
Endometrial Carcinoma: Molecular Cytogenetics and Transcriptomic Profile

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Material
2.2. G-Banding and Karyotyping
2.3. DNA and RNA Extraction
2.4. Genomic Imbalances
2.5. Pathogenic Variants
2.6. Microsatellite Instability (MSI) Status
2.7. Messenger RNA (mRNA) and MicroRNA (miRNA) Expression Profile
2.8. Statistical Analysis of mRNA and miRNA Expression Data
3. Results
3.1. G-Banding Analysis
3.2. Genomic Imbalances
3.3. Pathogenic Variants
3.4. Microsatellite Instability (MSI)
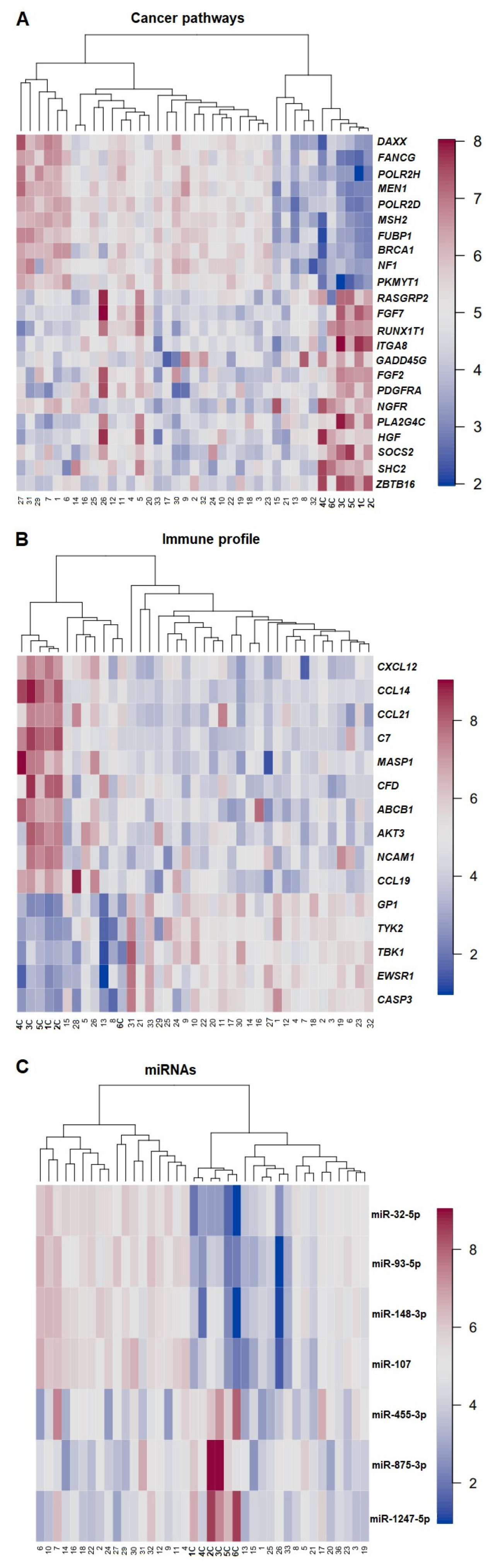
3.5. Gene Expression Profile—PCA, DGEA, and Hierarchical Clustering
3.6. Statistical Correlation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brüggmann, D.; Ouassou, K.; Klingelhöfer, D.; Bohlmann, M.K.; Jaque, J.; Groneberg, D.A. Endometrial cancer: Mapping the global landscape of research. J. Transl. Med. 2020, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.R.; Lax, S.F.; Lazar, A.J.; Longacre, T.A.; Malpica, A.; Matias-Guiu, X.; Nucci, M.; Oliva, E. Tumours of the Uterine Corpus in WHO Classification of Female Genital Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2020; Volume 4. [Google Scholar]
- Lu, K.H.; Broaddus, R.R. Endometrial Cancer. N. Engl. J. Med. 2020, 383, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Soslow, R.A.; Tornos, C.; Park, K.J.; Malpica, A.; Matias-Guiu, X.; Oliva, E.; Parkash, V.; Carlson, J.; McCluggage, W.G.; Gilks, C.B. Endometrial Carcinoma Diagnosis: Use of FIGO Grading and Genomic Subcategories in Clinical Practice: Recommendations of the International Society of Gynecological Pathologists. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2019, 38 (Suppl. S1), S64–S74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talhouk, A.; McAlpine, J.N. New classification of endometrial cancers: The development and potential applications of genomic-based classification in research and clinical care. Gynecol. Oncol. Res. Pract. 2016, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Gatius, S.; Cuevas, D.; Fernández, C.; Roman-Canal, B.; Adamoli, V.; Piulats, J.M.; Eritja, N.; Martin-Satue, M.; Moreno-Bueno, G.; Matias-Guiu, X. Tumor Heterogeneity in Endometrial Carcinoma: Practical Consequences. Pathobiology 2018, 85, 35–40. [Google Scholar] [CrossRef]
- Momeni-Boroujeni, A.; Nguyen, B.; Vanderbilt, C.M.; Ladanyi, M.; Abu-Rustum, N.R.; Aghajanian, C.; Ellenson, L.H.; Weigelt, B.; Soslow, R.A. Genomic landscape of endometrial carcinomas of no specific molecular profile. Mod. Pathol. 2022, 27, 1–10. [Google Scholar] [CrossRef]
