
Physiological Cell Culture Media Tune Mitochondrial Bioenergetics and Drug Sensitivity in Cancer Cell Models

 , , and
, , and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
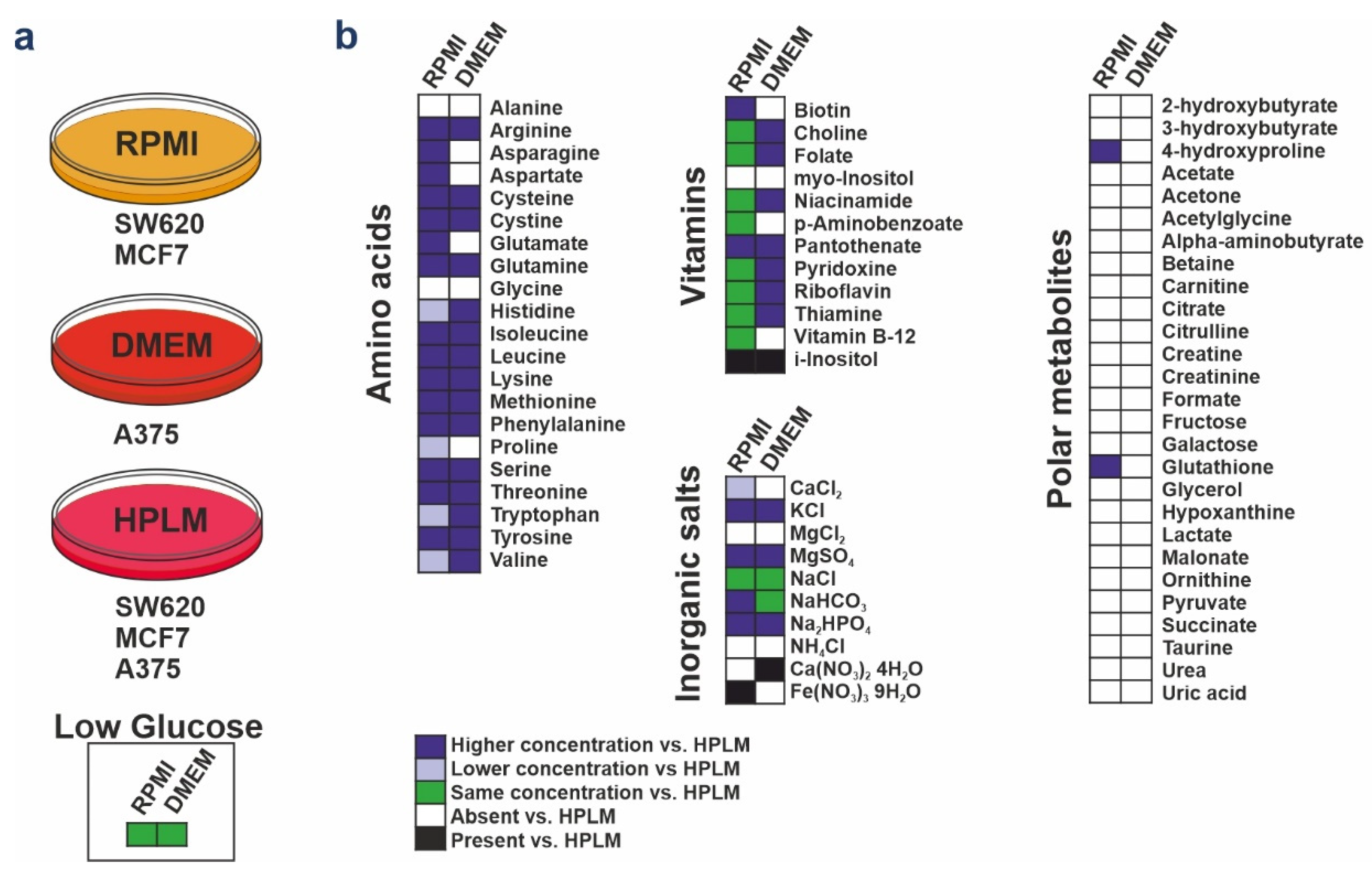
2.1. Cell Culture
2.2. Proliferation Assays
2.3. Sample Preparation and High-Resolution Respirometry
2.4. Statistical Analysis
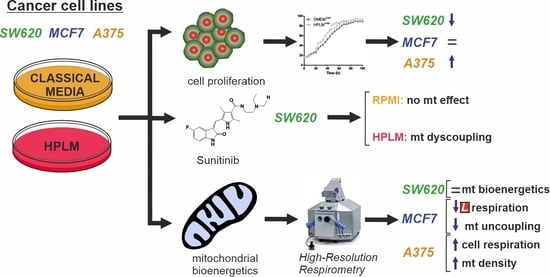
3. Results
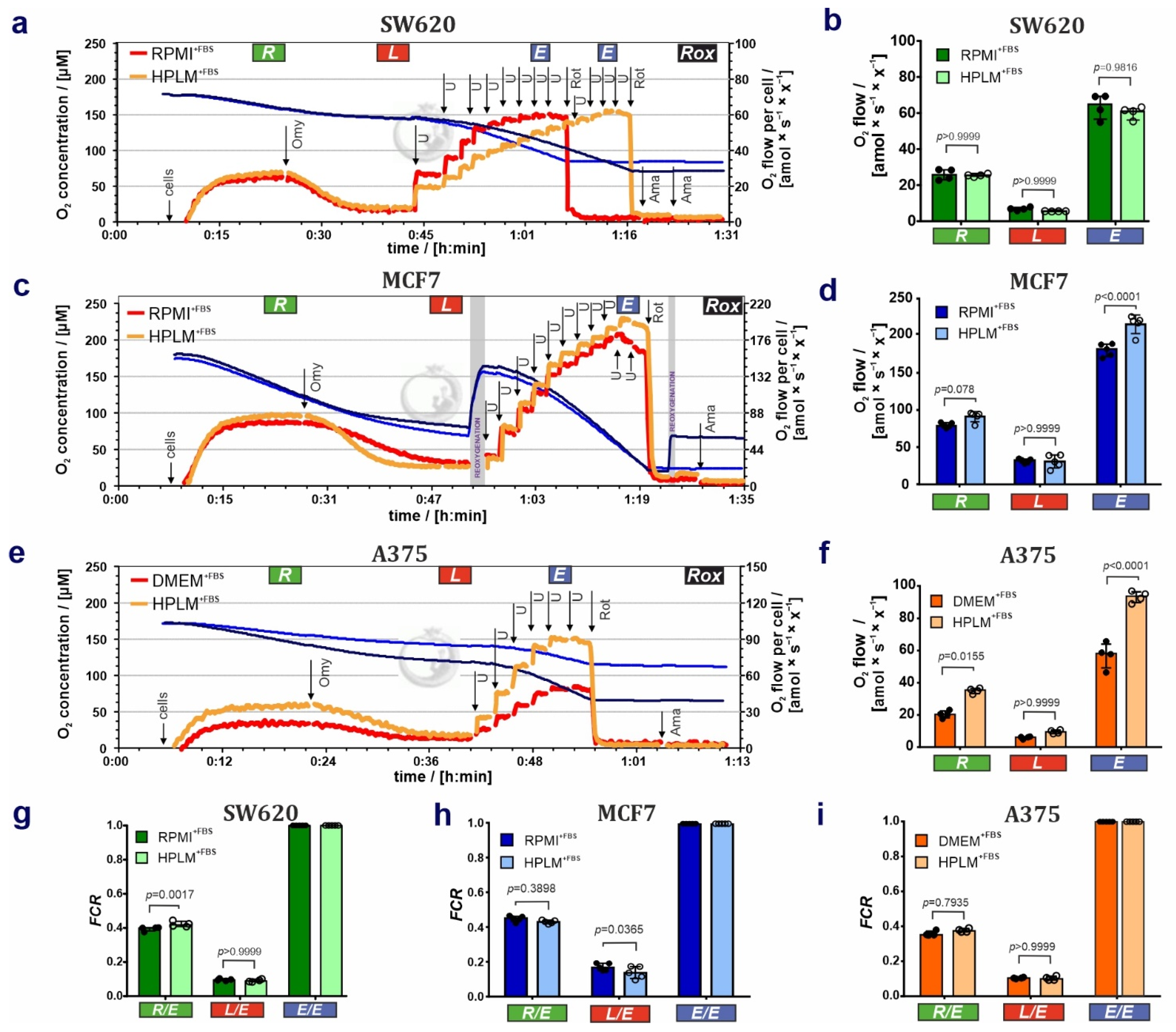
3.1. HPLM Influences Steady State Cancer Cell Proliferation and Mitochondrial Function
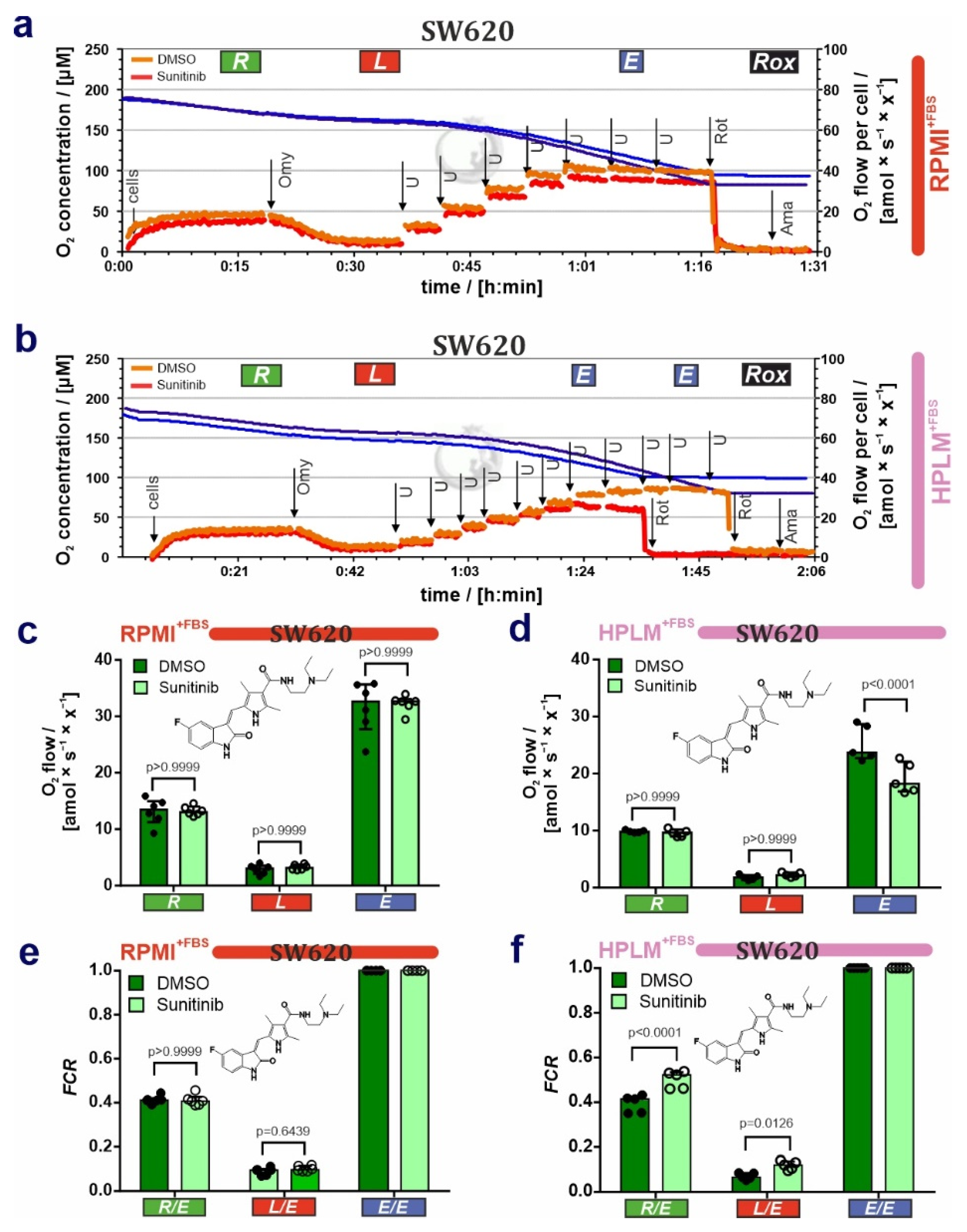
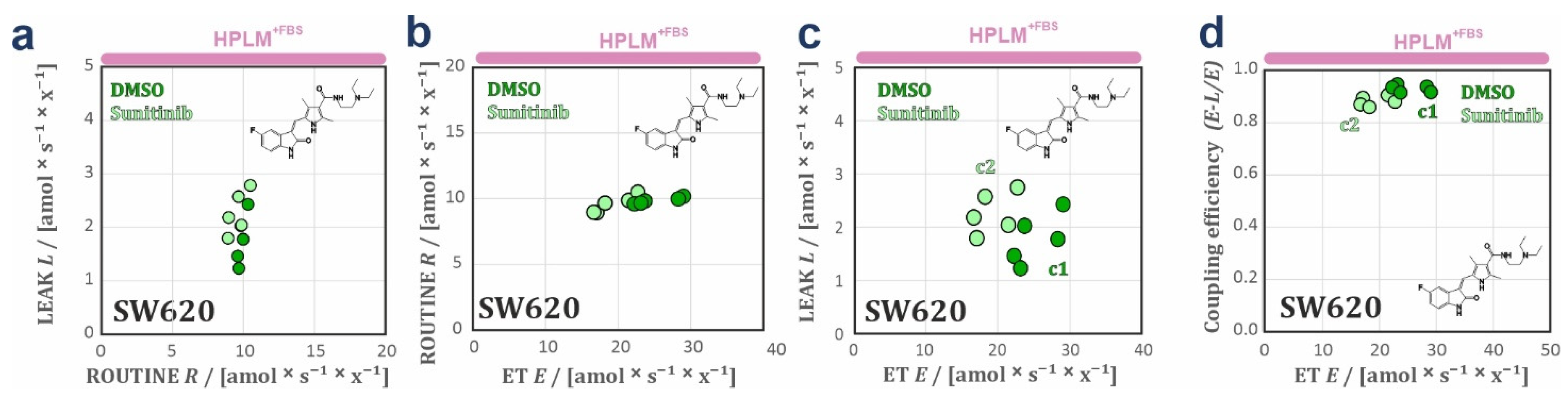
3.2. HPLM Modifies Mitochondrial Drug Sensitivity in Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.F.; Morton, H.J.; Parker, R.C. Nutrition of Animal Cells in Tissue Culture; Initial Studies on a Synthetic Medium. Proc. Soc. Exp. Biol. Med. 1950, 73, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H. Nutrition Needs of Mammalian Cells in Tissue Culture. Science 1955, 122, 501–504. [Google Scholar] [CrossRef]
- Eagle, H. Amino Acid Metabolism in Mammalian Cell Cultures. Science 1959, 130, 432–437. [Google Scholar] [CrossRef] [PubMed]
- E Moore, G.; E Gerner, R.; A Franklin, H. Culture of normal human leukocytes. JAMA 1967, 199, 519–524. [Google Scholar] [CrossRef]
- Cantor, J.R.; Abu-Remaileh, M.; Kanarek, N.; Freinkman, E.; Gao, X.; Louissaint, A., Jr.; Lewis, C.A.; Sabatini, D.M. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 2017, 169, 258–272.e17. [Google Scholar] [CrossRef]
- Vande Voorde, J.; Ackermann, T.; Pfetzer, N.; Sumpton, D.; Mackay, G.; Kalna, G.; Nixon, C.; Blyth, K.; Gottlieb, E.; Tardito, S. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv. 2019, 5, eaau7314. [Google Scholar] [CrossRef]
- Leney-Greene, M.A.; Boddapati, A.K.; Su, H.C.; Cantor, J.R.; Lenardo, M.J. Human Plasma-like Medium Improves T Lymphocyte Activation. iScience 2020, 23, 100759. [Google Scholar] [CrossRef]
