
Pathogenic Roles of RNA-Binding Proteins in Sarcomas


Abstract
:Simple Summary
Abstract
1. Introduction
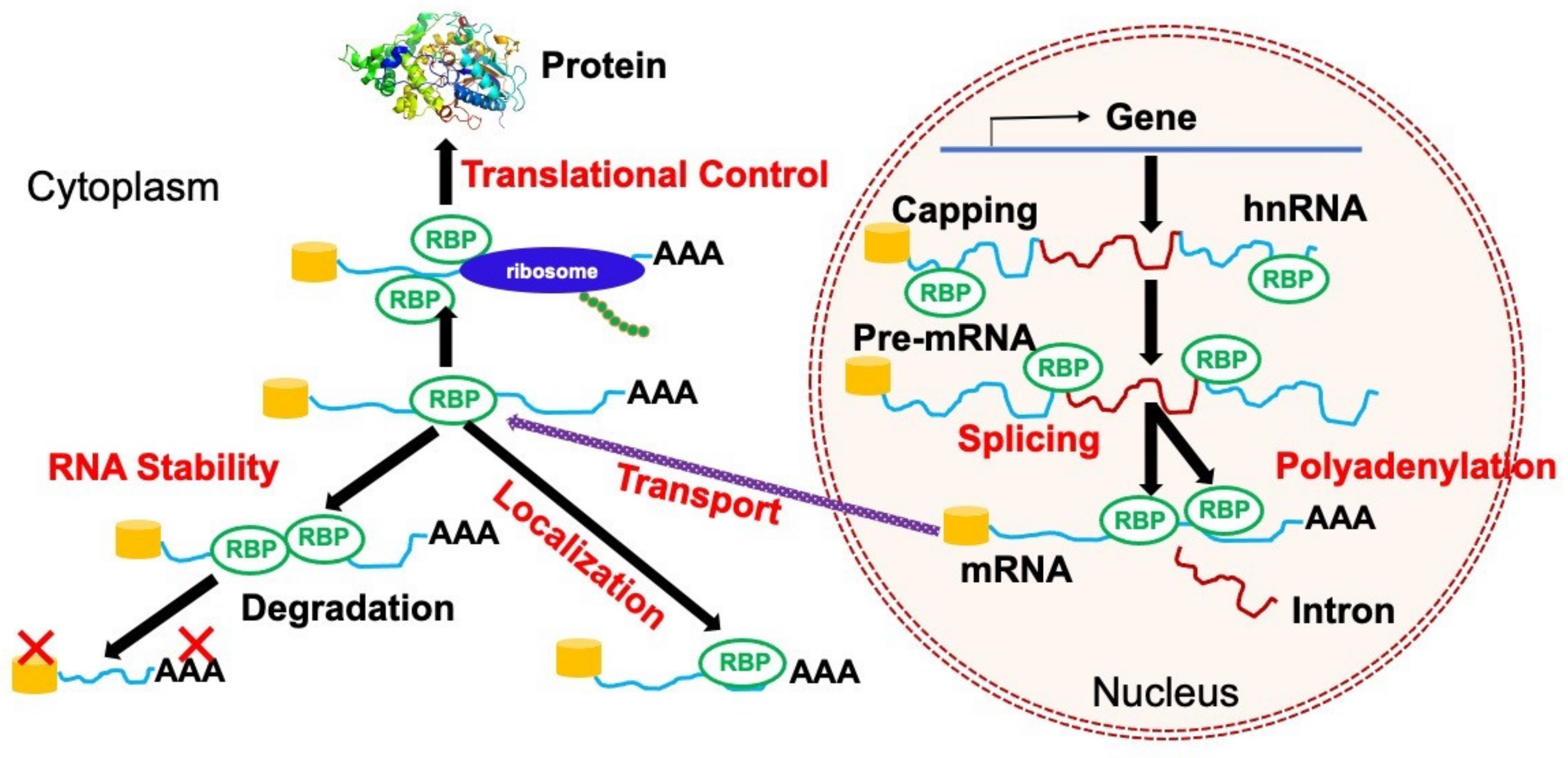
2. Regulations of RNA Metabolism by RBPs
2.1. Alternative Splicing
2.2. Alternative Polyadenylation
2.3. Stability
2.4. RNA Localization
2.5. Translation
2.6. Noncoding RNA Processing
3. Role of RBPs in Sarcomas
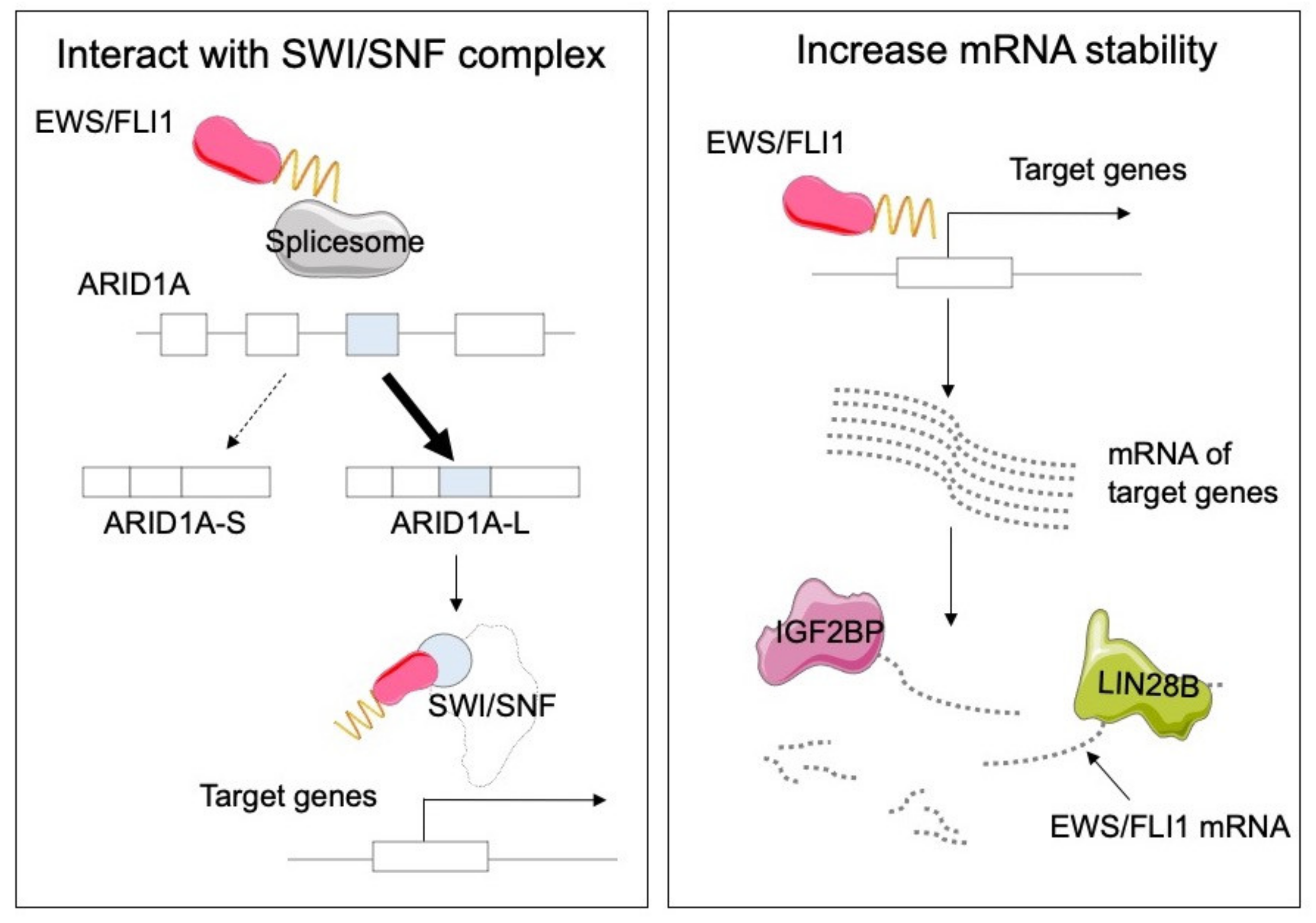
3.1. Ewing Sarcoma
3.2. Rhabdomyosarcoma
3.3. Synovial Sarcoma
3.4. Osteosarcoma
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hong, S. RNA Binding Protein as an Emerging Therapeutic Target for Cancer Prevention and Treatment. J. Cancer Prev. 2017, 22, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Baltz, A.G.; Munschauer, M.; Schwanhäusser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckmann, B.M.; Horos, R.; Fischer, B.; Castello, A.; Eichelbaum, K.; Alleaume, A.-M.; Schwarzl, T.; Curk, T.; Foehr, S.; Huber, W.; et al. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 2015, 6, 10127. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Lee, Y.; Lee, J.S. RNA-Binding Proteins in Cancer: Functional and Therapeutic Perspectives. Cancers 2020, 12, 2699. [Google Scholar] [CrossRef]
- Wang, S.; Sun, Z.; Lei, Z.; Zhang, H.-T. RNA-binding proteins and cancer metastasis. Semin. Cancer Biol. 2022, in press. [CrossRef] [PubMed]
- Bley, N.; Hmedat, A.; Müller, S.; Rolnik, R.; Rausch, A.; Lederer, M.; Hüttelmaier, S. Musashi-1-A Stemness RBP for Cancer Therapy? Biology 2021, 10, 407. [Google Scholar] [CrossRef]
- Liu, M.; Fu, X.; Jiang, L.; Ma, J.; Zheng, X.; Wang, S.; Guo, H.; Tian, T.; Nan, K.; Wang, W. Colon cancer cells secreted CXCL11 via RBP-Jκ to facilitated tumour-associated macrophage-induced cancer metastasis. J. Cell. Mol. Med. 2021, 25, 10575–10590. [Google Scholar] [CrossRef]
- Iino, K.; Mitobe, Y.; Ikeda, K.; Takayama, K.-I.; Suzuki, T.; Kawabata, H.; Suzuki, Y.; Horie-Inoue, K.; Inoue, S. RNA-binding protein NONO promotes breast cancer proliferation by post-transcriptional regulation of SKP2 and E2F8. Cancer Sci. 2020, 111, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.J.; Guo, H.; Jiang, L.L.; Jiao, M.; Wang, S.H.; Tian, T.; Fu, X.; Wang, W.J. Elevated RBP-Jκ and CXCL11 Expression in Colon Cancer is Associated with an Unfavorable Clinical Outcome. Cancer Manag. Res. 2021, 13, 3651–3661. [Google Scholar] [CrossRef]
