
Inflammation-Driven Regulation of PD-L1 and PD-L2, and Their Cross-Interactions with Protective Soluble TNFα Receptors in Human Triple-Negative Breast Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Stimulation by Pro-Inflammatory Cytokines
2.3. Flow Cytometry Analyses of PD-L1 and PD-L2 Expression
2.4. Analyses of Patient Datasets
2.5. Western Blot Analyses
2.6. Generating STAT1 and p65 KO Cells
2.7. ELISA Analyses
2.8. Analyses Using Recombinant Soluble TNFR1 and TNFR2
2.9. qPCR Analyses
2.10. Data Presentation and Statistical Analyses
3. Results
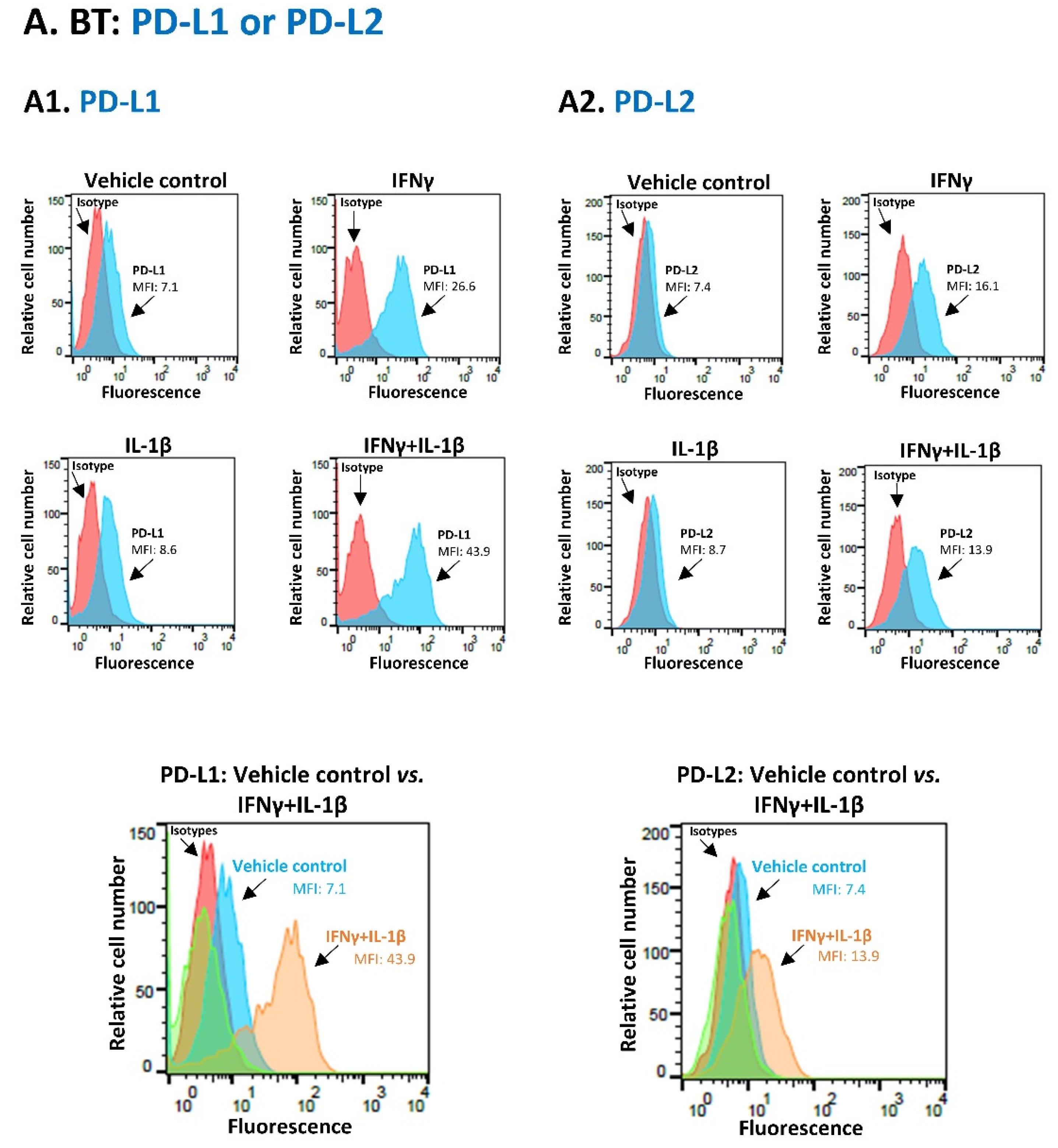
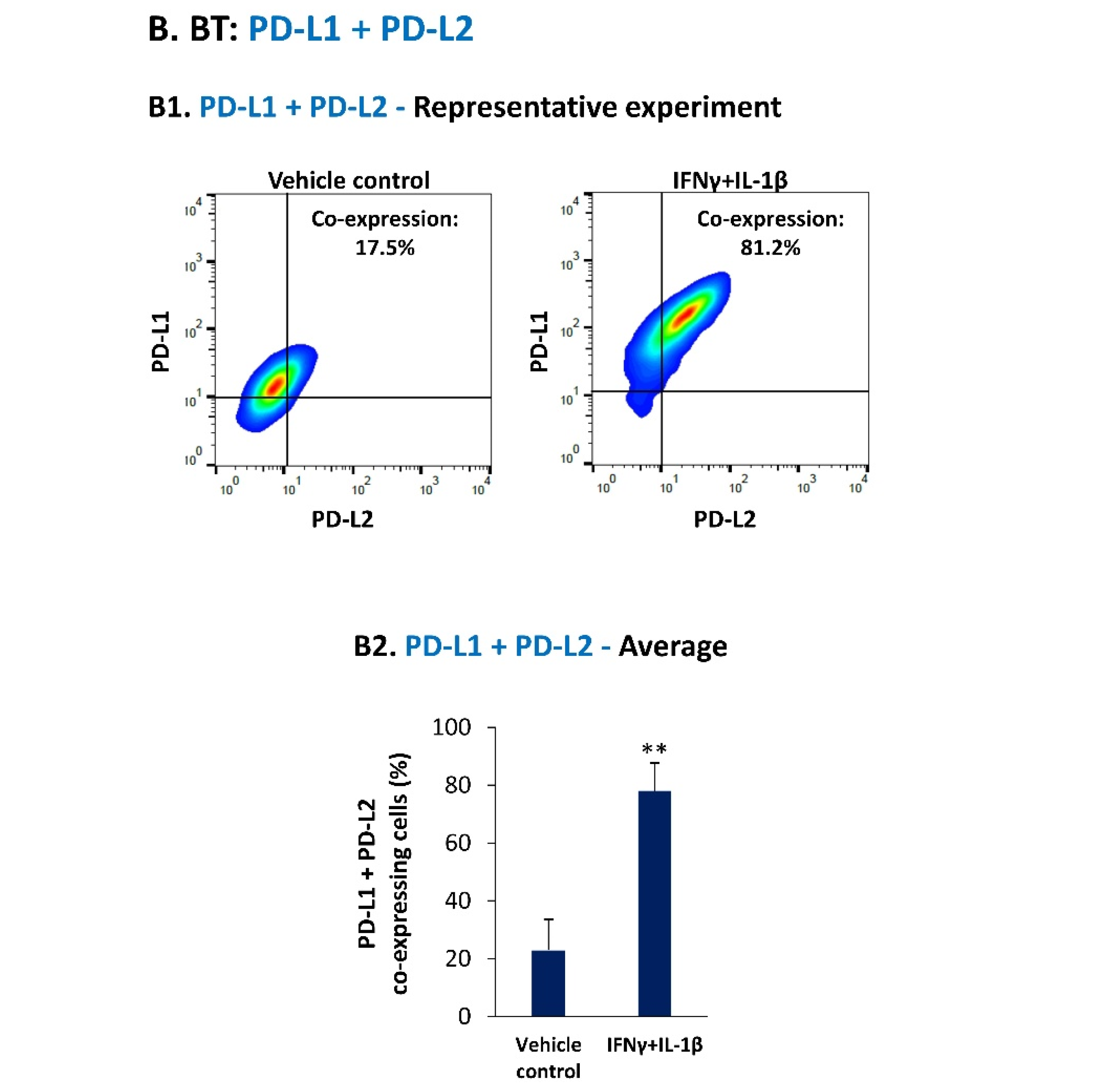
3.1. Pro-Inflammatory Cytokines Act in Cooperativity to Promote the Proportions of TNBC Cells Co-Expressing PD-L1 + PD-L2
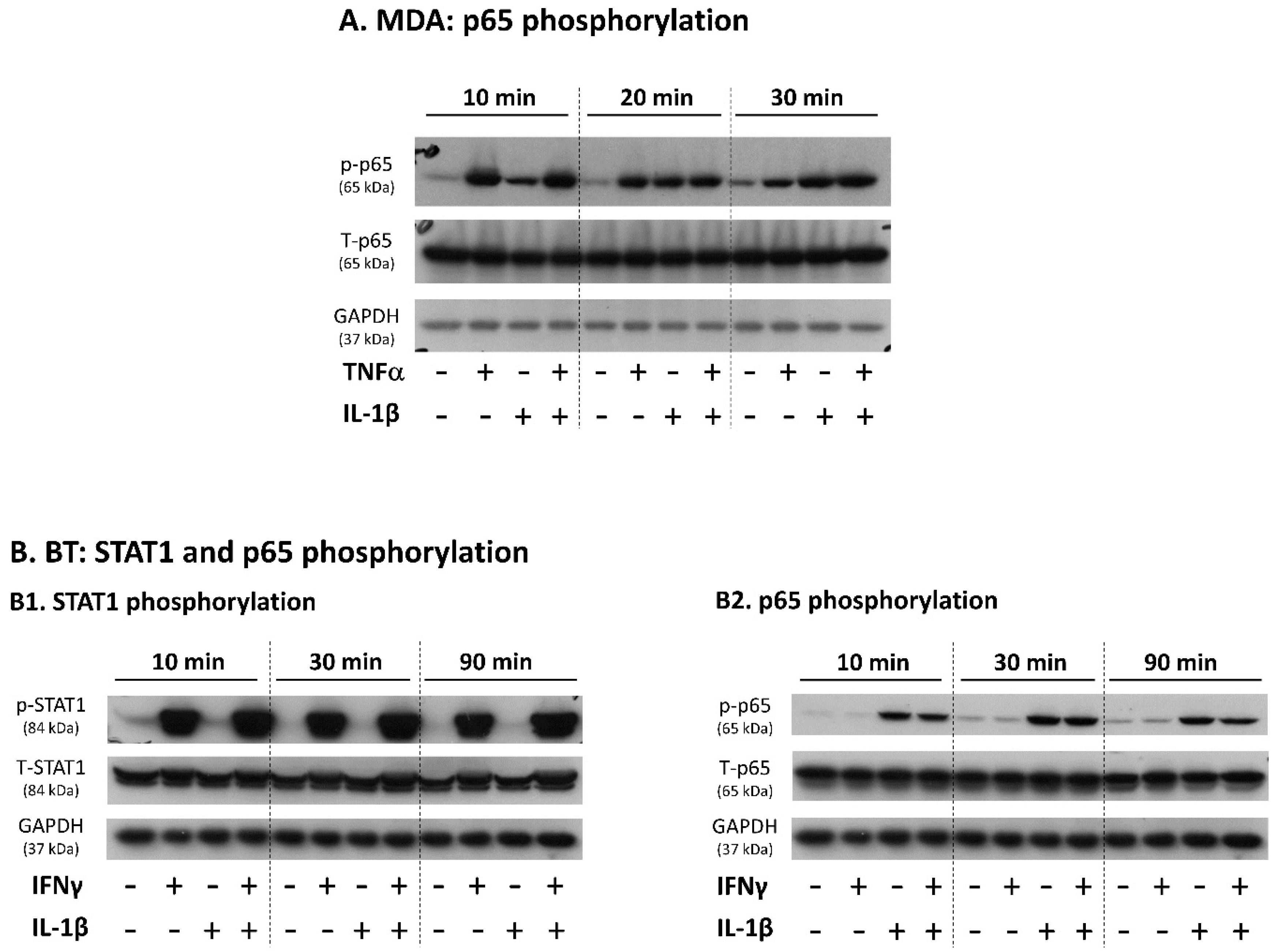
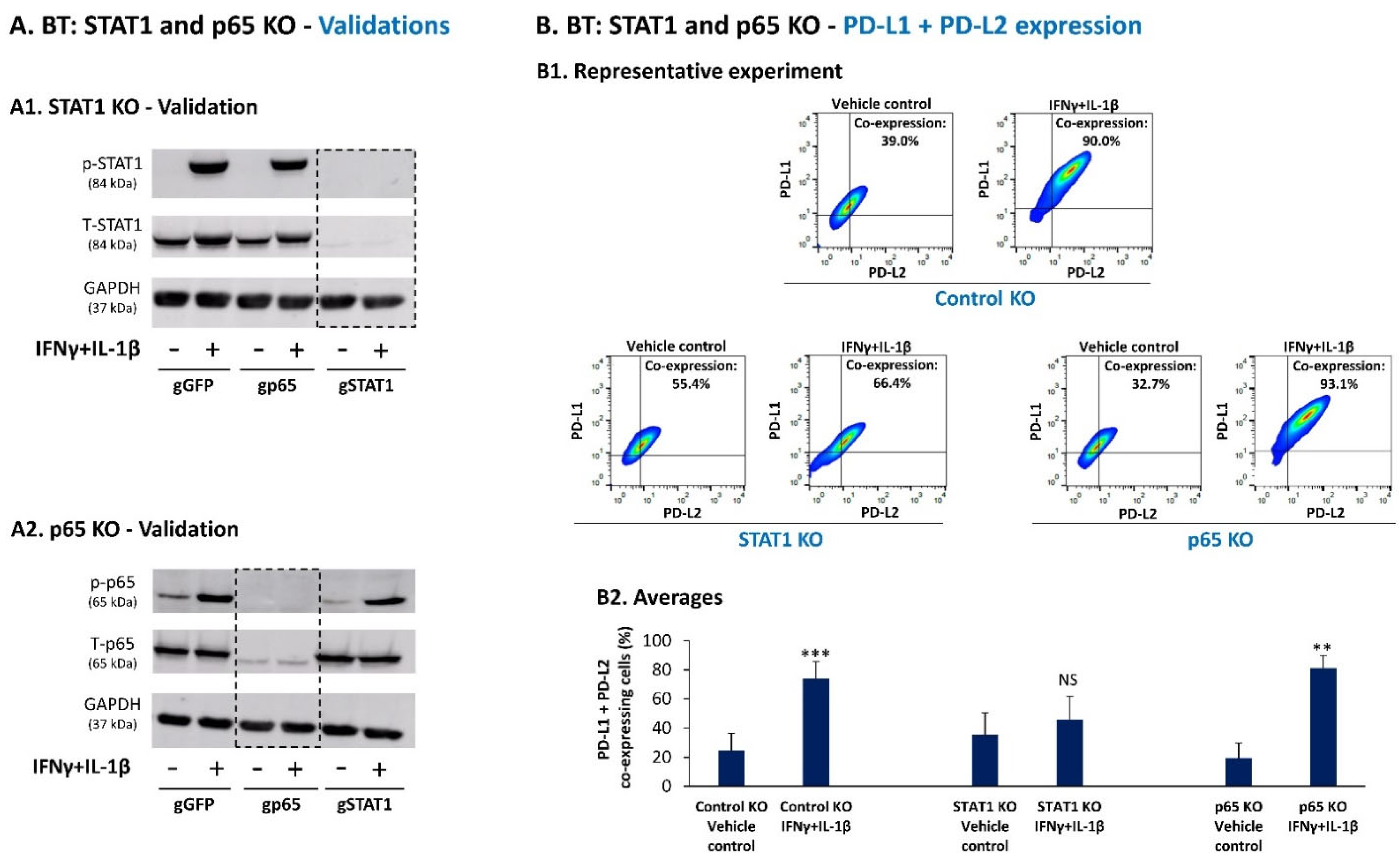
3.2. Upon IFNγ + IL-1β Stimulation of BT-549 Cells, Signals Are Channeled into STAT1-Mediated Induction of PD-L1 + PD-L2 Co-Expressing Cells