- Rossiter, N.J.; Huggler, K.S.; Adelmann, C.H.; Keys, H.R.; Soens, R.W.; Sabatini, D.M.; Cantor, J.R. CRISPR screens in physiologic medium reveal conditionally essential genes in human cells. Cell Metab. 2021, 33, 1248–1263.e9. [Google Scholar] [CrossRef]
- Parri, M.; Ippolito, L.; Cirri, P.; Ramazzotti, M.; Chiarugi, P. Metabolic Cell Communication within Tumour Microenvironment: Models, Methods and Perspectives. Curr. Opin. Biotechnol. 2020, 63, 210–219. [Google Scholar] [CrossRef]
- Kaambre, T.; Chekulayev, V.; Shevchuk, I.; Tepp, K.; Timohhina, N.; Varikmaa, M.; Bagur, R.; Klepinin, A.; Anmann, T.; Koit, A.; et al. Metabolic control analysis of respiration in human cancer tissue. Front. Physiol. 2013, 4, 151. [Google Scholar] [CrossRef] [PubMed]
- Amoêdo, N.D.; Rodrigues, M.F.; Pezzuto, P.; Galina, A.; da Costa, R.M.; de Almeida, F.C.L.; El-Bacha, T.; Rumjanek, F.D. Energy Metabolism in H460 Lung Cancer Cells: Effects of Histone Deacetylase Inhibitors. PLoS ONE 2011, 6, e22264. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, R.; Saavedra, E.; Rodríguez-Enríquez, S.; Gallardo-Pérez, J.C.; Quezada, H.; Westerhoff, H.V. Metabolic control analysis indicates a change of strategy in the treatment of cancer. Mitochondrion 2010, 10, 626–639. [Google Scholar] [CrossRef]
- Rinaldi, L.; Donne, R.D.; Borzacchiello, D.; Insabato, L.; Feliciello, A. The role of compartmentalized signaling pathways in the control of mitochondrial activities in cancer cells. Biochim. Biophys. Acta 2018, 1869, 293–302. [Google Scholar] [CrossRef]
- Sams-Dodd, F. Target-Based Drug Discovery: Is Something Wrong? Drug Discov. Today 2005, 10, 139–147. [Google Scholar] [CrossRef]