- Wang, X.Y.; Penalva, L.O.; Yuan, H.; Linnoila, R.I.; Lu, J.; Okano, H.; Glazer, R.I. Musashi1 regulates breast tumor cell proliferation and is a prognostic indicator of poor survival. Mol. Cancer 2010, 9, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Lee, S.C.; Hockemeyer, K.; Ghebrechristos, Y.E.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 369–384.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uehara, T.; Minoshima, Y.; Sagane, K.; Sugi, N.H.; Mitsuhashi, K.O.; Yamamoto, N.; Kamiyama, H.; Takahashi, K.; Kotake, Y.; Uesugi, M.; et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat. Chem. Biol. 2017, 13, 675–680. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, X. Posttranscriptional regulation of p53 and its targets by RNA-binding proteins. Curr. Mol. Med. 2008, 8, 845–849. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Pan, T.; Chen, L.; Wang, Q.; Chang, Z.; Zhou, W.; Li, X.; Xu, G.; Li, X.; Li, Y.; et al. Alternative splicing perturbation landscape identifies RNA binding proteins as potential therapeutic targets in cancer. Mol. Ther. Nucleic Acids 2021, 24, 792–806. [Google Scholar] [CrossRef]
- Climente-González, H.; Porta-Pardo, E.; Godzik, A.; Eyras, E. The Functional Impact of Alternative Splicing in Cancer. Cell Rep. 2017, 20, 2215–2226. [Google Scholar] [CrossRef] [Green Version]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and mechanisms of alternative splicing in cancer-implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Sciarrillo, R.; Wojtuszkiewicz, A.; Assaraf, Y.G.; Jansen, G.; Kaspers, G.J.L.; Giovannetti, E.; Cloos, J. The role of alternative splicing in cancer: From oncogenesis to drug resistance. Drug Resist. Updates 2020, 53, 100728. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.; Masshofer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; van de Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; The Cancer Genome Atlas Research Network; Buonamici, S.; Yu, L. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296.e4. [Google Scholar]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef] [Green Version]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Yoshimi, A.; Lin, K.-T.; Wiseman, D.H.; Rahman, M.A.; Pastore, A.; Wang, B.; Lee, S.C.-W.; Micol, J.