
Identification of a Novel Cuproptosis-Related Gene Signature for Prognostic Implication in Head and Neck Squamous Carcinomas

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Development and Validation of a Prognostic Cuproptosis-Related Gene Signature
2.3. Principal Component Analysis (PCA)
2.4. Construction of a Nomogram for OS Prediction
2.5. The Characteristic Distinction between the Low- and High-Risk Score Groups
2.6. Functional Enrichment Analysis
2.7. Gene Set Variation Analysis (GSVA)
2.8. PPI Network
2.9. Cell Culture
2.10. RNA Isolation and RT-qPCR
2.11. Statistical Analysis
3. Results
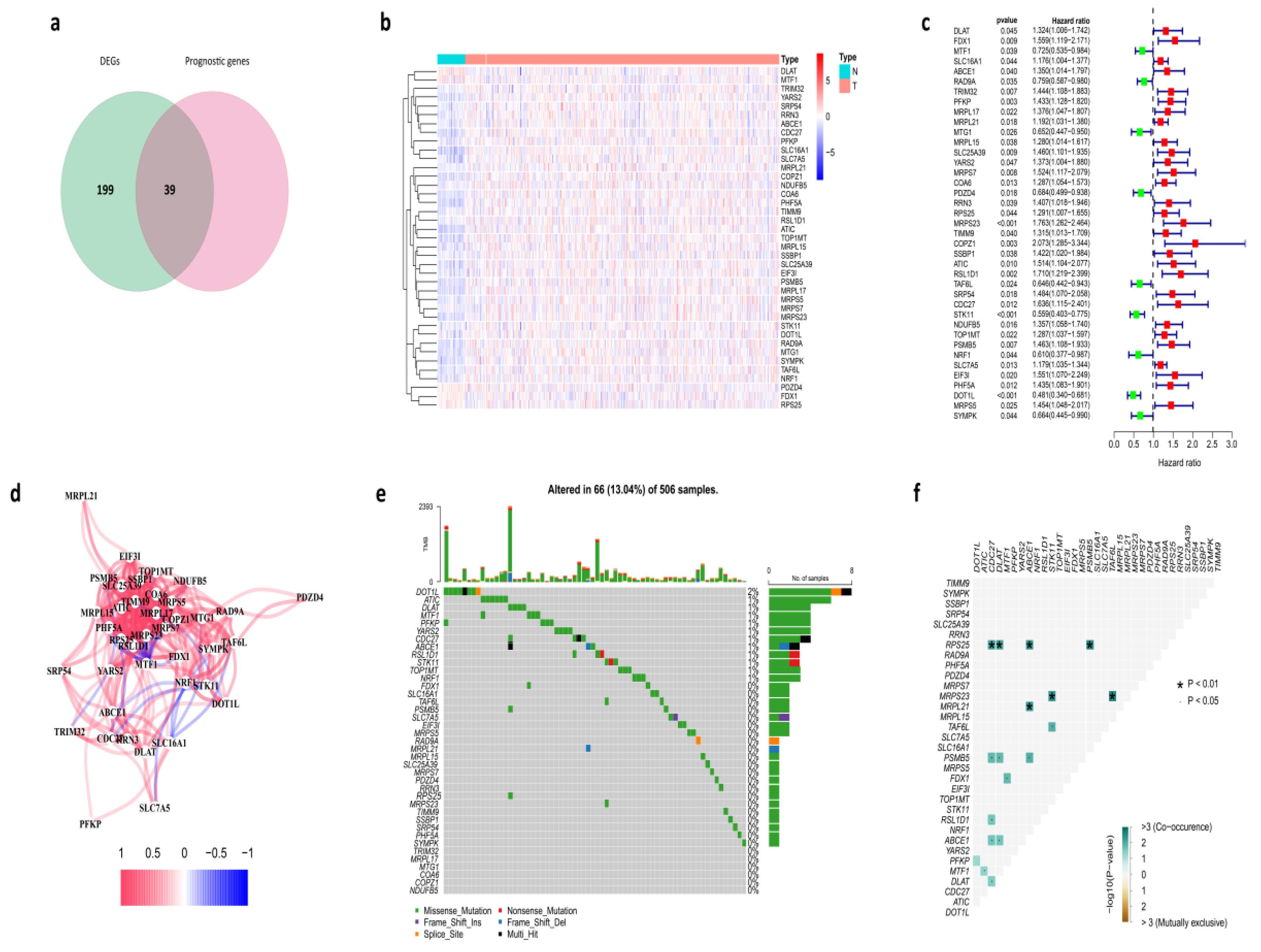
3.1. Identification of Prognostic Cuproptosis-Related DEGs in the TCGA Cohort
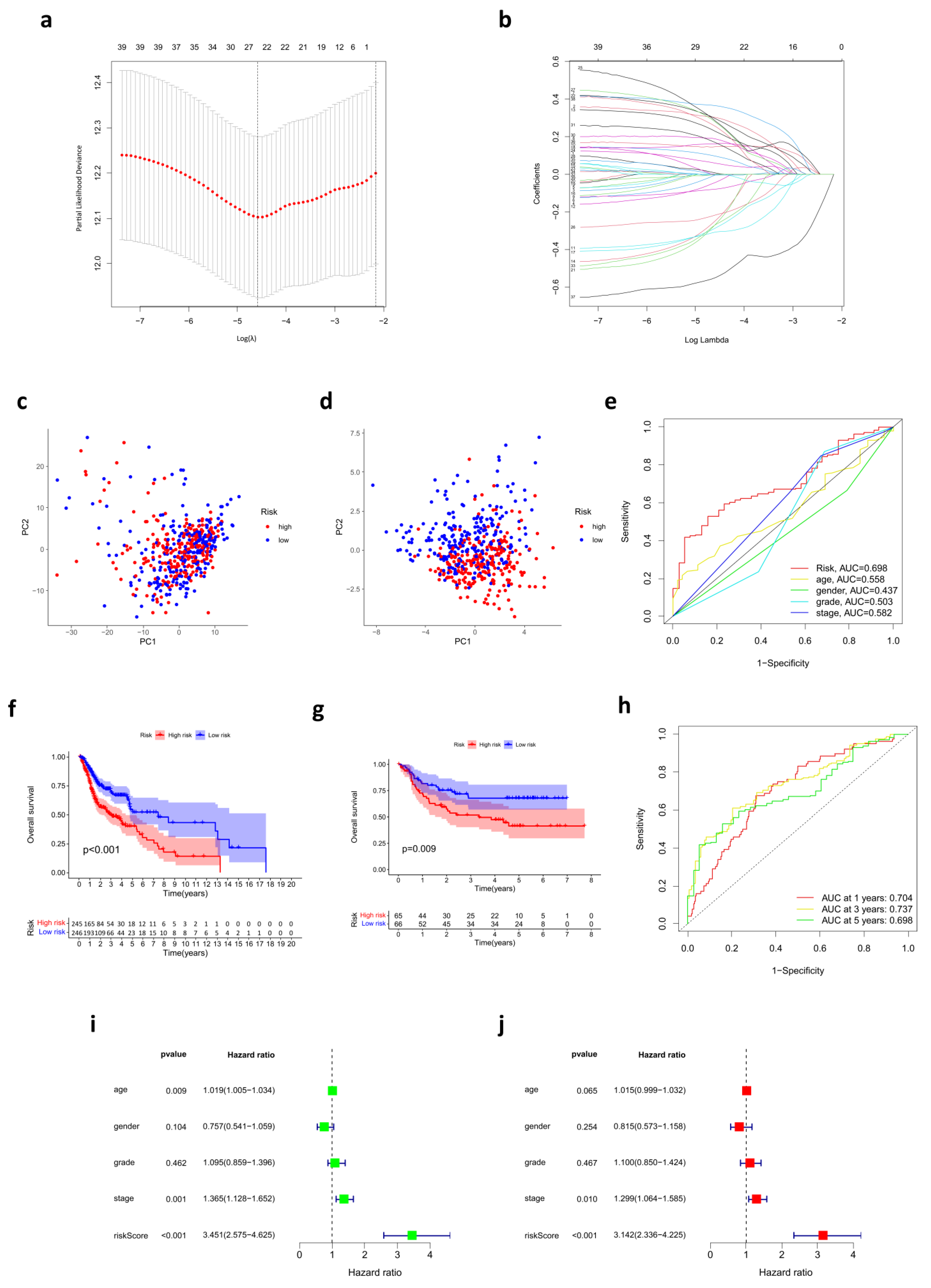
3.2. Prognostic Risk Score Model Developed in the TCGA Cohort
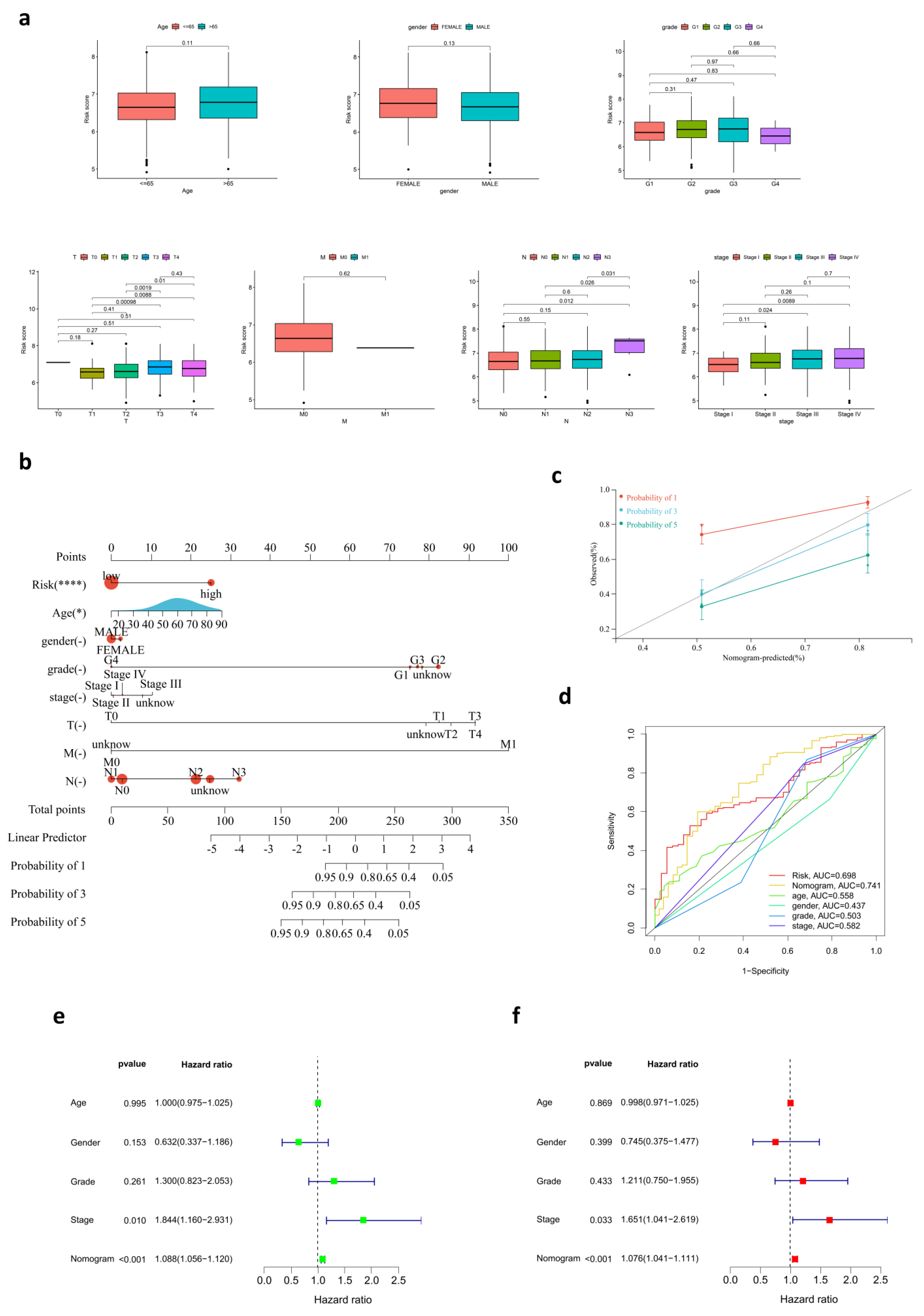
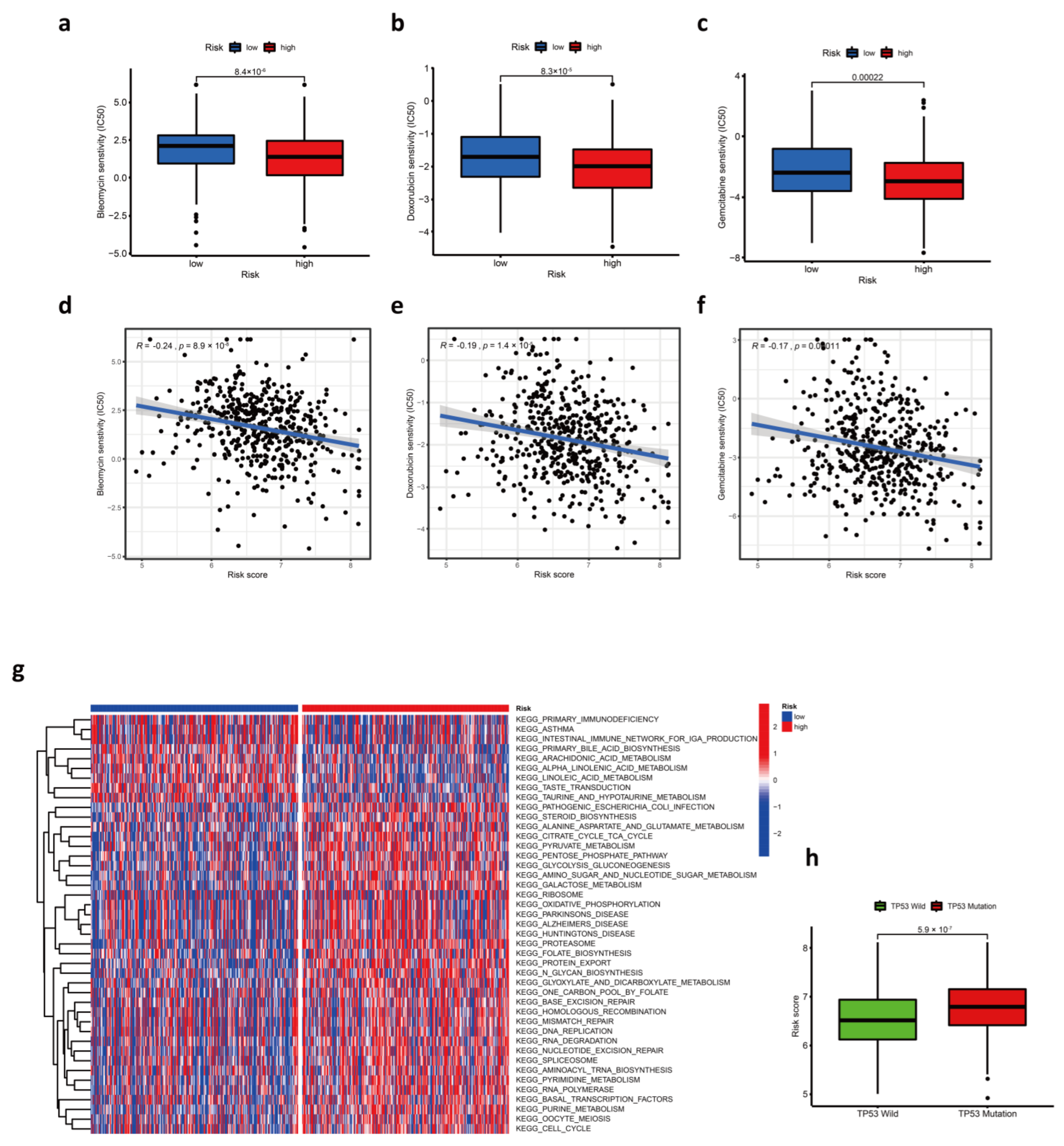
3.3. The Relationship between Risk Score and Clinical Characteristics
3.4. Development of a Nomogram for OS Prediction
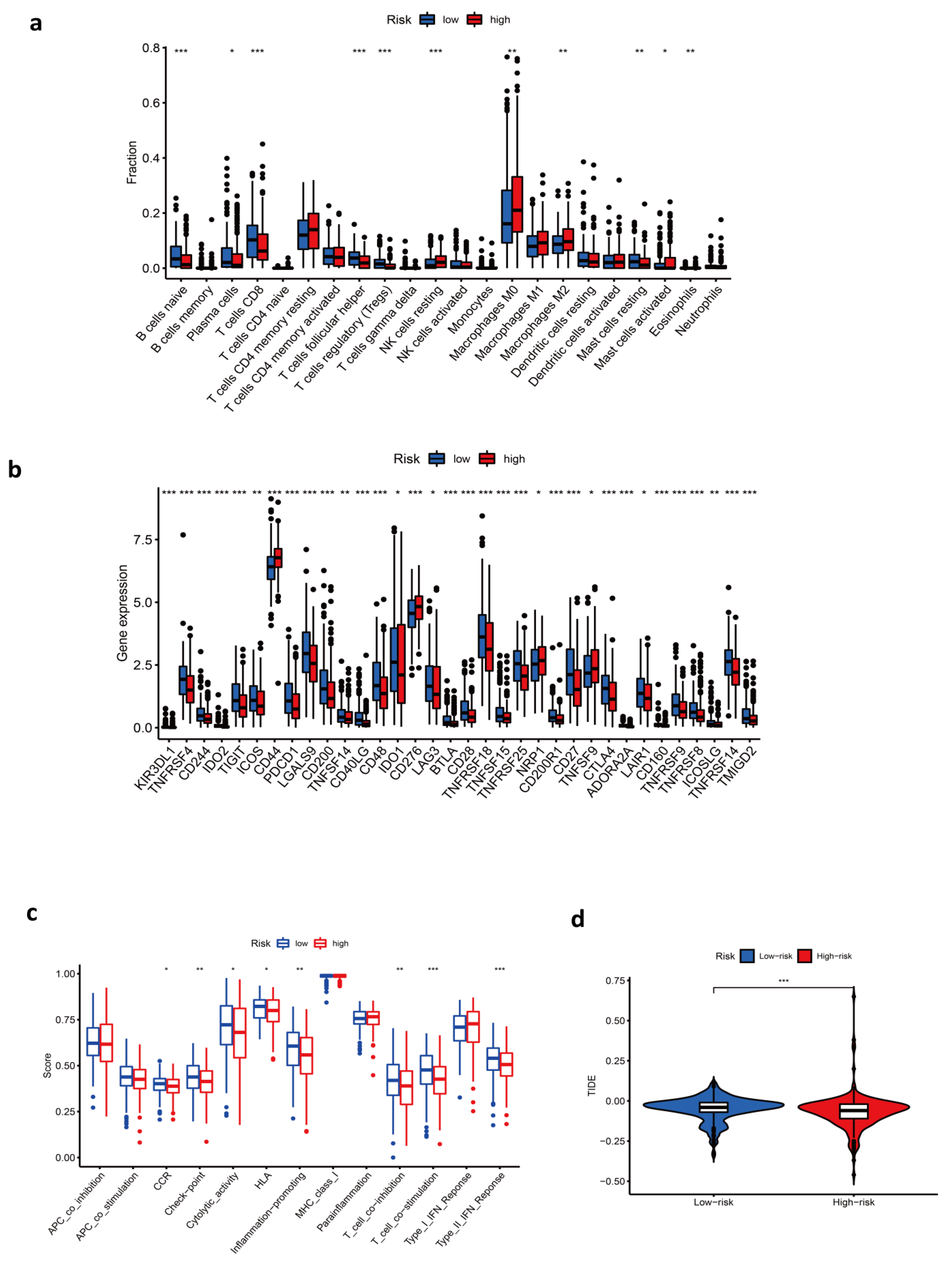
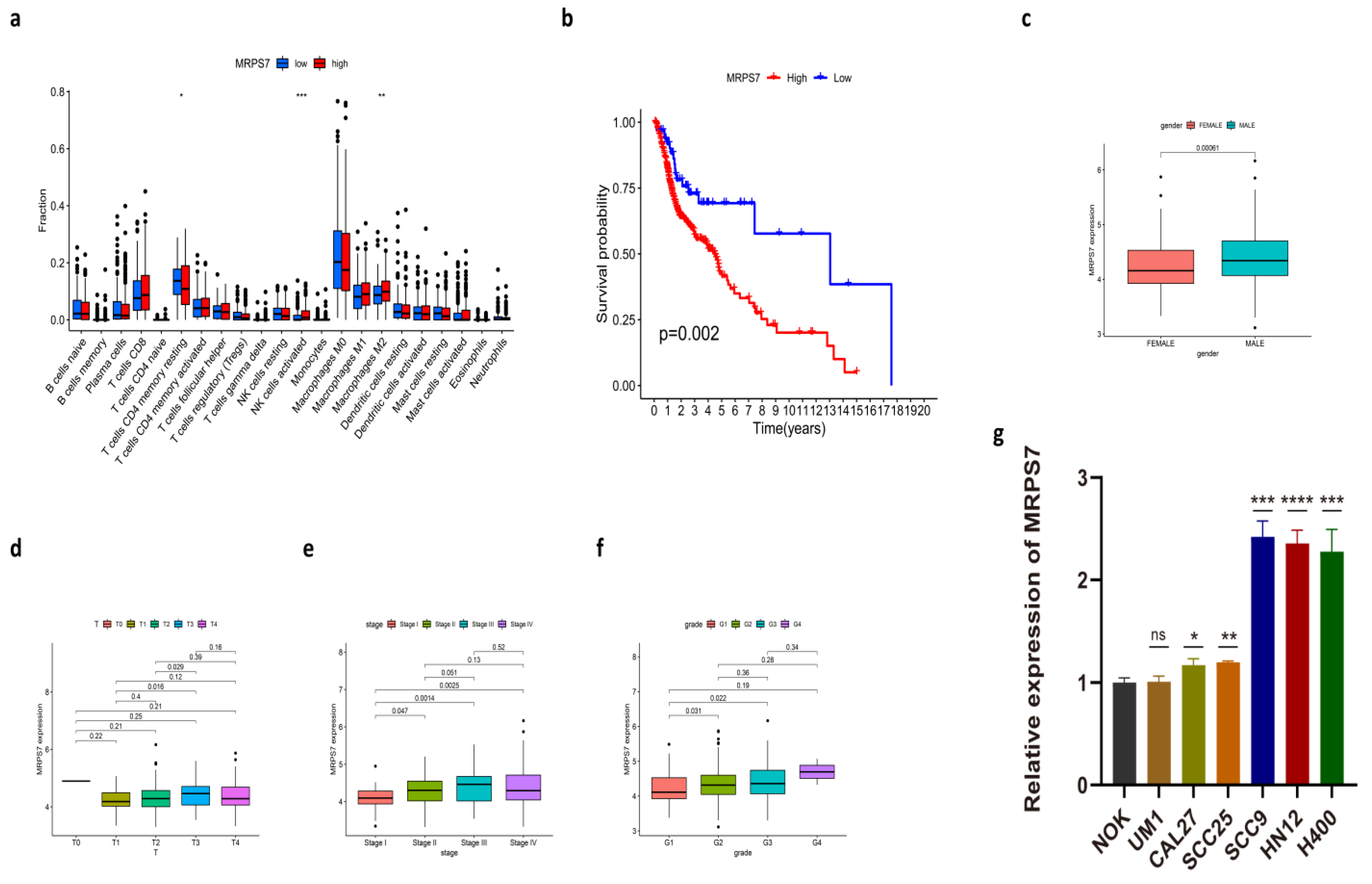
3.5. Immune-Related Characteristics in the Low- and High-Risk Score Groups
3.6. Response to Chemotherapy Drugs
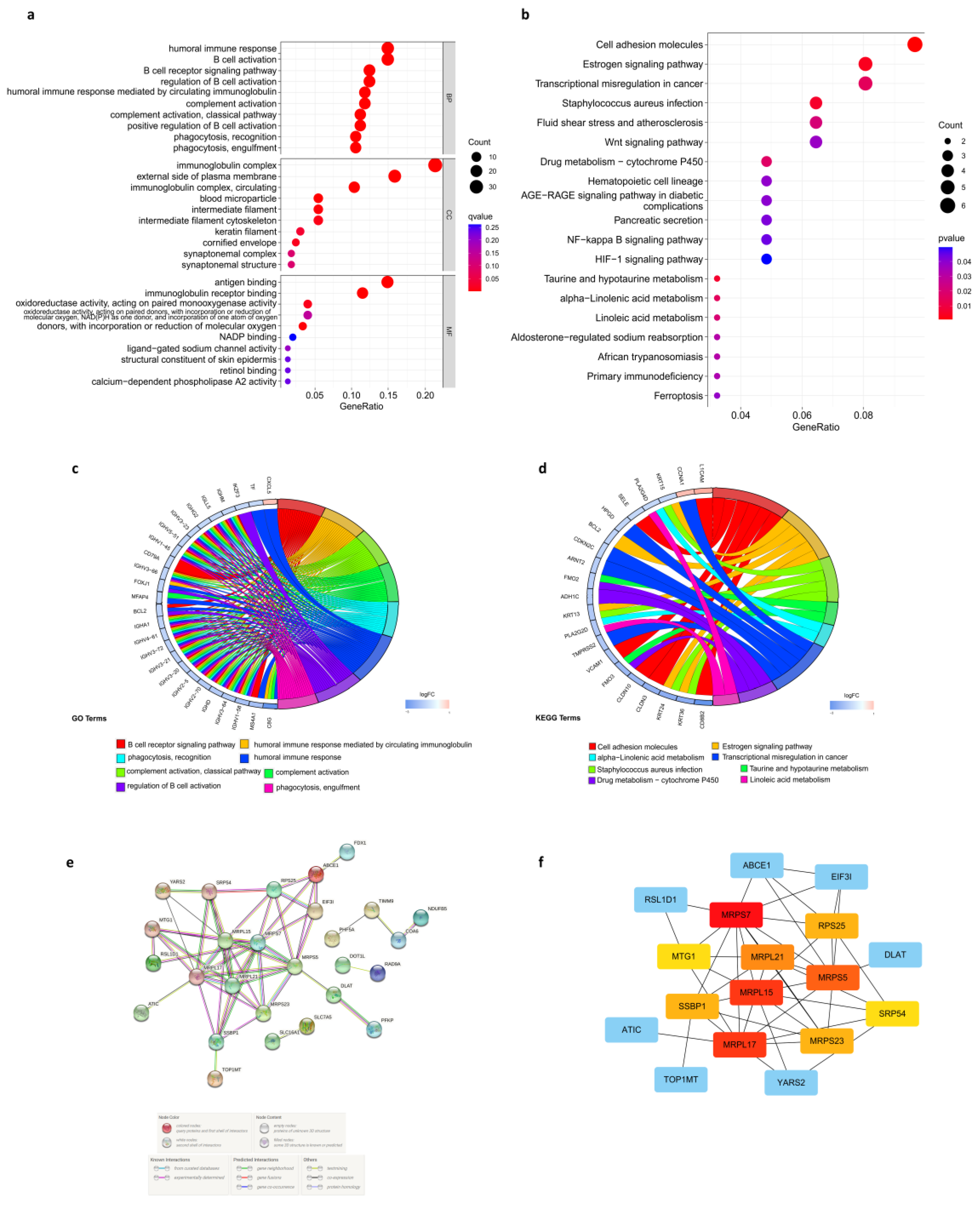
3.7. Enrichment Analysis in the Low- and High-Risk Score Groups
3.8. Protein-Protein Interaction (PPI) Network of Prognostic Cuproptosis-Related DEGs