- Moretto, J.; Chauffert, B.; Ghiringhelli, F.; Aldrich-Wright, J.R.; Bouyer, F. Discrepancy between in vitro and in vivo antitumor effect of a new platinum(II) metallointercalator. Investig. New Drugs 2010, 29, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Amoêdo, N.D.; Valencia, J.P.; Rodrigues, M.F.; Galina, A.; Rumjanek, F.D. How does the metabolism of tumour cells differ from that of normal cells. Biosci. Rep. 2013, 33, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Nadanaciva, S.; Will, Y. Current Concepts in Drug-Induced Mitochondrial Toxicity. Curr. Protoc. Toxicol. 2009, 40, 1–2. [Google Scholar] [CrossRef]
- Schöpf, B.; Schäfer, G.; Weber, A.; Talasz, H.; Eder, I.E.; Klocker, H.; Gnaiger, E. Oxidative phosphorylation and mitochondrial function differ between human prostate tissue and cultured cells. FEBS J. 2016, 283, 2181–2196. [Google Scholar] [CrossRef]
- Schöpf, B.; Weissensteiner, H.; Schäfer, G.; Fazzini, F.; Charoentong, P.; Naschberger, A.; Rupp, B.; Fendt, L.; Bukur, V.; Giese, I.; et al. OXPHOS remodeling in high-grade prostate cancer involves mtDNA mutations and increased succinate oxidation. Nat. Commun. 2020, 11, 1487. [Google Scholar] [CrossRef]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control. An Introduction to OXPHOS Analysis, 5th ed. Bioenerg. Commun. 2020, 2020-002. [Google Scholar]
- Gnaiger, E. Polarographic Oxygen Sensors, the Oxygraph and High-Resolution Respirometry to Assess Mitochondrial Function. In Drug-Induced Mitochondrial Dysfunction; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2008; pp. 327–352. [Google Scholar]
- Doerrier, C.; Garcia-Souza, L.F.; Krumschnabel, G.; Wohlfarter, Y.; Meszaros, A.T.; Gnaiger, E. High-Resolution Fluorespirometry and Oxphos Protocols for Human Cells, Permeabilized Fibers from Small Biopsies of Muscle, and Isolated Mitochondria. Methods Mol. Biol. 2018, 1782, 31–70. [Google Scholar] [PubMed]