-B.; Zhang, X.J.; De Botton, S.; et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019, 574, 273–277. [Google Scholar] [CrossRef]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, H.; Nishimura, K.; Yoshimi, A. Aberrant RNA splicing and therapeutic opportunities in cancers. Cancer Sci. 2022, 113, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, A.; Abdel-Wahab, O. Molecular Pathways: Understanding and Targeting Mutant Spliceosomal Proteins. Clin. Cancer Res. 2017, 23, 336–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Mir, C.; Garcia-Mayea, Y.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. RNA-binding proteins: Underestimated contributors in tumorigenesis. Semin. Cancer Biol. 2022. [Google Scholar] [CrossRef]
- Fernández-Miranda, G.; Méndez, R. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res. Rev. 2012, 11, 460–472. [Google Scholar] [CrossRef]
- Burns, D.M.; D’Ambrogio, A.; Nottrott, S.; Richter, J.D. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 2011, 473, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Burns, D.M.; Richter, J.D. CPEB regulation of human cellular senescence, energy metabolism, and p53 mRNA translation. Genes Dev. 2008, 22, 3449–3460. [Google Scholar] [CrossRef] [Green Version]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Garneau, N.L.; Wilusz, J.; Wilusz, C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007, 8, 113–126. [Google Scholar] [CrossRef]
- Yamauchi, T.; Masuda, T.; Canver, M.C.; Seiler, M.; Semba, Y.; Shboul, M.; Al-Raqad, M.; Maeda, M.; Schoonenberg, V.A.C.; Cole, M.A.; et al. Genome-wide CRISPR-Cas9 Screen Identifies Leukemia-Specific Dependence on a Pre-mRNA Metabolic Pathway Regulated by DCPS. Cancer Cell 2018, 33, 386–400.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimi, A.; Abdel-Wahab, O. Targeting mRNA Decapping in AML. Cancer Cell 2018, 33, 339–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Kiledjian, M. Functional link between the mammalian exosome and mRNA decapping. Cell 2001, 107, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, E.; le Hir, H.; Seraphin, B. DcpS can act in the 5′-3′ mRNA decay pathway in addition to the 3′-5′ pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 12081–12086. [Google Scholar] [CrossRef] [Green Version]
- Degrauwe, N.; Suvà, M.L.; Janiszewska, M.; Riggi, N.; Stamenkovic, I. IMPs: An RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016, 30, 2459–2474. [Google Scholar] [CrossRef] [Green Version]
- Suter, B. RNA localization and transport. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 938–951. [Google Scholar] [CrossRef]