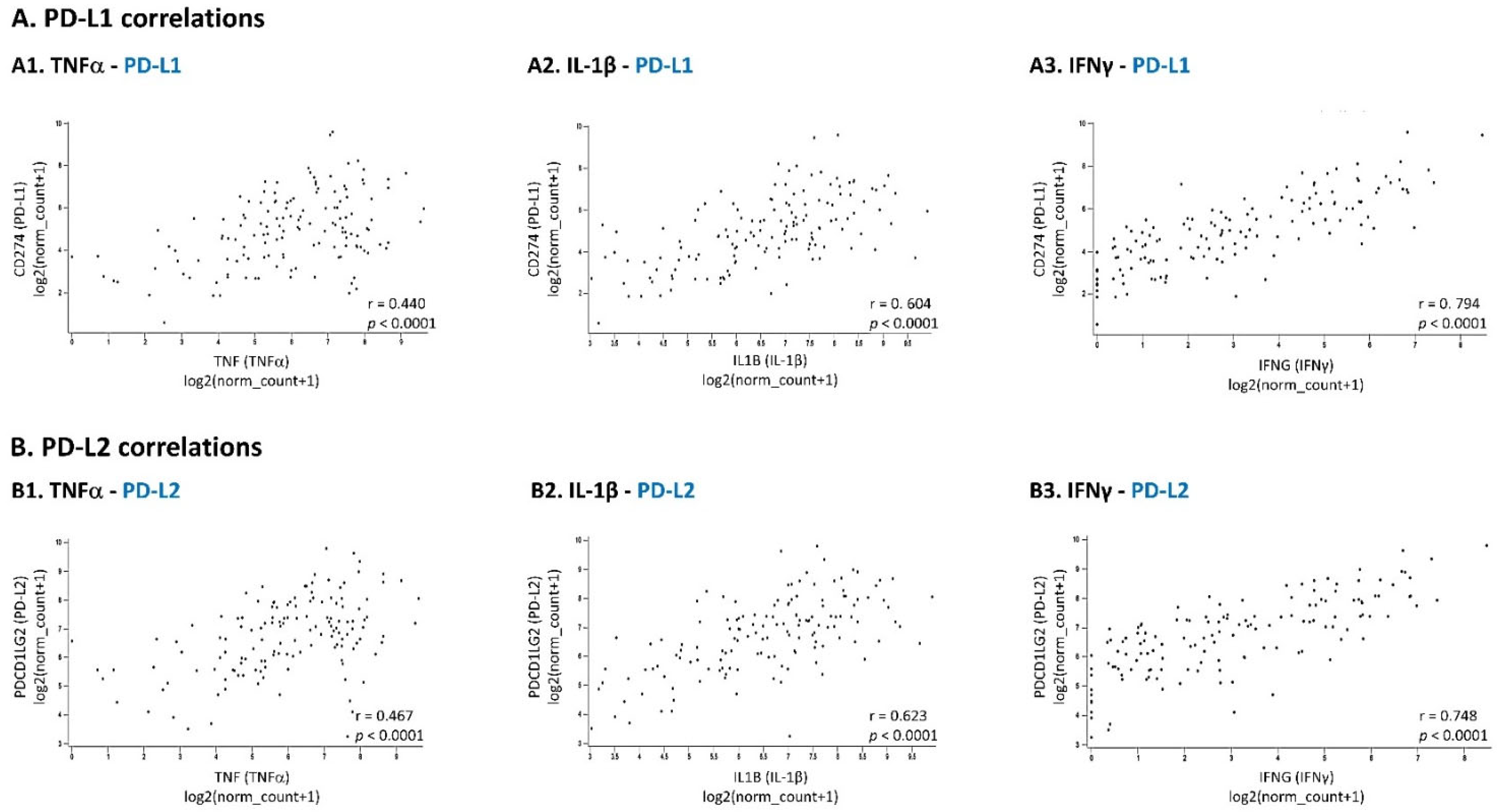
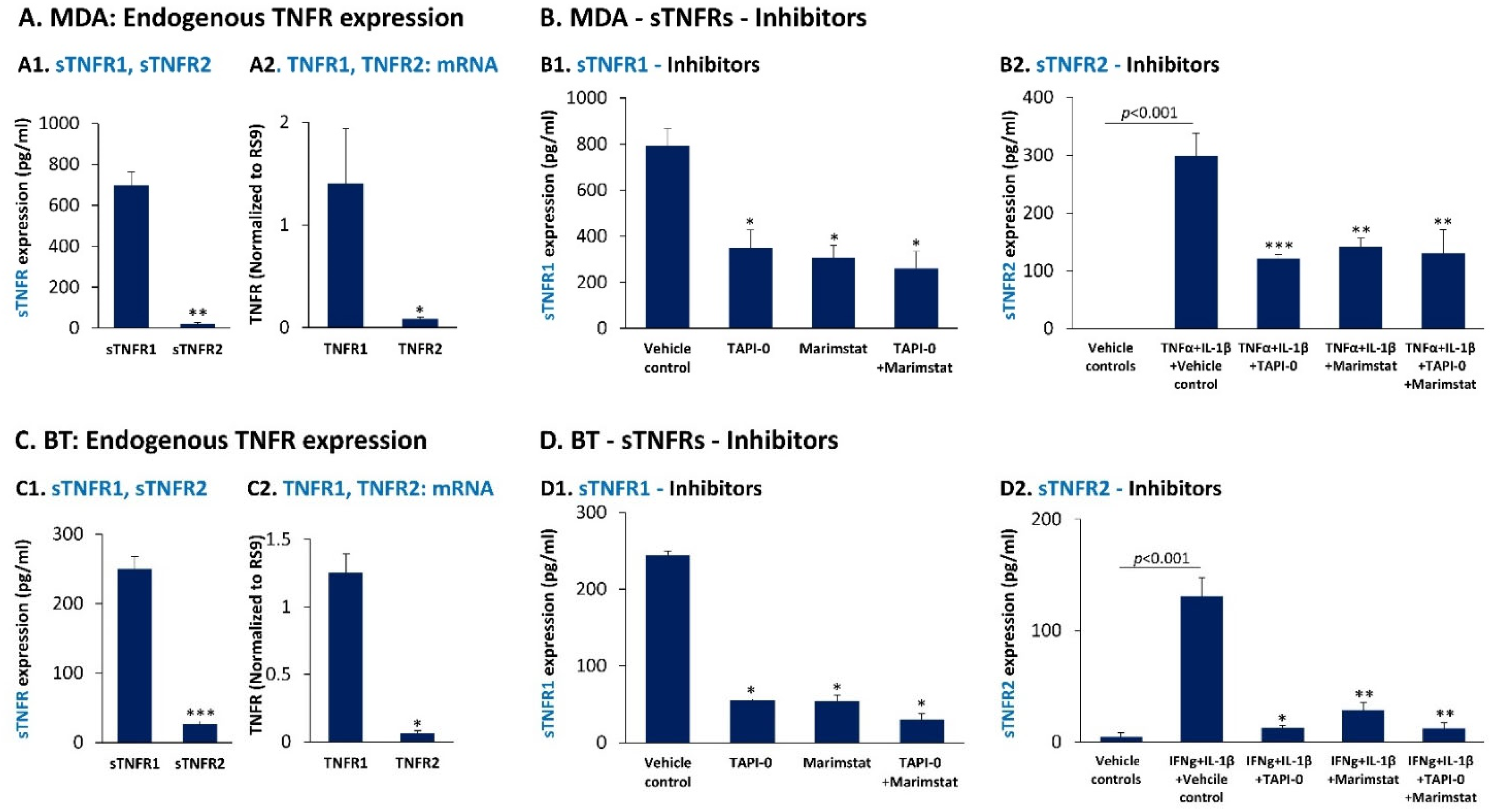
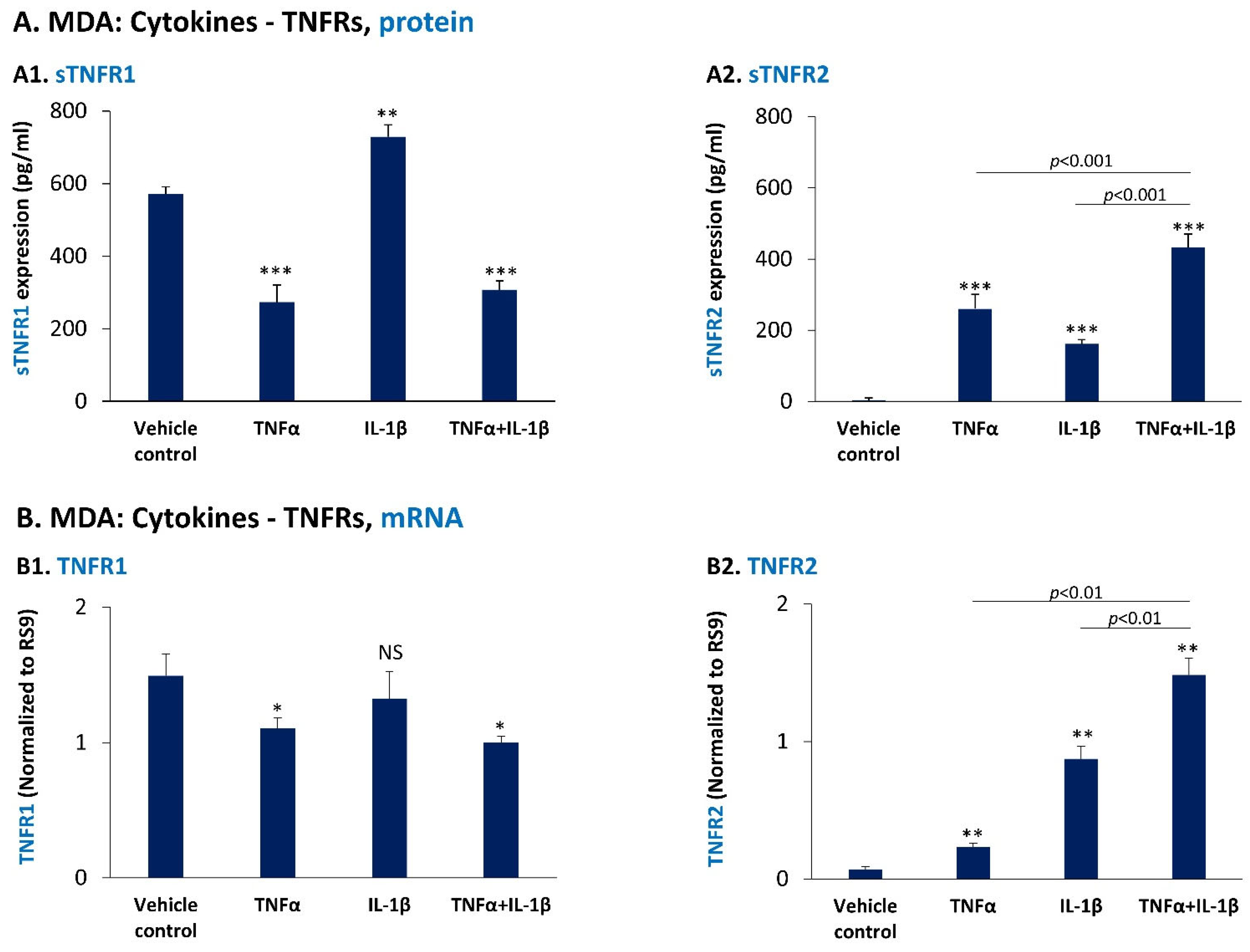
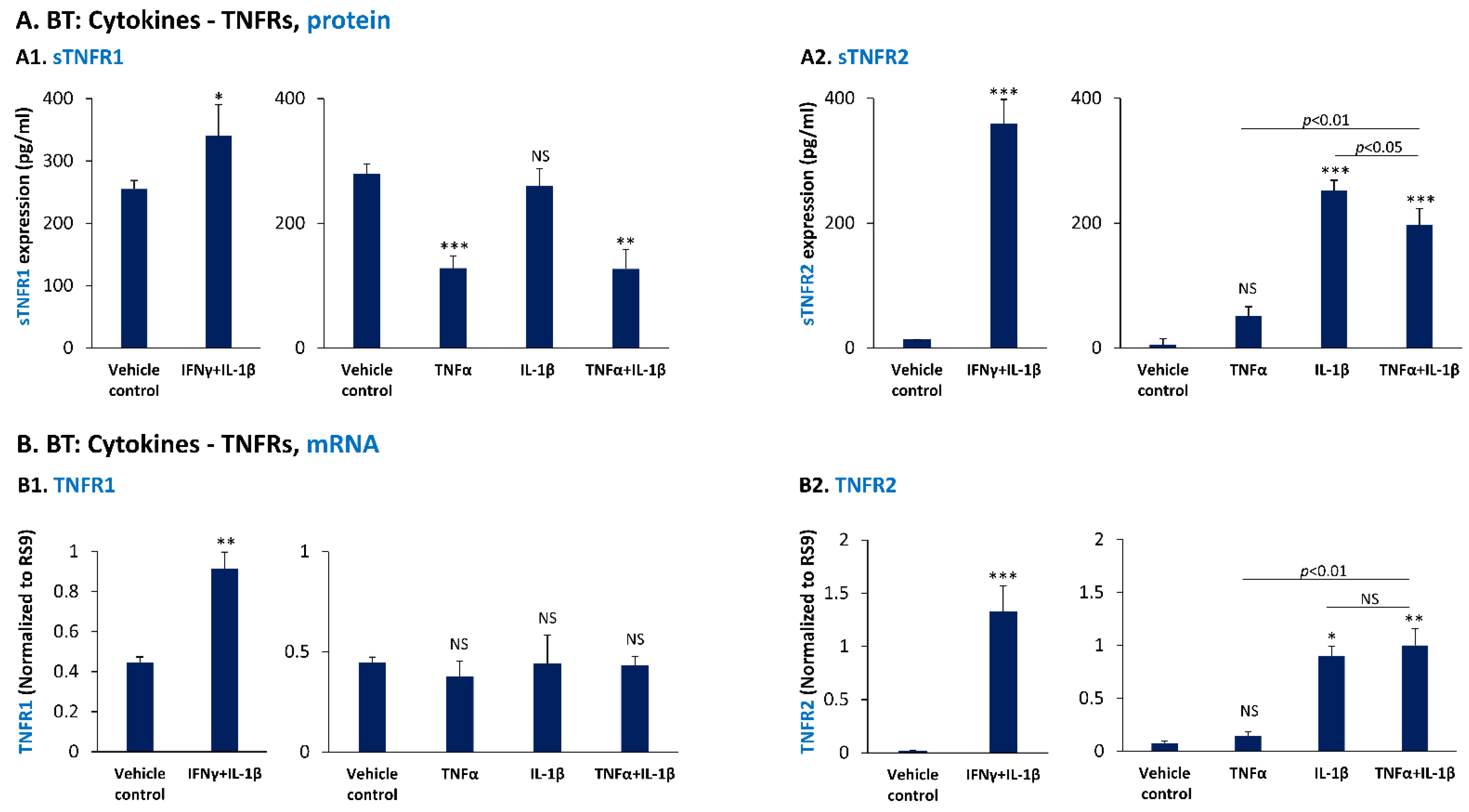
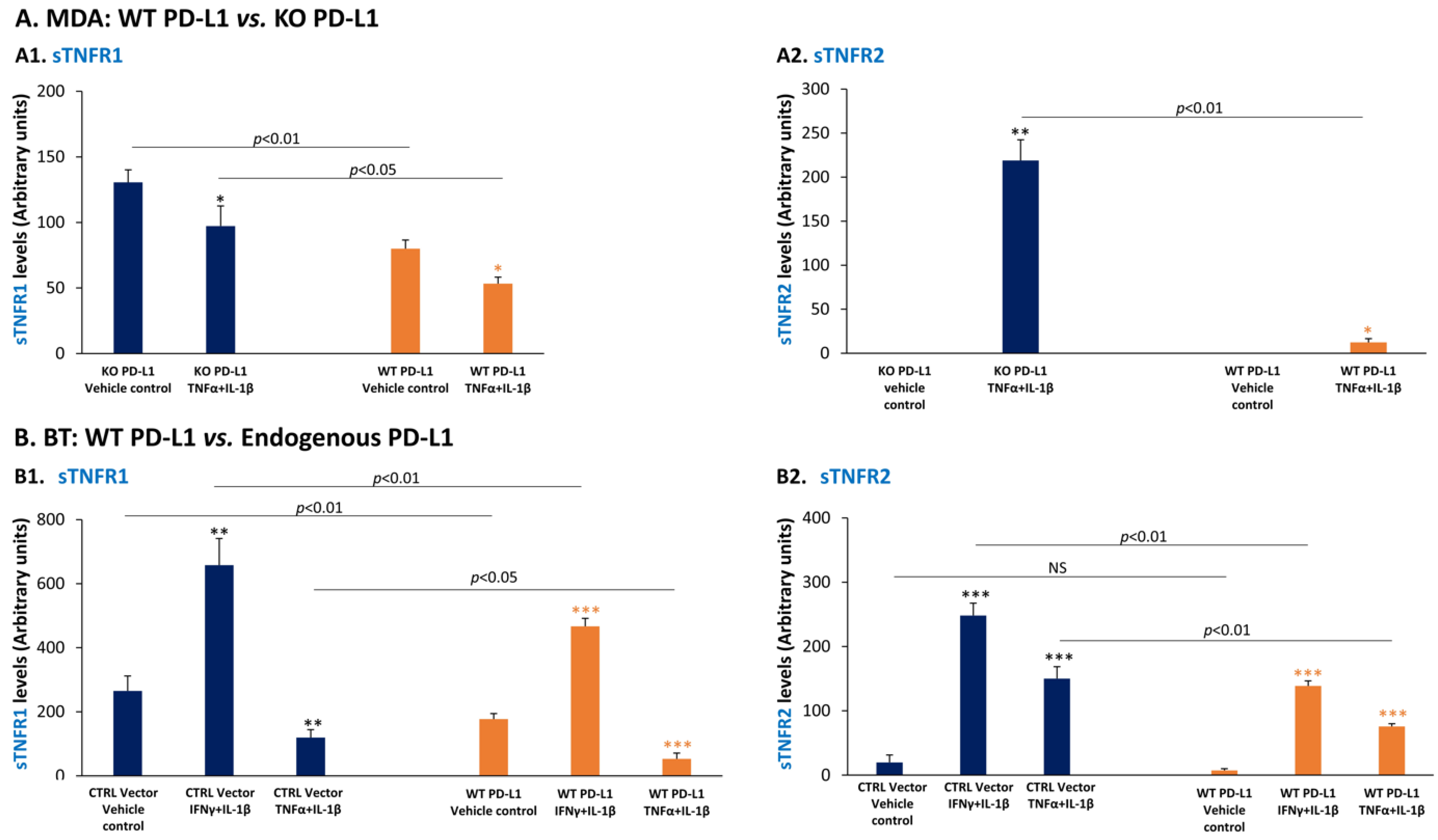
3.3. In TNBC Cells, Pro-Inflammatory Cytokines Regulate the Expression of sTNFR1 and sTNFR2 in Divergent Manners
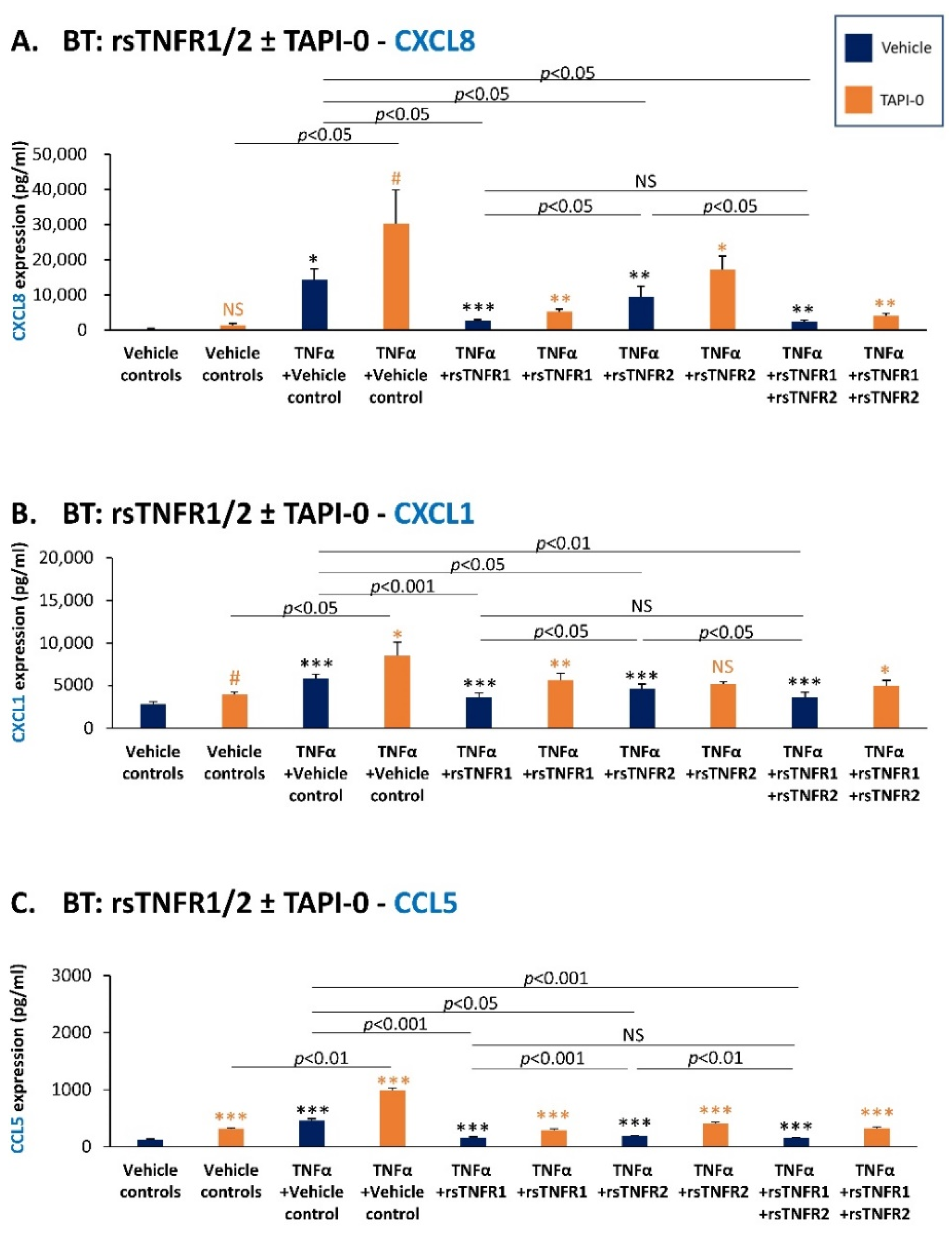
3.4. sTNFR1 and sTNFR2 Exhibit Potential Anti-Metastatic Functions by Inhibiting