with CVac, a Mucin 1 Dendritic Cell Therapy in a Randomized Phase 2 Trial. J. ImmunoTherapy Cancer 2016, 4, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.L.-L.; Kandalaft, L.E.; Tanyi, J.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Smith, L.; Nisenbaum, H.L.; et al. A Dendritic Cell Vaccine Pulsed with Autologous Hypochlorous Acid-Oxidized Ovarian Cancer Lysate Primes Effective Broad Antitumor Immunity: From Bench to Bedside. Clin. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.; Kim, Y.-M.; Kim, S.-B.; Kim, C.-S.; Kwon, S.-W.; Kim, Y.; Kim, H.; Lee, H. Therapeutic DC Vaccination with IL-2 as a Consolidation Therapy for Ovarian Cancer Patients: A Phase I/II Trial. Cell. Mol. Immunol. 2015, 12, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Morisaki, T.; Hikichi, T.; Onishi, H.; Morisaki, T.; Kubo, M.; Hirano, T.; Yoshimura, S.; Kiyotani, K.; Nakamura, Y. Intranodal Administration of Neoantigen Peptide-Loaded Dendritic Cell Vaccine Elicits Epitope-Specific T Cell Responses and Clinical Effects in a Patient with Chemorefractory Ovarian Cancer with Malignant Ascites. Immunol. Investig. 2020, 50, 562–579. [Google Scholar] [CrossRef]
- Block, M.S.; Dietz, A.B.; Gustafson, M.P.; Kalli, K.R.; Erskine, C.L.; Youssef, B.; Vijay, G.V.; Allred, J.B.; Pavelko, K.D.; Strausbauch, M.A.; et al. Th17-Inducing Autologous Dendritic Cell Vaccination Promotes Antigen-Specific Cellular and Humoral Immunity in Ovarian Cancer Patients. Nat. Commun. 2020, 11, 5173. [Google Scholar] [PubMed]
- Cannon, M.J.; Goyne, H.E.; Stone, P.J.B.; MacDonald, L.J.; James, L.E.; Cobos, E.; Chiriva-Internati, M. Modulation of P38 MAPK Signaling Enhances Dendritic Cell Activation of Human CD4+ Th17 Responses to Ovarian Tumor Antigen. Cancer Immunol. Immunother. 2013, 62, 839–849. [Google Scholar] [PubMed] [Green Version]
- Rahma, O.E.; Ashtar, E.; Czystowska, M.; Szajnik, M.E.; Wieckowski, E.; Bernstein, S.; Herrin, V.E.; Shams, M.A.; Steinberg, S.M.; Merino, M.; et al. A Gynecologic Oncology Group Phase II Trial of Two P53 Peptide Vaccine Approaches: Subcutaneous Injection and Intravenous Pulsed Dendritic Cells in High Recurrence Risk Ovarian Cancer Patients. Cancer Immunol. Immunother. 2012, 61, 373–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Lu, X.; Cui, P.; Piao, C.; Xiao, M.; Liu, X.; Wang, Y.; Wu, X.; Liu, J.; Yang, L. Phase I/II Clinical Trial of a Wilms’ Tumor 1-Targeted Dendritic Cell Vaccination-Based Immunotherapy in Patients with Advanced Cancer. Cancer Immunol. Immunother. 2019, 68, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Brossart, P.; Wirths, S.; Stuhler, G.; Reichardt, V.L.; Kanz, L.; Brugger, W. Induction of Cytotoxic T-Lymphocyte Responses in Vivo after Vaccinations with Peptide-Pulsed Dendritic Cells. Blood 2000, 96, 3102–3108. [Google Scholar] [CrossRef]
- Chu, C.S.; Boyer, J.; Schullery, D.S.; Gimotty, P.A.; Gamerman, V.; Bender, J.; Levine, B.L.; Coukos, G.; Rubin, S.C.; Morgan, M.A.; et al. Phase I/II Randomized Trial of Dendritic Cell Vaccination with or without Cyclophosphamide for Consolidation Therapy of Advanced Ovarian Cancer in First or Second Remission. Cancer Immunol. Immunother. 2012, 61, 629–641. [Google Scholar] [CrossRef]