- Gu, W.; Katz, Z.; Wu, B.; Park, H.Y.; Li, D.; Lin, S.; Wells, A.L.; Singer, R.H. Regulation of local expression of cell adhesion and motility-related mRNAs in breast cancer cells by IMP1/ZBP1. J. Cell Sci. 2012, 125, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Blagden, S.P.; Willis, A.E. The biological and therapeutic relevance of mRNA translation in cancer. Nat. Rev. Clin. Oncol. 2011, 8, 280–291. [Google Scholar] [CrossRef]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef]
- Chu, J.; Cargnello, M.; Topisirovic, I.; Pelletier, J. Translation Initiation Factors: Reprogramming Protein Synthesis in Cancer. Trends Cell Biol. 2016, 26, 918–933. [Google Scholar] [CrossRef]
- Forrest, A.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [PubMed] [Green Version]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Chen, Q.; Bei, M.; Shao, M.; Xu, J. Characterizing the tumor RBP-ncRNA circuits by integrating transcriptomics, interactomics and clinical data. Comput. Struct. Biotechnol. J. 2021, 19, 5235–5245. [Google Scholar] [CrossRef]
- Van Kouwenhove, M.; Kedde, M.; Agami, R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat. Rev. Cancer 2011, 11, 644–656. [Google Scholar] [CrossRef]
- Ustianenko, D.; Chiu, H.S.; Treiber, T.; Weyn-Vanhentenryck, S.M.; Treiber, N.; Meister, G.; Sumazin, P.; Zhang, C. LIN28 Selectively Modulates a Subclass of Let-7 MicroRNAs. Mol. Cell 2018, 71, 271–283.e5. [Google Scholar] [CrossRef] [Green Version]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Starke, S.; Jost, I.; Rossbach, O.; Schneider, T.; Schreiner, S.; Hung, L.H.; Bindereif, A. Exon circularization requires canonical splice signals. Cell Rep. 2015, 10, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, Y.; Wu, S.; Zhou, Z.; Ding, X.; Shi, R.; Thorne, R.F.; Zhang, X.D.; Hu, W.; Wu, M. CircACC1 Regulates Assembly and Activation of AMPK Complex under Metabolic Stress. Cell Metab. 2019, 30, 157–173.e7. [Google Scholar] [CrossRef]
- Chen, J.; Wu, Y.; Luo, X.; Jin, D.; Zhou, W.; Ju, Z.; Wang, D.; Meng, Q.; Wang, H.; Fu, X.; et al. Circular RNA circRHOBTB3 represses metastasis by regulating the HuR-mediated mRNA stability of PTBP1 in colorectal cancer. Theranostics 2021, 11, 7507–7526. [Google Scholar] [CrossRef]