- Kasius, C.J.; Pijnenborg, J.M.A.; Lindemann, K.; Forsse, D.; van Zwol, J.; Kristensen, G.B.; Krakstad, C.; Werner, H.M.J.; Amant, F. Risk Stratification of Endometrial Cancer Patients: FIGO Stage, Biomarkers and Molecular Classification. Cancers 2021, 13, 5848. [Google Scholar] [CrossRef]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Homepage. Available online: http://cancergenome.nih.gov/abouttcga (accessed on 15 January 2022).
- Available online: https://mitelmandatabase.isb-cgc.org/ (accessed on 1 February 2022).
- Mandahl, N. Methods in Solid Tumor Cytogenetics, 3rd ed.; Rooney, D.E., Ed.; Oxford, UK, 2001. [Google Scholar]
- McGowan-Jordan, J.; Shaffer, L.G.; Schmid, M. An International System for Human Cytogenomic Nomenclature (2016) ISCN 2016; Karger: Berlin, Germany, 2016. [Google Scholar]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef]
- Brunetti, M.; Panagopoulos, I.; Kostolomov, I.; Davidson, B.; Heim, S.; Micci, F. Mutation analysis and genomic imbalances of cells found in effusion fluids from patients with ovarian cancer. Oncol. Lett. 2020, 20, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 30 March 2022).
- Shapiro, S.S.; Wilk, M.B. An analysis of variance test for normality (complete samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B-Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Johnson, R.A.; Wichern, D.W. Applied Multivariate Statistical Analysis, 6th ed.; Pearson: Upper Saddle River, NJ, USA, 2019; pp. 18–773. [Google Scholar]
- Maimon, O.; Rokach, L. Clustering Methods. In Data Mining and Knowledge Discovery Handbook; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Ward, J.H., Jr. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Micci, F.; Teixeira, M.R.; Haugom, L.; Kristensen, G.; Abeler, V.M.; Heim, S. Genomic aberrations in carcinomas of the uterine corpus. Genes Chromosom. Cancer 2004, 40, 229–246. [Google Scholar] [CrossRef]
- Elman, J.S.; Ni, T.K.; Mengwasser, K.E.; Jin, D.; Wronski, A.; Elledge, S.J.; Kuperwasser, C. Identification of FUBP1 as a Long Tail Cancer Driver and Widespread Regulator of Tumor Suppressor and Oncogene Alternative Splicing. Cell Rep. 2019, 28, 3435–3449.e5. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.J.; Wei, Z.; Sun, J.; Bhattacharya, A.; Savage, D.J.; Serda, R.; Mackeyev, Y.; Curley, S.A.; Bu, P.; Wang, L.; et al. A recellularized human colon model identifies cancer driver genes. Nat. Biotechnol. 