3.9. The Expression of MRPS7 in HNSC Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Dai, Y.; Xing, X.; Sun, X. Identification and validation of a prognostic signature and combination drug therapy for immunotherapy of head and neck squamous cell carcinoma. Comput. Struct. Biotechnol. J. 2021, 19, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Ge, H.; Wu, Y.; Zhang, Y.; Guo, S.; Zhang, P.; Cheng, J.; Wang, Y. Identification of an autophagy-related prognostic signature in head and neck squamous cell carcinoma. J. Oral Pathol. Med. 2021, 50, 1040–1049. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Gholipour, M.; Taheri, M.; Shirvani Farsani, Z. MicroRNA profile in the squamous cell carcinoma: Prognostic and diagnostic roles. Heliyon 2020, 6, e05436. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, G. The Potential of Oxidative Stress Related Genes as Prognostic Biomarkers in Head and Neck Squamous Cell Carcinoma. Comb. Chem. High Throughput Screen. 2021. [Google Scholar] [CrossRef]
- Tsang, T.; Davis, C.I.; Brady, D.C. Copper biology. Curr. Biol. 2021, 31, R421–R427. [Google Scholar] [CrossRef]
- McAuslan, B.R.; Reilly, W. Endothelial cell phagokinesis in response to specific metal ions. Exp. Cell Res. 1980, 130, 147–157. [Google Scholar] [CrossRef]
- Sen, C.K.; Khanna, S.; Venojarvi, M.; Trikha, P.; Ellison, E.C.; Hunt, T.K.; Roy, S. Copper-induced vascular endothelial growth factor expression and wound healing. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1821-7. [Google Scholar] [CrossRef]
- Jiang, Y.; Huo, Z.; Qi, X.; Zuo, T.; Wu, Z. Copper-induced tumor cell death mechanisms and antitumor theragnostic applications of copper complexes. Nanomedicine 2022, 17, 303–324. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M.J. Copper metabolism as a unique vulnerability in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118893. [Google Scholar] [CrossRef]