- Riggi, N.; Suva, M.L.; Stamenkovic, I. Ewing’s Sarcoma. N. Engl. J. Med. 2021, 384, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, H.; Zhang, K.; Fox-Walsh, K.; Yang, Y.; Zhang, C.; Huang, J.; Li, H.; Zhou, Y.; Fu, X.D. The RNA binding protein EWS is broadly involved in the regulation of pri-miRNA processing in mammalian cells. Nucleic Acids Res. 2017, 45, 12481–12495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, S.; Graham, T.G.W.; Dugast-Darzacq, C.; Dailey, G.M.; Darzacq, X.; Tjian, R. Tuning levels of low-complexity domain interactions to modulate endogenous oncogenic transcription. Mol. Cell 2022, 82, 2084–2097.e5. [Google Scholar] [CrossRef]
- Lin, L.; Huang, M.; Shi, X.; Mayakonda, A.; Hu, K.; Jiang, Y.Y.; Guo, X.; Chen, L.; Pang, B.; Doan, N.; et al. Super-enhancer-associated MEIS1 promotes transcriptional dysregulation in Ewing sarcoma in co-operation with EWS-FLI1. Nucleic Acids Res. 2019, 47, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.L.; Vallurupalli, M.; Chen, L.; Crompton, B.; Cowley, G.; Vazquez, F.; Weir, B.A.; Tsherniak, A.; Parasuraman, S.; Kim, S.; et al. Functional, chemical genomic, and super-enhancer screening identify sensitivity to cyclin D1/CDK4 pathway inhibition in Ewing sarcoma. Oncotarget 2015, 6, 30178–30193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schonegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef] [Green Version]
- Boulay, G.; Volorio, A.; Iyer, S.; Broye, L.C.; Stamenkovic, I.; Riggi, N.; Rivera, M.N. Epigenome editing of microsatellite repeats defines tumor-specific enhancer functions and dependencies. Genes Dev. 2018, 32, 1008–1019. [Google Scholar] [CrossRef] [Green Version]
- Vibert, J.; Saulnier, O.; Collin, C.; Petit, F.; Borgman, K.J.E.; Vigneau, J.; Gautier, M.; Zaidi, S.; Pierron, G.; Watson, S.; et al. Oncogenic chimeric transcription factors drive tumor-specific transcription, processing, and translation of silent genomic regions. Mol. Cell 2022, 82, 2458–2471.e9. [Google Scholar] [CrossRef] [PubMed]
- Selvanathan, S.P.; Graham, G.T.; Grego, A.R.; Baker, T.M.; Hogg, J.R.; Simpson, M.; Batish, M.; Crompton, B.; Stegmaier, K.; Tomazou, E.M.; et al. EWS-FLI1 modulated alternative splicing of ARID1A reveals novel oncogenic function through the BAF complex. Nucleic Acids Res. 2019, 47, 9619–9636. [Google Scholar] [CrossRef] [PubMed]