2016, 34, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Monte, N.M.; Webster, K.A.; Neuberg, D.; Dressler, G.R.; Mutter, G.L. Joint Loss of PAX2 and PTEN Expression in Endometrial Precancers and Cancer. Cancer Res. 2010, 70, 6225–6232. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Bueno, G.; Hardisson, D.; Sarrió, D.; Sánchez, C.; Cassia, R.; Prat, J.; Herman, J.G.; Esteller, M.; Matías-Guiu, X.; Palacios, J. Abnormalities of E- and P-cadherin and catenin (β-, γ-catenin, and p120ctn) expression in endometrial cancer and endometrial atypical hyperplasia. J. Pathol. 2003, 199, 471–478. [Google Scholar] [CrossRef]
- Mutter, G.L.; Ince, T.A.; Baak, J.P.; A Kust, G.; Zhou, X.P.; Eng, C. Molecular identification of latent precancers in histologically normal endometrium. Cancer Res. 2001, 61, 4311–4314. [Google Scholar] [PubMed]
- Brooks, R.A.; Fleming, G.F.; Lastra, R.R.; Lee, N.K.; Moroney, J.W.; Son, C.H.; Tatebe, K.; Veneris, J.L. Current recommendations and recent progress in endometrial cancer. CA A Cancer J. Clin. 2019, 69, 258–279. [Google Scholar] [CrossRef]
- Concin, N.; Matias-Guiu, X.; Vergote, I.; Cibula, D.; Mirza, M.R.; Marnitz, S.; Ledermann, J.; Bosse, T.; Chargari, C.; Fagotti, A.; et al. ESGO/ESTRO/ESP guidelines for the management of patients with endometrial carcinoma. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2021, 31, 12–39. [Google Scholar] [CrossRef] [PubMed]
- Raffone, A.; Travaglino, A.; Raimondo, D.; Neola, D.; Renzulli, F.; Santoro, A.; Insabato, L.; Casadio, P.; Zannoni, G.F.; Zullo, F.; et al. Prognostic value of myometrial invasion and TCGA groups of endometrial carcinoma. Gynecol. Oncol. 2021, 162, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Raffone, A.; Travaglino, A.; Raimondo, D.; Neola, D.; Maletta, M.; Santoro, A.; Insabato, L.; Casadio, P.; Fanfani, F.; Zannoni, G.F.; et al. Lymphovascular space invasion in endometrial carcinoma: A prognostic factor independent from molecular signature. Gynecol. Oncol. 2022, 165, 192–197. [Google Scholar] [CrossRef]
- Micci, F.; Heim, S. Tumors of the female genital organs. Cancer Cytogenet. 2015, 144, 447–480. [Google Scholar] [CrossRef]
- Milatovich, A.; Heerema, N.A.; Palmer, C.G. Cytogenetic studies of endometrial malignancies. Cancer Genet. Cytogenet. 1990, 46, 41–53. [Google Scholar] [CrossRef]
- Available online: www.sanger.ac.uk/genetics/CGP/cosmic (accessed on 15 January 2022).