- Loveland, B.E.; Zhao, A.; White, S.; Gan, H.; Hamilton, K.; Xing, P.-X.; Pietersz, G.A.; Apostolopoulos, V.; Vaughan, H.; Karanikas, V.; et al. Mannan-MUC1–Pulsed Dendritic Cell Immunotherapy: A Phase I Trial in Patients with Adenocarcinoma. Clin. Cancer Res. 2006, 12, 869–877. [Google Scholar] [CrossRef] [Green Version]
- Hernando, J.J.; Park, T.-W.; Fischer, H.-P.; Zivanovic, O.; Braun, M.; Pölcher, M.; Grünn, U.; Leutner, C.; Pötzsch, B.; Kuhn, W. Vaccination with Dendritic Cells Transfected with MRNA-Encoded Folate-Receptor-α for Relapsed Metastatic Ovarian Cancer. Lancet Oncol. 2007, 8, 451–454. [Google Scholar] [CrossRef]
- Coosemans, A.; Vanderstraeten, A.; Tuyaerts, S.; Verschuere, T.; Moerman, P.; Berneman, Z.; Vergote, I.; Amant, F.; van Gool, S.W. Immunological Response after WT1 MRNA-Loaded Dendritic Cell Immunotherapy in Ovarian Carcinoma and Carcinosarcoma. Anticancer Res 2013, 33, 3855–3859. [Google Scholar]
- Kandalaft, L.E.; Powell, D.J.; Chiang, C.L.; Tanyi, J.; Kim, S.; Bosch, M.; Montone, K.; Mick, R.; Levine, B.L.; Torigian, D.A.; et al. Autologous Lysate-Pulsed Dendritic Cell Vaccination Followed by Adoptive Transfer of Vaccine-Primed Ex Vivo Co-Stimulated t Cells in Recurrent Ovarian Cancer. OncoImmunology 2013, 2, e22664. [Google Scholar] [CrossRef] [Green Version]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized Cancer Vaccine Effectively Mobilizes Antitumor T Cell Immunity in Ovarian Cancer. Sci. Transl. Med. 2018, 10, eaao5931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanyi, J.L.; Chiang, C.L.-L.; Chiffelle, J.; Thierry, A.-C.; Baumgartener, P.; Huber, F.; Goepfert, C.; Tarussio, D.; Tissot, S.; Torigian, D.A.; et al. Personalized Cancer Vaccine Strategy Elicits Polyfunctional T Cells and Demonstrates Clinical Benefits in Ovarian Cancer. npj Vaccines 2021, 6, 1–14. [Google Scholar]
- Hernando, J.J.; Park, T.-W.; Kubler, K.; Offergeld, R.; Schlebusch, H.; Baukneckt, T. Vaccination with Autologous Tumour Antigen-Pulsed Dendritic Cells in Advanced Gynaecological Malignancies: Clinical and Immunological Evaluation of a Phase I Trial. Cancer Immunol. Immunother. 2002, 51, 45–52. [Google Scholar] [CrossRef]
- Homma, S.; Sagawa, Y.; Ito, M.; Ohno, T.; Toda, G. Cancer Immunotherapy Using Dendritic/Tumour-Fusion Vaccine Induces Elevation of Serum Anti-Nuclear Antibody with Better Clinical Responses. Clin. Exp. Immunol. 2006, 144, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Peethambaram, P.P.; Melisko, M.E.; Rinn, K.J.; Alberts, S.R.; Provost, N.M.; Jones, L.A.; Sims, R.B.; Lin, L.R.C.; Frohlich, M.W.; Park, J.W. A Phase I Trial of Immunotherapy with Lapuleucel-T (APC8024) in Patients with Refractory Metastatic Tumors That Express HER-2/Neu. Clin. Cancer Res. 2009, 15, 5937–5944. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.L.; Quinn, M.A.; Grant, P.T.; Allen, D.G.; Jobling, T.W.; White, S.C.; Zhao, A.; Karanikas, V.; Vaughan, H.; Pietersz, G.; et al. A Phase 2, Single-Arm Study of an Autologous Dendritic Cell Treatment against Mucin 1 in Patients with Advanced Epithelial Ovarian Cancer. J. ImmunoTherapy Cancer 2014, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Chiba, A.; Izawa, H.; Yanagida, E.; Okamoto, M.; Shimodaira, S.; Yonemitsu, Y.; Shibamoto, Y.; Suzuki, N.; Nagaya, M. The Feasibility and Clinical Effects of Dendritic Cell-Based Immunotherapy Targeting Synthesized Peptides for Recurrent Ovarian Cancer. J. Ovarian Res. 2014, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bapsy, P.P.; Sharan, B.; Kumar, C.; Das, R.P.; Rangarajan, B.; Jain, M.; Suresh Attili, V.S.; Subramanian, S.; Aggarwal, S.; Srivastava, M.; et al. Open-Label, Multi-Center, Non-Randomized, Single-Arm Study to Evaluate the Safety and Efficacy of Dendritic Cell Immunotherapy in Patients with Refractory Solid Malignancies, on Supportive Care. Cytotherapy 2014, 16, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Cibula, D.; Rob, L.; Mallmann, P.; Knapp, P.; Klat, J.; Chovanec, J.; Minar, L.; Melichar, B.; Hein, A.; Kieszko, D.; et al. Dendritic Cell-Based Immunotherapy (DCVAC/OvCa) Combined with Second-Line Chemotherapy in Platinum-Sensitive Ovarian Cancer (SOV02): A Randomized, Open-Label, Phase 2 Trial. Gynecol. Oncol. 2021, 162, 652–660. [Google Scholar] [CrossRef]
- DuPage, M.; Mazumdar, C.; Schmidt, L.M.; Cheung, A.F.; Jacks, T. Expression of Tumour-Specific Antigens Underlies Cancer Immunoediting. Nature 2012, 482, 405–409. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; van Gool, S.W.; Agostinis, P. Dendritic Cell Vaccines Based on Immunogenic Cell Death Elicit Danger Signals and T Cell–Driven Rejection of High-Grade Glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef] [PubMed]
- Montico, B.; Lapenta, C.; Ravo, M.; Martorelli, D.; Muraro, E.; Zeng, B.; Comaro, E.; Spada, M.; Donati, S.; Santini, S.M.; et al. Exploiting a New Strategy to Induce Immunogenic Cell Death to Improve Dendritic Cell-Based Vaccines for Lymphoma Immunotherapy. Oncoimmunology 2017, 6, e1356964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, D.A.; Webb, J.R.; Nielsen, J.S.; Martin, S.D.; Kroeger, D.R.; Milne, K.; Castellarin, M.; Twumasi-Boateng, K.; Watson, P.H.; Holt, R.A.; et al. Surveillance of the Tumor Mutanome by T Cells during Progression from Primary to Recurrent Ovarian Cancer. Clin. Cancer Res. 2014, 20, 1125–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehata, M.; Mukherjee, A.; Deen, S.; Al-Attar, A.; Durrant, L.G.; Chan, S. Human Leukocyte Antigen Class I Expression Is an Independent Prognostic Factor in Advanced Ovarian Cancer Resistant to First-Line Platinum Chemotherapy. Br. J. Cancer 2009, 101, 1321–1328. [Google Scholar] [CrossRef] [Green Version]
- Han, L.Y.; Fletcher, M.S.; Urbauer, D.L.; Mueller, P.; Landen, C.N.; Kamat, A.A.; Lin, Y.G.; Merritt, W.M.; Spannuth, W.A.; Deavers, M.T.; et al. HLA Class I Antigen Processing Machinery Component Expression and Intratumoral T-Cell Infiltrate as Independent Prognostic Markers in Ovarian Carcinoma. Clin. Cancer Res. 2008, 14, 3372–3379. [Google Scholar] [CrossRef] [Green Version]