- Keskin, T.; Bakaric, A.; Waszyk, P.; Boulay, G.; Torsello, M.; Cornaz-Buros, S.; Chevalier, N.; Geiser, T.; Martin, P.; Volorio, A.; et al. LIN28B Underlies the Pathogenesis of a Subclass of Ewing Sarcoma LIN28B Control of EWS-FLI1 Stability. Cell Rep. 2020, 30, 4567–4583. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, C.; Pasello, M.; Ventura, S.; Grilli, A.; Calzolari, L.; Toracchio, L.; Lollini, P.L.; Donati, D.M.; Picci, P.; Ferrari, S.; et al. Insulin-Like Growth Factor 2 mRNA-Binding Protein 3 is a Novel Post-Transcriptional Regulator of Ewing Sarcoma Malignancy. Clin. Cancer Res. 2018, 24, 3704–3716. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Primers 2019, 5, 1. [Google Scholar] [CrossRef]
- Arndt, C.A.; Stoner, J.A.; Hawkins, D.S.; Rodeberg, D.A.; Hayes-Jordan, A.A.; Paidas, C.N.; Parham, D.M.; Teot, L.A.; Wharam, M.D.; Breneman, J.C.; et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: Children’s oncology group study D9803. J. Clin. Oncol. 2009, 27, 5182–5188. [Google Scholar] [CrossRef] [Green Version]
- Pacenta, H.L.; Allen-Rhoades, W.; Langenau, D.; Houghton, P.J.; Keller, C.; Heske, C.M.; Deel, M.D.; Linardic, C.M.; Shern, J.F.; Stewart, E.; et al. Prioritization of Novel Agents for Patients with Rhabdomyosarcoma: A Report from the Children’s Oncology Group (COG) New Agents for Rhabdomyosarcoma Task Force. J. Clin. Med. 2021, 10, 1416. [Google Scholar] [CrossRef]
- Martins, A.S.; Olmos, D.; Missiaglia, E.; Shipley, J. Targeting the insulin-like growth factor pathway in rhabdomyosarcomas: Rationale and future perspectives. Sarcoma 2011, 2011, 209736. [Google Scholar] [CrossRef] [Green Version]
- Khurshid, S.; Montes, M.; Comiskey, D.F.; Shane, B.; Matsa, E.; Jung, F.; Brown, C.; Bid, H.K.; Wang, R.; Houghton, P.J.; et al. Splice-switching of the insulin receptor pre-mRNA alleviates tumorigenic hallmarks in rhabdomyosarcoma. NPJ Precis. Oncol. 2022, 6, 1. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.X.; Rao, X.; Liu, Y. Modulator-Dependent RBPs Changes Alternative Splicing Outcomes in Kidney Cancer. Front. Genet. 2020, 11, 265. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Van Tine, B.A. Synovial Sarcoma: Current Concepts and Future Perspectives. J. Clin. Oncol. 2018, 36, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, R.T.; Pulice, J.L.; Valencia, A.M.; McBride, M.J.; McKenzie, Z.M.; Gillespie, M.A.; Ku, W.L.; Teng, M.; Cui, K.; Williams, R.T.; et al. SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat. Genet. 