- Oliveri, V. Selective Targeting of Cancer Cells by Copper Ionophores: An Overview. Front. Mol. Biosci. 2022, 9, 841814. [Google Scholar] [CrossRef]
- Vyas, A.; Duvvuri, U.; Kiselyov, K. Copper-dependent ATP7B up-regulation drives the resistance of TMEM16A-overexpressing head-and-neck cancer models to platinum toxicity. Biochem. J. 2019, 476, 3705–3719. [Google Scholar] [CrossRef]
- Ressnerova, A.; Raudenska, M.; Holubova, M.; Svobodova, M.; Polanska, H.; Babula, P.; Masarik, M.; Gumulec, J. Zinc and Copper Homeostasis in Head and Neck Cancer: Review and Meta-Analysis. Curr. Med. Chem. 2016, 23, 1304–1330. [Google Scholar] [CrossRef]
- Chen, F.; Wang, J.; Chen, J.; Yan, L.; Hu, Z.; Wu, J.; Bao, X.; Lin, L.; Wang, R.; Cai, L.; et al. Serum copper and zinc levels and the risk of oral cancer: A new insight based on large-scale case-control study. Oral Dis. 2019, 25, 80–86. [Google Scholar] [CrossRef]
- Grassi, I.; Nanni, C.; Cicoria, G.; Blasi, C.; Bunkheila, F.; Lopci, E.; Colletti, P.M.; Rubello, D.; Fanti, S. Usefulness of 64Cu-ATSM in head and neck cancer: A preliminary prospective study. Clin. Nucl. Med. 2014, 39, e59–e63. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef]
- Prime, S.S.; Nixon, S.V.; Crane, I.J.; Stone, A.; Matthews, J.B.; Maitland, N.J.; Remnant, L.; Powell, S.K.; Game, S.M.; Scully, C. The behaviour of human oral squamous cell carcinoma in cell culture. J. Pathol. 1990, 160, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Gothelf, A.; Mir, L.M.; Gehl, J. Electrochemotherapy: Results of cancer treatment using enhanced delivery of bleomycin by electroporation. Cancer Treat. Rev. 2003, 29, 371–387. [Google Scholar] [CrossRef]