- Norell, H.; Carlsten, M.; Ohlum, T.; Malmberg, K.-J.; Masucci, G.; Schedvins, K.; Altermann, W.; Handke, D.; Atkins, D.; Seliger, B.; et al. Frequent Loss of HLA-A2 Expression in Metastasizing Ovarian Carcinomas Associated with Genomic Haplotype Loss and HLA-A2-Restricted HER-2/Neu-Specific Immunity. Cancer Res. 2006, 66, 6387–6394. [Google Scholar] [CrossRef] [Green Version]
- Buzzi, S.; Rubboli, D.; Buzzi, G.; Buzzi, A.M.; Morisi, C.; Pironi, F. CRM197 (Nontoxic Diphtheria Toxin): Effects on Advanced Cancer Patients. Cancer Immunol. Immunother. 2004, 53, 1041–1048. [Google Scholar] [CrossRef]
- Kamboj, K.K.; Kirchner, H.L.; Kimmel, R.; Greenspan, N.S.; Schreiber, J.R. Significant Variation in Serotype-Specific Immunogenicity of the Seven-Valent Streptococcus Pneumoniae Capsular Polysaccharide-CRM197 Conjugate Vaccine Occurs despite Vigorous T Cell Help Induced by the Carrier Protein. J. Infect. Dis. 2003, 187, 1629–1638. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Fang, X.; Wang, H.; Li, D.; Wang, X. Ovarian Cancer-Intrinsic Fatty Acid Synthase Prevents Anti-Tumor Immunity by Disrupting Tumor-Infiltrating Dendritic Cells. Front. Immunol. 2018, 9, 2927. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.A.; Baer, J.M.; Knolhoff, B.L.; Nywening, T.M.; Panni, R.Z.; Su, X.; Weilbaecher, K.N.; Hawkins, W.G.; Ma, C.; Fields, R.C.; et al. Breast and Pancreatic Cancer Interrupt IRF8-Dependent Dendritic Cell Development to Overcome Immune Surveillance. Nat. Commun. 2018, 9, 1–19. [Google Scholar] [CrossRef]
- Krempski, J.; Karyampudi, L.; Behrens, M.D.; Erskine, C.L.; Hartmann, L.; Dong, H.; Goode, E.L.; Kalli, K.R.; Knutson, K.L. Tumor-Infiltrating Programmed Death Receptor-1+ Dendritic Cells Mediate Immune Suppression in Ovarian Cancer. J. Immunol. 2011, 186, 6905–6913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Li, Y.; Zhang, D.; Ma, J. Axl Inhibition Induces the Antitumor Immune Response Which Can Be Further Potentiated by PD-1 Blockade in the Mouse Cancer Models. Oncotarget 2017, 8, 89761–89774. [Google Scholar] [CrossRef] [Green Version]
- Tesone, A.J.; Rutkowski, M.R.; Brencicova, E.; Svoronos, N.; Perales-Puchalt, A.; Stephen, T.L.; Allegrezza, M.J.; Payne, K.K.; Nguyen, J.M.; Wickramasinghe, J.; et al. Satb1 Overexpression Drives Tumor-Promoting Activities in Cancer-Associated Dendritic Cells. Cell Rep. 2016, 14, 1774–1786. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Engleman, E.G. Mechanisms of Action of Dendritic Cell Vaccines for the Treatment of Cancer. Drug Discov. Today Dis. Mech. 2006, 3, 213–218. [Google Scholar] [CrossRef]
- Ferris, S.T.; Ohara, R.A.; Ou, F.; Wu, R.; Huang, X.; Kim, S.; Chen, J.; Liu, T.-T.; Schreiber, R.D.; Murphy, T.L.; et al. CDC1 Vaccines Drive Tumor Rejection by Direct Presentation Independently of Host CDC1. Cancer Immunol. Res. 2022, 10, 920–931. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-Infiltrating DCs Suppress Nucleic Acid–Mediated Innate Immune Responses through Interactions between the Receptor TIM-3 and the Alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Xu, M.M.; Pu, Y.; Han, D.; Shi, Y.; Cao, X.; Liang, H.; Chen, X.; Li, X.-D.; Deng, L.; Chen, Z.J.; et al. Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-Priming through Signal Regulatory Protein α Signaling. Immunity 2017, 47, 363–373.e5. [Google Scholar] [CrossRef]