- Gnaiger, E. Bioenergetic Cluster Analysis—Mitochondrial Respiratory Control in Human Fibroblasts. MitoFit Prepr. 2021, 2021, 8. [Google Scholar]
- Gnaiger, E. Capacity of oxidative phosphorylation in human skeletal muscle: New perspectives of mitochondrial physiology. Int. J. Biochem. Cell Biol. 2009, 41, 1837–1845. [Google Scholar] [CrossRef]
- Gnaiger, E.; Aasander, F.E.; Abdul Karim, N.; Abdel-Rahman, E.A.; Abumrad Nada, A.; Acuna-Castroviejo, D.; Adiele Reginald, C.; Ahn, B.; Alencar, M.B.; Ali Sameh, S.; et al. Mitochondrial Physiology- Mitoeagle Task Group. Bioenerg. Commun. 2020, 2020, 1. [Google Scholar]
- Zdrazilova, L.; Hansikova, H.; Gnaiger, E. Comparable respiratory activity in attached and suspended human fibroblasts. PLoS ONE 2022, 17, e0264496. [Google Scholar] [CrossRef]
- Chiusa, M.; Hool, S.-L.; Truetsch, P.; Djafarzadeh, S.; Jakob, S.M.; Seifriz, F.; Scherer, S.J.; Suter, T.M.; Zuppinger, C.; Zbinden, S. Cancer therapy modulates VEGF signaling and viability in adult rat cardiac microvascular endothelial cells and cardiomyocytes. J. Mol. Cell. Cardiol. 2012, 52, 1164–1175. [Google Scholar]
- Goodman, V.L.; Rock, E.P.; Dagher, R.; Ramchandani, R.P.; Abraham, S.; Gobburu, J.V.; Booth, B.P.; Verbois, S.L.; Morse, D.E.; Liang, C.Y.; et al. Approval Summary: Sunitinib for the Treatment of Imatinib Refractory or Intolerant Gastrointestinal Stromal Tumors and Advanced Renal Cell Carcinoma. Clin. Cancer Res. 2007, 13, 1367–1373. [Google Scholar] [CrossRef]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control. An Introduction to OXPHOS Analysis, 4th ed.; Mitochondr Physiol Network, Ed.; Oroboros MiPNet Publications: Innsbruck, Austria, 2014; Volume 19, ISBN 978-3-9502399-8-0. [Google Scholar]
- Hütter, E.; Renner, K.; Pfister, G.; Stöckl, P.; Jansen-Dürr, P.; Gnaiger, E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem. J. 2004, 380, 919–928. [Google Scholar] [CrossRef]