- Zakelj, M.N.; Prevc, A.; Kranjc, S.; Cemazar, M.; Todorovic, V.; Savarin, M.; Scancar, J.; Kosjek, T.; Groselj, B.; Strojan, P.; et al. Electrochemotherapy of radioresistant head and neck squamous cell carcinoma cells and tumor xenografts. Oncol. Rep. 2019, 41, 1658–1668. [Google Scholar] [CrossRef] [PubMed]
- Enokida, T.; Tahara, M. Electrochemotherapy in the Treatment of Head and Neck Cancer: Current Conditions and Future Directions. Cancers 2021, 13, 1418. [Google Scholar] [CrossRef]
- Wang, H.-H.; Fu, Z.-G.; Li, W.; Li, Y.-X.; Zhao, L.-S.; Wen, L.; Zhang, J.-J.; Wen, N. The synthesis and application of nano doxorubicin- indocyanine green matrix metalloproteinase-responsive hydrogel in chemophototherapy for head and neck squamous cell carcinoma. Int. J. Nanomed. 2019, 14, 623–638. [Google Scholar] [CrossRef]
- Soundararajan, A.; Dodd, G.D.; Bao, A.; Phillips, W.T.; McManus, L.M.; Prihoda, T.J.; Goins, B.A. Chemoradionuclide therapy with 186Re-labeled liposomal doxorubicin in combination with radiofrequency ablation for effective treatment of head and neck cancer in a nude rat tumor xenograft model. Radiology 2011, 261, 813–823. [Google Scholar] [CrossRef]
- Schmoldt, A.; Benthe, H.F.; Haberland, G. Digitoxin metabolism by rat liver microsomes. Biochem. Pharmacol. 1975, 24, 1639–1641. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, L.; Hu, G.-Q.; Zhang, N.; Zhu, X.-D.; Yang, K.-Y.; Jin, F.; Shi, M.; Chen, Y.-P.; Hu, W.-H.; et al. Gemcitabine and Cisplatin Induction Chemotherapy in Nasopharyngeal Carcinoma. N. Engl. J. Med. 2019, 381, 1124–1135. [Google Scholar] [CrossRef]