- Ogawa, F.; Amano, H.; Eshima, K.; Ito, Y.; Matsui, Y.; Hosono, K.; Kitasato, H.; Iyoda, A.; Iwabuchi, K.; Kumagai, Y.; et al. Prostanoid Induces Premetastatic Niche in Regional Lymph Nodes. J. Clin. Investig. 2014, 124, 4882–4894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Ning, F.; Cole, C.B.; Annunziata, C.M. Driving Immune Responses in the Ovarian Tumor Microenvironment. Front. Oncol. 2021, 10, 604084. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Jin, Y.; Tian, Y.; Zhang, H.; Wu, J.; Lu, W.; Lu, X. Regulatory B Cells Contribute to the Impaired Antitumor Immunity in Ovarian Cancer Patients. Tumor Biol. 2016, 37, 6581–6588. [Google Scholar] [CrossRef]
- Nowak, M.; Klink, M. The Role of Tumor-Associated Macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cells 2020, 9, 1299. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Sasano, T.; Komura, N. Targeting Myeloid-Derived Suppressor Cells in Ovarian Cancer. Cells 2021, 10, 329. [Google Scholar] [CrossRef]
- Coosemans, A.; Baert, T.; Ceusters, J.; Busschaert, P.; Landolfo, C.; Verschuere, T.; van Rompuy, A.S.; Vanderstichele, A.; Froyman, W.; Neven, P.; et al. Myeloid-Derived Suppressor Cells at Diagnosis May Discriminate between Benign and Malignant Ovarian Tumors. Int. J. Gynecol. Cancer 2019, 29, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Santegoets, S.J.A.M.; de Groot, A.F.; Dijkgraaf, E.M.; Simões, A.M.C.; van der Noord, V.E.; van Ham, J.J.; Welters, M.J.P.; Kroep, J.R.; van der Burg, S.H. The Blood MMDSC to DC Ratio Is a Sensitive and Easy to Assess Independent Predictive Factor for Epithelial Ovarian Cancer Survival. OncoImmunology 2018, 7, e1465166. [Google Scholar] [CrossRef] [Green Version]
- Bakdash, G.; Buschow, S.I.; Gorris, M.A.; Halilovic, A.; Hato, S.V.; Sköld, A.E.; Schreibelt, G.; Sittig, S.P.; Torensma, R.; Duiveman-de Boer, T.; et al. Expansion of a BDCA1+CD14+ Myeloid Cell Population in Melanoma Patients May Attenuate the Efficacy of Dendritic Cell Vaccines. Cancer Res. 2016, 76, 4332–4346. [Google Scholar] [CrossRef] [Green Version]
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Teijeira, A.; Kandalaft, L.E.; Romero, P.; Coukos, G.; et al. The Clinical Application of Cancer Immunotherapy Based on Naturally Circulating Dendritic Cells. J. ImmunoTherapy Cancer 2019, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, P.A.; Liu, S.; Yeh, J.E.; Ye, D.Q.; Barbuto, J.A.M.; Frank, D.A.; Toniolo, P.A.; Liu, S.; Yeh, J.E.; Ye, D.Q.; et al. Deregulation of SOCS5 Suppresses Dendritic Cell Function in Chronic Lymphocytic Leukemia. Oncotarget 2016, 7, 46301–46314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wculek, S.K.; Amores-Iniesta, J.; Conde-Garrosa, R.; Khouili, S.C.; Melero, I.; Sancho, D. Effective Cancer Immunotherapy by Natural Mouse Conventional Type-1 Dendritic Cells Bearing Dead Tumor Antigen. J. ImmunoTherapy Cancer 2019, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cueto, F.J.; Sancho, D. The Flt3L/Flt3 Axis in Dendritic Cell Biology and Cancer Immunotherapy. Cancers 2021, 13, 1525. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.; Hou, Y.; Rivas, A.; Benike, C.; Yuen, A.; Fisher, G.A.; Davis, M.M.; Engleman, E.G. Altered Peptide Ligand Vaccination with Flt3 Ligand Expanded Dendritic Cells for Tumor Immunotherapy. Proc. Natl. Acad. Sci. USA 2001, 98, 8809–8814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.F.; Grimm, A.J.; Chiang, C.L.-L.; Mookerjee, A.; Flies, D.; Jean, S.; McCann, G.A.; Michaux, J.; Pak, H.; Huber, F.; et al. Rapid Tumor Vaccine Using Toll-like Receptor-Activated Ovarian Cancer Ascites Monocytes. J. ImmunoTherapy Cancer 2020, 8, e000875. [Google Scholar] [CrossRef] [PubMed]
- de Bruyn, C.; Ceusters, J.; Landolfo, C.; Baert, T.; Thirion, G.; Claes, S.; Vankerckhoven, A.; Wouters, R.; Schols, D.; Timmerman, D.; et al. Neo-Adjuvant Chemotherapy Reduces, and Surgery Increases Immunosuppression in First-Line Treatment for Ovarian Cancer. Cancers 2021, 13, 5899. [Google Scholar] [CrossRef] [PubMed]
- Caminschi, I.; Maraskovsky, E.; Heath, W.R. Targeting Dendritic Cells in Vivo for Cancer Therapy. Front. Immunol. 2012, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Tacken, P.J.; de Vries, I.J.M.; Torensma, R.; Figdor, C.G. Dendritic-Cell Immunotherapy: From Ex Vivo Loading to In Vivo Targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef]
- Baldin, A.V.; Savvateeva, L.V.; Bazhin, A.V.; Zamyatnin, A.A. Dendritic Cells in Anticancer Vaccination: Rationale for Ex Vivo Loading or In Vivo Targeting. Cancers 2020, 12, 590. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of Antigen-Specific Immunity with a Vaccine Targeting NY-ESO-1 to the Dendritic Cell Receptor DEC-205. Sci. Transl. Med. 2014, 6, 232ra51. [Google Scholar] [CrossRef]
- Morse, M.A.; Chapman, R.; Powderly, J.; Blackwell, K.; Keler, T.; Green, J.; Riggs, R.; He, L.-Z.; Ramakrishna, V.; Vitale, L.; et al. Phase I Study Utilizing a Novel Antigen-Presenting Cell–Targeted Vaccine with Toll-like Receptor Stimulation to Induce Immunity to Self-Antigens in Cancer Patients. Clin. Cancer Res. 2011, 17, 4844–4853. [Google Scholar] [CrossRef] [Green Version]
- Fucikova, J.; Palova-Jelinkova, L.; Bartunkova, J.; Spisek, R. Induction of Tolerance and Immunity by Dendritic Cells: Mechanisms and Clinical Applications. Front. Immunol. 2019, 10, 2393. [Google Scholar] [CrossRef]
- Byrne, K.T.; Vonderheide, R.H. CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep. 2016, 15, 2719–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornburg, M.; Desbois, M.; Lu, S.; Guan, Y.; Lo, A.A.; Kaufman, S.; Elrod, A.; Lotstein, A.; DesRochers, T.M.; Munoz-Rodriguez, J.L.; et al. Single-Cell Dissection of Cellular Components and Interactions Shaping the Tumor Immune Phenotypes in Ovarian Cancer. Cancer Cell 2021, 39, 928–944.e6. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Hori, S.; Ohnishi, S.; Toritsuka, M.; Fujii, T.; Shimizu, T.; Owari, T.; Morizawa, Y.; Gotoh, D.; Itami, Y.; et al. Supplementary Granulocyte Macrophage Colony-stimulating Factor to Chemotherapy and Programmed Death-ligand 1 Blockade Decreases Local Recurrence after Surgery in Bladder Cancer. Cancer Sci. 2019, 110, 3315–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.X.; Chan, S.; Kwek, S.; Lewis, J.; Dao, V.; Zhang, L.; Cooperberg, M.R.; Ryan, C.J.; Lin, A.M.; Friedlander, T.W.; et al. Systemic GM-CSF Recruits Effector T Cells into the Tumor Microenvironment in Localized Prostate Cancer. Cancer Immunol. Res. 2016, 4, 948–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulley, J.L.; Madan, R.A.; Schlom, J. Impact of Tumour Volume on the Potential Efficacy of Therapeutic Vaccines. Curr. Oncol. 2011, 18, 150–157. [Google Scholar] [CrossRef] [Green Version]


| Status | NCT Number | Phase | Treatment|Combinations |
|---|---|---|---|
| Active, not recruiting | NCT00799110 | Phase 2 | DC/tumor fusion vaccine|Imiquimod|GM-CSF |
| Active, not recruiting | NCT02033616 | Phase 2 | DC vaccine|PBMCs |
| Active, not recruiting | NCT02111941 | Early Phase 1 | DC vaccine |
| Not yet recruiting | NCT05270720 | Phase 1 | DC vaccine |
| Not yet recruiting | NCT03735589 | Phase 1|Phase 2 | DC vaccine|Autologous NK Cell-like CTLs |
| Recruiting | NCT00703105 | Phase 2 | DC vaccine |
| Recruiting | NCT04614051 | Phase 1 | DC vaccine |
| Recruiting | NCT04739527 | Phase 1 | DC vaccine |
| Recruiting | NCT04834544 | Phase 2 | DC vaccine |
| Completed | NCT01617629 | Phase 2 | DC vaccine |
| Completed | NCT01068509 | Phase 2 | DC vaccine |
| Completed | NCT01132014 | Early Phase 1 | DC vaccine |
| Completed | NCT00478452 | Phase 1 | DC vaccine|DC vaccine with Cyclophosphamide |
| Completed | NCT00683241 | Phase 1 | DC vaccine |
| Completed | NCT00005956 | - | HER-2/neu intracellular domain protein|DC vaccine |
| Completed | NCT03657966 | Phase 2 | DC vaccine|Standard-of-care chemotherapy |
| Completed | NCT00004604 | Phase 1 | DC vaccine |
| Completed | NCT00027534 | Phase 1 | DC vaccine |
| Completed | NCT00019084 | Phase 2 | Aldesleukin|DC vaccine|Ras peptide cancer vaccine|Sargramostim|Autologous lymphocytes|Therapeutic tumor-infiltrating lymphocytes |
| Completed | NCT02179515 | Phase 1 | DC vaccine |
| Completed | NCT01132014 | Early phase 1 | DC vaccine |
| Completed | NCT02107950 | Phase 2 | DC vaccine in parallel with chemotherapy|Standard-of-care |
| Completed | NCT02107937 | Phase 2 | DC vaccine with Standard-of-care|DC vaccine after chemotherapy|Standard-of-care |
| Unknown status | NCT01456065 | Phase 1 | DC vaccine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caro, A.A.; Deschoemaeker, S.; Allonsius, L.; Coosemans, A.; Laoui, D. Dendritic Cell Vaccines: A Promising Approach in the Fight against Ovarian Cancer. Cancers 2022, 14, 4037. https://doi.org/10.3390/cancers14164037
Caro AA, Deschoemaeker S, Allonsius L, Coosemans A, Laoui D. Dendritic Cell Vaccines: A Promising Approach in the Fight against Ovarian Cancer. Cancers. 2022; 14(16):4037. https://doi.org/10.3390/cancers14164037
Chicago/Turabian StyleCaro, Aarushi Audhut, Sofie Deschoemaeker, Lize Allonsius, An Coosemans, and Damya Laoui. 2022. "Dendritic Cell Vaccines: A Promising Approach in the Fight against Ovarian Cancer" Cancers 14, no. 16: 4037. https://doi.org/10.3390/cancers14164037