2017, 49, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Neftel, C.; Shore, M.E.; Weisman, H.R.; Mathewson, N.D.; McBride, M.J.; Haas, B.; Izar, B.; Volorio, A.; Boulay, G.; et al. Opposing immune and genetic mechanisms shape oncogenic programs in synovial sarcoma. Nat. Med. 2021, 27, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Grossi, E.; Raimondi, I.; Goni, E.; Gonzalez, J.; Marchese, F.P.; Chapaprieta, V.; Martin-Subero, J.I.; Guo, S.; Huarte, M. A lncRNA-SWI/SNF complex crosstalk controls transcriptional activation at specific promoter regions. Nat. Commun. 2020, 11, 936. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.; Gorlick, R. Advancing therapy for osteosarcoma. Nat. Rev. Clin. Oncol. 2021, 18, 609–624. [Google Scholar] [CrossRef]
- Cortés-Ciriano, I.; Lee, J.J.-K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.-Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zhang, C.; Zhang, Y.; Hong, C.S.; Chen, W.; Shen, W.; Wang, H.; He, J.; Chen, P.; Zhou, Y.; et al. Down regulation of human positive coactivator 4 suppress tumorigenesis and lung metastasis of osteosarcoma. Oncotarget 2017, 8, 53210–53225. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulos, I.; Mencinger, M.; Dietrich, C.U.; Bjerkehagen, B.; Saeter, G.; Mertens, F.; Mandahl, N.; Heim, S. Fusion of the RBP56 and CHN genes in extraskeletal myxoid chondrosarcomas with translocation t(9;17)(q22;q11). Oncogene 1999, 18, 7594–7598. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Sarcoma | RNA Binding Proteins | T-Statistic | p-Value |
|---|---|---|---|
| Ewing sarcoma | EWSR1 | −13.16812526 | 1.03 × 10−36 |
| IGF2BP1 | −12.2904187 | 1.70 × 10−32 | |
| STAG1 | −11.52248821 | 5.57 × 10−29 | |
| Rhabdomyosarcoma | QKI | −8.65279311 | 2.29 × 10−17 |
| Synovial sarcoma | SRSF2 | −5.668685512 | 2.09 × 10−8 |
| LSM14B | −4.679259416 | 3.26 × 10−6 | |
| Osteosarcoma | SUB1 | −5.501544322 | 4.92 × 10−8 |
| ASCC3 | −4.133181793 | 3.91 × 10−5 | |
| Liposarcoma | DAZAP1 | −5.33256277 | 1.19 × 10−7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hai, Y.; Kawachi, A.; He, X.; Yoshimi, A. Pathogenic Roles of RNA-Binding Proteins in Sarcomas. Cancers 2022, 14, 3812. https://doi.org/10.3390/cancers14153812
Hai Y, Kawachi A, He X, Yoshimi A. Pathogenic Roles of RNA-Binding Proteins in Sarcomas. Cancers. 2022; 14(15):3812. https://doi.org/10.3390/cancers14153812
Chicago/Turabian StyleHai, Yu, Asuka Kawachi, Xiaodong He, and Akihide Yoshimi. 2022. "Pathogenic Roles of RNA-Binding Proteins in Sarcomas" Cancers 14, no. 15: 3812. https://doi.org/10.3390/cancers14153812