- Shewach, D.S.; Lawrence, T.S. Antimetabolite radiosensitizers. J. Clin. Oncol. 2007, 25, 4043–4050. [Google Scholar] [CrossRef]
- Huo, R.-X.; Jin, Y.-Y.; Zhuo, Y.-X.; Ji, X.-T.; Cui, Y.; Wu, X.-J.; Wang, Y.-J.; Zhang, L.; Zhang, W.-H.; Cai, Y.-M.; et al. Concurrent chemoradiotherapy using gemcitabine and nedaplatin in recurrent or locally advanced head and neck squamous cell carcinoma. World J. Clin. Cases 2022, 10, 3414–3425. [Google Scholar] [CrossRef]
- Aguilar-Ponce, J.; Granados-García, M.; Villavicencio, V.; Poitevin-Chacón, A.; Green, D.; Dueñas-González, A.; Herrera-Gómez, A.; Luna-Ortiz, K.; Alvarado, A.; Martínez-Said, H.; et al. Phase II trial of gemcitabine concurrent with radiation for locally advanced squamous cell carcinoma of the head and neck. Ann. Oncol. 2004, 15, 301–306. [Google Scholar] [CrossRef]
- Hsieh, C.-Y.; Lien, M.-Y.; Lin, C.-Y.; Lo, W.-J.; Hua, C.-H.; Chang, W.-C.; Chiu, C.-F.; Lin, C.-C. Rituximab in combination with gemcitabine plus cisplatin in patients with recurrent and metastatic head and neck squamous cell carcinoma: A phase I trial. BMC Cancer 2022, 22, 169. [Google Scholar] [CrossRef]
- Benasso, M.; Corvò, R.; Ponzanelli, A.; Sanguineti, G.; Ricci, I.; Pallestrini, E.; Santelli, A.; Vitale, V.; Rosso, R. Alternating gemcitabine and cisplatin with gemcitabine and radiation in stage IV squamous cell carcinoma of the head and neck. Ann. Oncol. 2004, 15, 646–652. [Google Scholar] [CrossRef]
- Leroy, B.; Fournier, J.L.; Ishioka, C.; Monti, P.; Inga, A.; Fronza, G.; Soussi, T. The TP53 website: An integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 2013, 41, D962-9. [Google Scholar] [CrossRef]