- Astashkina, A.; Mann, B.; Grainger, D.W. A critical evaluation of in vitro cell culture models for high-throughput drug screening and toxicity. Pharmacol. Ther. 2012, 134, 82–106. [Google Scholar] [CrossRef]
- Gordon, J.L.; Brown, M.A.; Reynolds, M.M. Cell-Based Methods for Determination of Efficacy for Candidate Therapeutics in the Clinical Management of Cancer. Diseases 2018, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Röck, R.; Mayrhofer, J.E.; Torres-Quesada, O.; Enzler, F.; Raffeiner, A.; Raffeiner, P.; Feichtner, A.; Huber, R.G.; Koide, S.; Taylor, S.S.; et al. BRAF inhibitors promote intermediate BRAF(V600E) conformations and binary interactions with activated RAS. Sci. Adv. 2019, 5, eaav8463. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.; Engqvist, H.; Biermann, J.; Rönnerman, E.W.; Forssell-Aronsson, E.; Kovács, A.; Karlsson, P.; Helou, K.; Parris, T.Z. Optimization of cell viability assays to improve replicability and reproducibility of cancer drug sensitivity screens. Sci. Rep. 2020, 10, 5798. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, J.E.; Enzler, F.; Feichtner, A.; Röck, R.; Fleischmann, J.; Raffeiner, A.; Tschaikner, P.; Ogris, E.; Huber, R.G.; Hartl, M.; et al. Mutation-oriented profiling of autoinhibitory kinase conformations predicts RAF inhibitor efficacies. Proc. Natl. Acad. Sci. USA 2020, 117, 31105–31113. [Google Scholar] [CrossRef]
- Feinberg, K.; Kolaj, A.; Wu, C.; Grinshtein, N.; Krieger, J.R.; Moran, M.F.; Rubin, L.; Miller, F.D.; Kaplan, D.R. A neuroprotective agent that inactivates prodegenerative TrkA and preserves mitochondria. J. Cell Biol. 2017, 216, 3655–3675. [Google Scholar] [CrossRef]