- León-Castillo, A.; Britton, H.; McConechy, M.K.; McAlpine, J.N.; Nout, R.; Kommoss, S.; Brucker, S.Y.; Carlson, J.W.; Epstein, E.; Rau, T.T.; et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J. Pathol. 2020, 250, 323–335. [Google Scholar] [CrossRef]
- McConechy, M.K.; Talhouk, A.; Leung, S.; Chiu, D.S.; Yang, W.; Senz, J.; Reha-Krantz, L.J.; Lee, C.-H.; Huntsman, D.G.; Gilks, C.B.; et al. Endometrial Carcinomas with POLE Exonuclease Domain Mutations Have a Favorable Prognosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 2865–2873. [Google Scholar] [CrossRef] [Green Version]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic Review of Microsatellite Instability and Colorectal Cancer Prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef]
- Sinicrope, F.A.; Sargent, D.J. Molecular Pathways: Microsatellite Instability in Colorectal Cancer: Prognostic, Predictive, and Therapeutic Implications. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1506–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evrard, C.; Alexandre, J. Predictive and Prognostic Value of Microsatellite Instability in Gynecologic Cancer (Endometrial and Ovarian). Cancers 2021, 13, 2434. [Google Scholar] [CrossRef] [PubMed]
- Stelloo, E.; Jansen, A.M.L.; Osse, E.M.; Nout, R.A.; Creutzberg, C.L.; Ruano, D.; Church, D.N.; Morreau, H.; Smit, V.T.H.B.M.; van Wezel, T.; et al. Practical guidance for mismatch repair-deficiency testing in endometrial cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 96–102. [Google Scholar] [CrossRef]
- Kurnit, K.C.; Westin, S.N.; Coleman, R.L. Microsatellite instability in endometrial cancer: New purpose for an old test. Cancer 2019, 125, 2154–2163. [Google Scholar] [CrossRef] [PubMed]
- McMeekin, D.S.; Tritchler, D.L.; Cohn, D.E.; Mutch, D.G.; Lankes, H.A.; Geller, M.A.; Powell, M.A.; Backes, F.J.; Landrum, L.M.; Zaino, R.; et al. Clinicopathologic Significance of Mismatch Repair Defects in Endometrial Cancer: An NRG Oncology/Gynecologic Oncology Group Study. J. Clin. Oncol. 2016, 34, 3062–3068. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, I.; Martín-Arruti, M.; Lopez-Lopez, E.; Garcia-Orad, A. Lack of association between deficient mismatch repair expression and outcome in endometrial carcinomas of the endometrioid type. Gynecol. Oncol. 2014, 134, 20–23. [Google Scholar] [CrossRef]
- di Rorà, A.G.L.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Laitinen, T.; East, M.P. PKMYT1: A forgotten member of the WEE1 family. Nat. Rev. Drug Discov. 2020, 19, 157. [Google Scholar] [CrossRef] [Green Version]