- Del Giudice, L.; Alifano, P.; Calcagnile, M.; Di Schiavi, E.; Bertapelle, C.; Aletta, M.; Pontieri, P. Mitochondrial ribosomal protein genes connected with Alzheimer’s and tellurite toxicity. Mitochondrion 2022, 64, 45–58. [Google Scholar] [CrossRef]
- Tapiero, H.; Townsend, D.M.; Tew, K.D. Trace elements in human physiology and pathology. Copper. Biomed. Pharmacother. 2003, 57, 386–398. [Google Scholar] [CrossRef]
- Kim, B.-E.; Nevitt, T.; Thiele, D.J. Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 2008, 4, 176–185. [Google Scholar] [CrossRef]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef]
- Leary, S.C.; Cobine, P.A.; Nishimura, T.; Verdijk, R.M.; de Krijger, R.; de Coo, R.; Tarnopolsky, M.A.; Winge, D.R.; Shoubridge, E.A. COX19 mediates the transduction of a mitochondrial redox signal from SCO1 that regulates ATP7A-mediated cellular copper efflux. Mol. Biol. Cell 2013, 24, 683–691. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Kozyreva, T.; Zovo, K.; Palumaa, P. Affinity gradients drive copper to cellular destinations. Nature 2010, 465, 645–648. [Google Scholar] [CrossRef]
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological relevance and mechanisms. Arch. Toxicol. 2014, 88, 1929–1938. [Google Scholar] [CrossRef]
- Barber, R.G.; Grenier, Z.A.; Burkhead, J.L. Copper Toxicity Is Not Just Oxidative Damage: Zinc Systems and Insight from Wilson Disease. Biomedicines 2021, 9, 316. [Google Scholar] [CrossRef]
- Doguer, C.; Ha, J.-H.; Collins, J.F. Intersection of Iron and Copper Metabolism in the Mammalian Intestine and Liver. Compr. Physiol. 2018, 8, 1433–1461. [Google Scholar] [CrossRef]