- Grzes, K.M.; Sanin, D.E.; Kabat, A.M.; Stanczak, M.A.; Edwards-Hicks, J.; Matsushita, M.; Hackl, A.; Hässler, F.; Knoke, K.; Zahalka, S.; et al. Plasmacytoid dendritic cell activation is dependent on coordinated expression of distinct amino acid transporters. Immunity 2021, 54, 2514–2530.e7. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1–10. [Google Scholar] [CrossRef]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of Oxidative Mitochondrial Metabolism in Epithelial Cancer Cells in Situ: Visualizing the Therapeutic Effects of Metformin in Tumor Tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef]
- Yu, M. Generation, Function and Diagnostic Value of Mitochondrial DNA Copy Number Alterations in Human Cancers. Life Sci. 2011, 89, 65–71. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.H. Mitochondrial Fusion Supports Increased Oxidative Phosphorylation During Cell Proliferation. eLife 2019, 8, e41351. [Google Scholar] [CrossRef] [PubMed]
- Reznik, E.; Miller, M.L.; Senbabaoglu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA Copy Number Variation across Human Cancers. eLife 2016, 5, e10769. [Google Scholar] [CrossRef] [PubMed]
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 2016, 36, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.E.; Short, S.P.; Williams, C.S. Colorectal Cancer and Metabolism. Curr. Colorectal Cancer Rep. 2018, 14, 226–241. [Google Scholar] [CrossRef]
- Shen, C.; Xuan, B.; Yan, T.; Ma, Y.; Xu, P.; Tian, X.; Zhang, X.; Cao, Y.; Ma, D.; Zhu, X.; et al. m6A-dependent glycolysis enhances colorectal cancer progression. Mol. Cancer 2020, 19, 72. [Google Scholar] [CrossRef]
- Klepinina, L.; Klepinin, A.; Truu, L.; Chekulayev, V.; Vija, H.; Kuus, K.; Teino, I.; Pook, M.; Maimets, T.; Kaambre, T. Colon cancer cell differentiation by sodium butyrate modulates metabolic plasticity of Caco-2 cells via alteration of phosphotransfer network. PLoS ONE 2021, 16, e0245348. [Google Scholar] [CrossRef]