- Owusu, B.Y.; Galemmo, R.; Janetka, J.; Klampfer, L. Hepatocyte Growth Factor, a Key Tumor-Promoting Factor in the Tumor Microenvironment. Cancers 2017, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Nakamura, T.; Kramer, R.H. Hepatocyte growth factor/scatter factor induces tyrosine phosphorylation of focal adhesion kinase (p125FAK) and promotes migration and invasion by oral squamous cell carcinoma cells. J. Biol. Chem. 1994, 269, 31807–31813. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nakamura, T. Hepatocyte growth factor and the Met system as a mediator of tumor–stromal interactions. Int. J. Cancer 2006, 119, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Kataoka, H.; Kawano, H.; Yokogami, K.; Nakano, S.; Goya, T.; Uchino, H.; Koono, M.; Wakisaka, S. Comparative analysis of expression of hepatocyte growth factor and its receptor, c-met, in gliomas, meningiomas and schwannomas in humans. Cancer Lett. 1998, 124, 149–155. [Google Scholar] [CrossRef]
- Appleman, L.; Parikh, R.A.; Wang, P.; Beumer, J.H.; Chu, E. The potential roles of hepatocyte growth factor (HGF)-MET pathway inhibitors in cancer treatment. OncoTargets Ther. 2014, 7, 969–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosavi, F.; Giovannetti, E.; Saso, L.; Firuzi, O. HGF/MET pathway aberrations as diagnostic, prognostic, and predictive biomarkers in human cancers. Crit. Rev. Clin. Lab. Sci. 2019, 56, 533–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Hong, S.H.; Kim, J.Y.; Kim, I.-C.; Park, Y.-W.; Lee, S.-J.; Song, S.-W.; Park, G.; Kim, T.M.; Kim, Y.-H.; et al. Preclinical development of a humanized neutralizing antibody targeting HGF. Exp. Mol. Med. 2017, 49, e309. [Google Scholar] [CrossRef]
- Cretu, A.; Sha, X.; Tront, J.; Hoffman, B.; Liebermann, D.A. Stress sensor Gadd45 genes as therapeutic targets in cancer. Cancer Ther. 2009, 7, 268–276. [Google Scholar]
- Tamura, R.E.; De Vasconcellos, J.F.; Sarkar, D.; Libermann, T.A.; Fisher, P.B.; Zerbini, L.F. GADD45 Proteins: Central Players in Tumorigenesis. Curr. Mol. Med. 2012, 12, 634–651. [Google Scholar] [CrossRef]
- Ying, J.; Srivastava, G.; Hsieh, W.-S.; Gao, Z.; Murray, P.; Liao, S.-K.; Ambinder, R.; Tao, Q. The Stress-Responsive Gene GADD45G Is a Functional Tumor Suppressor, with Its Response to Environmental Stresses Frequently Disrupted Epigenetically in Multiple Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 6442–6449. [Google Scholar] [CrossRef] [Green Version]
- Van Veen, M.; Matas-Rico, E.; Van De Wetering, K.; Leyton-Puig, D.; Kedziora, K.M.; De Lorenzi, V.; Stijf-Bultsma, Y.; van den Broek, B.; Jalink, K.; Sidenius, N.; et al. Negative regulation of urokinase receptor activity by a GPI-specific phospholipase C in breast cancer cells. eLife 2017, 6, e23649. [Google Scholar] [CrossRef]
- Han, J.; Deng, X.; Sun, R.; Luo, M.; Liang, M.; Gu, B.; Zhang, T.; Peng, Z.; Lu, Y.; Tian, C.; et al. GPI Is a Prognostic Biomarker and Correlates With Immune Infiltrates in Lung Adenocarcinoma. Front. Oncol. 2021, 11, 752642. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.I.; Reis, R.M.; Calin, G.A. MicroRNA history: Discovery, recent applications, and next frontiers. Mutat. Res. Mol. Mech. Mutagen. 