- Urso, E.; Maffia, M. Behind the Link between Copper and Angiogenesis: Established Mechanisms and an Overview on the Role of Vascular Copper Transport Systems. J. Vasc. Res. 2015, 52, 172–196. [Google Scholar] [CrossRef]
- Bharathi Devi, S.R.; Dhivya, M.A.; Sulochana, K.N. Copper transporters and chaperones: Their function on angiogenesis and cellular signalling. J. Biosci. 2016, 41, 487–496. [Google Scholar] [CrossRef]
- Li, Y. Copper homeostasis: Emerging target for cancer treatment. IUBMB Life 2020, 72, 1900–1908. [Google Scholar] [CrossRef]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: ‘Copper That Cancer’. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef]
- Babak, M.V.; Ahn, D. Modulation of Intracellular Copper Levels as the Mechanism of Action of Anticancer Copper Complexes: Clinical Relevance. Biomedicines 2021, 9, 852. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.K. Role of trace elements in cancer. Cancer Res. 1975, 35, 3481–3487. [Google Scholar] [PubMed]
- Lelièvre, P.; Sancey, L.; Coll, J.-L.; Deniaud, A.; Busser, B. The Multifaceted Roles of Copper in Cancer: A Trace Metal Element with Dysregulated Metabolism, but Also a Target or a Bullet for Therapy. Cancers 2020, 12, 3594. [Google Scholar] [CrossRef] [PubMed]
- Kadu, P.; Sawant, B.; Kale, P.P.; Prabhavalkar, K. Copper-lowering agents as an adjuvant in chemotherapy. Indian J. Pharmacol. 2021, 53, 221–225. [Google Scholar] [CrossRef]
- Öhrvik, H.; Thiele, D.J. The role of Ctr1 and Ctr2 in mammalian copper homeostasis and platinum-based chemotherapy. J. Trace Elem. Med. Biol. 2015, 31, 178–182. [Google Scholar] [CrossRef]