- Meng, L.; Chen, D.; Meng, G.; Lu, L.; Han, C. Dysregulation of the Sirt5/Idh2 Axis Contributes to Sunitinib Resistance in Human Renal Cancer Cells. FEBS Open Bio 2021, 11, 921–931. [Google Scholar] [CrossRef]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Heiden, M.G.V. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef]
- Bouitbir, J.; Alshaikhali, A.; Panajatovic, M.V.; Abegg, V.F.; Paech, F.; Krahenbuhl, S. Mitochondrial Oxidative Stress Plays a Critical Role in the Cardiotoxicity of Sunitinib: Running Title: Sunitinib and Oxidative Stress in Hearts. Toxicology 2019, 426, 152281. [Google Scholar] [CrossRef]
- Paech, F.; Abegg, V.F.; Duthaler, U.; Terracciano, L.; Bouitbir, J.; Krähenbühl, S. Sunitinib induces hepatocyte mitochondrial damage and apoptosis in mice. Toxicology 2018, 409, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Errington, T.M.; Denis, A.; Perfito, N.; Iorns, E.; A Nosek, B. Challenges for assessing replicability in preclinical cancer biology. eLife 2021, 10, e67995. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Quesada, O.; Doerrier, C.; Strich, S.; Gnaiger, E.; Stefan, E. Physiological Cell Culture Media Tune Mitochondrial Bioenergetics and Drug Sensitivity in Cancer Cell Models. Cancers 2022, 14, 3917. https://doi.org/10.3390/cancers14163917
Torres-Quesada O, Doerrier C, Strich S, Gnaiger E, Stefan E. Physiological Cell Culture Media Tune Mitochondrial Bioenergetics and Drug Sensitivity in Cancer Cell Models. Cancers. 2022; 14(16):3917. https://doi.org/10.3390/cancers14163917
Chicago/Turabian StyleTorres-Quesada, Omar, Carolina Doerrier, Sophie Strich, Erich Gnaiger, and Eduard Stefan. 2022. "Physiological Cell Culture Media Tune Mitochondrial Bioenergetics and Drug Sensitivity in Cancer Cell Models" Cancers 14, no. 16: 3917. https://doi.org/10.3390/cancers14163917