2011, 717, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Tang, C.; Chen, B.; Lei, P.; Song, J.; Xin, G.; Wang, Z.; Hui, Y.; Yao, W.; Wang, G.; et al. miR-32-5p suppresses the proliferation and migration of pancreatic adenocarcinoma cells by targeting TLDC1. Mol. Med. Rep. 2021, 24, 1–9. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Zhou, H.-G.; Chen, L.-H.; Qu, D.-C.; Wang, C.-J.; Xia, Z.-Y.; Zheng, J.-H. MiR-32-5p regulates the proliferation and metastasis of cervical cancer cells by targeting HOXB8. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 87–95. [Google Scholar] [CrossRef]
- Fu, X.; Liu, M.; Qu, S.; Ma, J.; Zhang, Y.; Shi, T.; Wen, H.; Yang, Y.; Wang, S.; Wang, J.; et al. Exosomal microRNA-32-5p induces multidrug resistance in hepatocellular carcinoma via the PI3K/Akt pathway. J. Exp. Clin. Cancer Res. 2018, 37, 52. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Distribution |
|---|---|
| Post-operative histology | Endometrioid G1 (n = 8) Endometrioid G2 (n = 11) Endometrioid G3 (n = 5) |
| Clear Cell (n = 1) Serous (n = 3) Mixed (n = 3) Adenocarcinoma NOS (n = 2) | |
| FIGO Stage 2009 | IA (n = 13) IB (n = 9) II (n = 2) IIIA (n = 2) IIIB (n = 1) IIIC1 (n = 2) IVB (n = 3) |
| Post-operative treatment | none (n = 20) Platinum based chemotherapy (n = 12) Chemotherapy + radiation therapy (n = 1) |
| Recurrence | yes (n = 10) no (n = 23) |
| Site of recurrence | vagina (n = 3) distant (n = 2) multiple sites (n = 4) isolated nodal pelvic (n = 1) |
| Age at diagnosis | average years 70 (range 54 to 94) |
| Deaths | n = 9 |
| OS | average months 44.5 |
| Name | Sequence | Gene | Position on GRch37/hg19 Assembly |
|---|---|---|---|
| M13-POLEint9FW | 5′-TGTAAAACGACGGCCAGTAGTCTTAGGGTCCTTCTCCCA-3′ | POLE | chr12 − 132676775 132676795 |
| M13-POLEint10R | 5′-CAGGAAACAGCTATGACCTGTGTGGATTCCCACTCGAAA-3′ | POLE | chr12 + 132676361 132676381 |
| M13-POLEint12FW3 | 5′-TGTAAAACGACGGCCAGTGGGGTTCCCGGGCTGCATGTTA-3′ | POLE | chr12 − 132673744 132673765 |
| M13-POLEint13REV3 | 5′-CAGGAAACAGCTATGACCCTCCGTGGCCATCTGGATGCGT-3′ | POLE | chr12 + 132673513 132673534 |
| M13-POLEint13F | 5′-TGTAAAACGACGGCCAGTACACACGTGTTTTGTCCTGTG-3′ | POLE | chr12 − 132673397 132673417 |
| M13-POLEint14R | 5′-CAGGAAACAGCTATGACCCAGGGCCAGAGAATTCCCAA-3′ | POLE | chr12 + 132672996 132673015 |
| Sample | Karyotype | Subgroup * |
|---|---|---|
| 1 | 46,XX,dup(1)(q21q32)[15]/46,XX[1] | B |
| 2 | 48,XX,+i(1)(q10),+?10[10]/46,XX[8] | C |
| 3 | 47,XX,+i(1)(q10),t(8;16)(p21;p13)[cp11] | C |
| 4 | 48~49,XX,+i(1)(q10),+2,inc[cp4]/46,XX[17] | C |
| 5 | 49~50,XX,+X,+i(1)(q(10),+10[cp4]/46,XX[2] | C |
| 6 | 47,XX,+i(1)(q10)[3] | A |
| 7 | 53-55,XX,+del(1)(p31),+i(1)(q10),+3,+6,+7,+8,+8,+9,−13,−15,+2mar[cp10] | C |
| 8 | 47,XX,+der(1;10)(q10;q10)[9]/46,XX[1] | C |
| 9 | 41~42,der(19)t(1;19)(q21;q13),inc[cp2]/46,XX[6] | C |
| 10 | 46,XX,add(3)(q21)[cp2]/46,XX[23] | D |
| 11 | 47,XX,+i(1)(q10)[7]/46,XX[3] | A |
| 12 | 46,XX,del(16)(q12)[11]/46,XX[4] | D |
| 13 | 46,XX,dup(1)(q23q32)[cp8]/46,XX[2] | B |