- Inkol, J.M.; Poon, A.C.; Mutsaers, A.J. Inhibition of copper chaperones sensitizes human and canine osteosarcoma cells to carboplatin chemotherapy. Vet. Comp. Oncol. 2020, 18, 559–569. [Google Scholar] [CrossRef]
- Percival, S.S. Copper and immunity. Am. J. Clin. Nutr. 1998, 67, 1064S–1068S. [Google Scholar] [CrossRef]
- Voli, F.; Valli, E.; Lerra, L.; Kimpton, K.; Saletta, F.; Giorgi, F.M.; Mercatelli, D.; Rouaen, J.R.C.; Shen, S.; Murray, J.E.; et al. Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef]
- Menezes, M.J.; Guo, Y.; Zhang, J.; Riley, L.G.; Cooper, S.T.; Thorburn, D.R.; Li, J.; Dong, D.; Li, Z.; Glessner, J.; et al. Mutation in mitochondrial ribosomal protein S7 (MRPS7) causes congenital sensorineural deafness, progressive hepatic and renal failure and lactic acidemia. Hum. Mol. Genet. 2015, 24, 2297–2307. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, S.; Zhao, L.; Wu, X.-B.; Wang, Z.; Cai, L.-Y.; Pan, D.; Li, Y.; Zhou, Y.; Shen, Y. Identification of a Novel Cuproptosis-Related Gene Signature for Prognostic Implication in Head and Neck Squamous Carcinomas. Cancers 2022, 14, 3986. https://doi.org/10.3390/cancers14163986
Tang S, Zhao L, Wu X-B, Wang Z, Cai L-Y, Pan D, Li Y, Zhou Y, Shen Y. Identification of a Novel Cuproptosis-Related Gene Signature for Prognostic Implication in Head and Neck Squamous Carcinomas. Cancers. 2022; 14(16):3986. https://doi.org/10.3390/cancers14163986
Chicago/Turabian StyleTang, Shouyi, Li Zhao, Xing-Bo Wu, Zhen Wang, Lu-Yao Cai, Dan Pan, Ying Li, Yu Zhou, and Yingqiang Shen. 2022. "Identification of a Novel Cuproptosis-Related Gene Signature for Prognostic Implication in Head and Neck Squamous Carcinomas" Cancers 14, no. 16: 3986. https://doi.org/10.3390/cancers14163986