| 14 | 50~100,+i(1)(q10),+7,inc[cp9]/46,XX[1] | C |
| 15 | 48,XX,+9,+10,der(15)t(1;15)(q11;p13)[8]/46,XX[2] | C |
| 16 | 46~47,XX,+i(1)(q10)[cp6]/46,XX[4] | A |
| 17 | 45,XX,t(12;14)(q15;q24),-22[6]/43~45,idem,+r,inc[cp2]/46,XX[2] | D |
| 18 | 42~46,XX,+7,-21[cp7]/91~93,idemx2[cp3]/46,XX[4] | D |
| 19 | 47,XX,+X[11] | D |
| 20 | 47,XX,+1,der(1;13)(q10;q10),+8[10] | C |
| 21 | 47,XX,+add(1)(p11)[5]/46,XX[5] | A |
| 22 | 47,XX,+10,del(16)(q13)[3]/46,XX[8] | D |
| 23 | 46,XX,r(13)[10] | D |
| 24 | 41~70,i(1)(q10),+mar,inc[cp7]/46,XX[1] | C |
| 25 | 46,XX,t(2;12)(q37;q?13)[3]/46,XX[9] | D |
| 26 | 46,XX,?t(4;6)(q?28;p22),add(11)(q22)[6]/46,XX[5] | D |
| 27 | 52,XX,i(1)(q10),+3,+8,+8,+10,+10[10] | C |
| 28 | 38~75,inv(1)(p13p36),add(2)(q33),add(7)(q32),inc[cp7]/46,XX[5] | D |
| 29 | 36~74,dup(1)(q21q23),der(?)t(1;?)(p13;?),der(?)t(1;?)(q12;?),add(11)(q23),inc[cp7/46,XX[4] | B |
| 30 | 44~46,XX,der(1)t(1;1)(p36;q21~q23)[cp7]/46,XX[3] | A |
| 31 | 44~46,add(1)(p13),+i(1)(q10),del(3)(q13),inc[cp13] | C |
| 32 | 48~49,XX,+del(1)(p22),+12,inc[cp6] | C |
| 33 | 45~47,XX,der(3)t(1;3)(q11;p25)[cp3]/46,XX[7] | C |
| Sample | Overall Call | Markers Unstable | Markers Stable |
|---|---|---|---|
| 1 | MSS | 0 | 13 |
| 2 | MSI-Low | 1 | 12 |
| 3 | MSI-High | 5 | 8 |
| 4 | MSI-High | 10 | 3 |
| 5 | MSI-High | 6 | 7 |
| 6 | MSI-High | 5 | 8 |
| 7 | MSI-Low | 2 | 11 |
| 8 | MSI-High | 5 | 8 |
| 9 | MSI-Low | 1 | 12 |
| 10 | MSS | 0 | 13 |
| 11 | MSI-High | 12 | 1 |
| 12 | MSI-High | 6 | 7 |
| 13 | MSI-High | 4 | 9 |
| 14 | MSS | 0 | 13 |
| 15 | MSS | 0 | 13 |
| 16 | MSS | 0 | 13 |
| 17 | MSI-High | 7 | 6 |
| 18 | MSI-High | 10 | 3 |
| 19 | MSI-High | 7 | 6 |
| 20 | MSI-High | 4 | 9 |
| 21 | MSI-High | 7 | 6 |
| 22 | MSI-High | 6 | 7 |
| 23 | MSS | 0 | 13 |
| 24 | MSS | 0 | 13 |
| 25 | MSS | 0 | 13 |
| 26 | MSS | 0 | 13 |
| 27 | MSS | 0 | 13 |
| 28 | MSS | 0 | 13 |
| 29 | MSS | 0 | 13 |
| 30 | MSI-High | 7 | 6 |
| 31 | MSS | 0 | 13 |
| 32 | MSS | 0 | 13 |
| 33 | MSS | 0 | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunetti, M.; Panagopoulos, I.; Vitelli, V.; Andersen, K.; Hveem, T.S.; Davidson, B.; Eriksson, A.G.Z.; Trent, P.K.B.; Heim, S.; Micci, F. Endometrial Carcinoma: Molecular Cytogenetics and Transcriptomic Profile. Cancers 2022, 14, 3536. https://doi.org/10.3390/cancers14143536
Brunetti M, Panagopoulos I, Vitelli V, Andersen K, Hveem TS, Davidson B, Eriksson AGZ, Trent PKB, Heim S, Micci F. Endometrial Carcinoma: Molecular Cytogenetics and Transcriptomic Profile. Cancers. 2022; 14(14):3536. https://doi.org/10.3390/cancers14143536
Chicago/Turabian StyleBrunetti, Marta, Ioannis Panagopoulos, Valeria Vitelli, Kristin Andersen, Tarjei S. Hveem, Ben Davidson, Ane Gerda Z. Eriksson, Pernille Kristina Bjerre Trent, Sverre Heim, and Francesca Micci. 2022. "Endometrial Carcinoma: Molecular Cytogenetics and Transcriptomic Profile" Cancers 14, no. 14: 3536. https://doi.org/10.3390/cancers14143536