TNFα-Induced Production of Inflammatory Pro-Metastatic Chemokines by TNBC Cells
3.5. PD-L1 Down-Regulates the Expression of the Anti-Metastatic Factors sTNFR1 and sTNFR2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Naimi, A.; Mohammed, R.N.; Raji, A.; Chupradit, S.; Yumashev, A.V.; Suksatan, W.; Shalaby, M.N.; Thangavelu, L.; Kamrava, S.; Shomali, N.; et al. Tumor immunotherapies by immune checkpoint in-hibitors (ICIs); the pros and cons. Cell Commun. Signal. 2022, 20, 44. [Google Scholar] [CrossRef] [PubMed]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response; resistance; and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Mediratta, K.; El-Sahli, S.; D’Costa, V.; Wang, L. Current Progresses and Challenges of Immunotherapy in Triple-Negative Breast Cancer. Cancers 2020, 12, 3529. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Galdiero, M.R.; Marone, G.; Mantovani, A. Cancer Inflammation and Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Propper, D.J.; Balkwill, F.R. Harnessing cytokines and chemokines for cancer therapy. Nat. Rev. Clin. Oncol. 2022, 19, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Chen, Z.; Chen, K.; Liao, F.T.; Chung, C.E.; Liu, X.; Lin, Y.C.; Keohavong, P.; Leikauf, G.D.; Di, Y.P. Lipopolysaccharide-Mediated Chronic Inflammation Promotes Tobacco Carcinogen-Induced Lung Cancer and Determines the Efficacy of Immunotherapy. Cancer Res. 2021, 81, 144–157. [Google Scholar] [CrossRef]
- Santoni, M.; Romagnoli, E.; Saladino, T.; Foghini, L.; Guarino, S.; Capponi, M.; Giannini, M.; Cognigni, P.D.; Ferrara, G.; Battelli, N. Triple negative breast cancer: Key role of Tumor-Associated Macrophages in regulating the activity of anti-PD-1/PD-L1 agents. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2018, 1869, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Tartey, S.; Neale, G.; Vogel, P.; Malireddi, R.K.S.; Kanneganti, T.D. A MyD88/IL1R Axis Regulates PD-1 Expression on Tumor-Associated Macrophages and Sustains Their Immunosuppressive Function in Melanoma. Cancer Res. 2021, 81, 2358–2372. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, M.F.; Bruni, S.; Elizalde, P.V.; Schillaci, R. Tumor Necrosis Factor alpha Blockade: An Op-portunity to Tackle Breast Cancer. Front. Oncol. 2020, 10, 584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [Green Version]
- Cruceriu, D.; Baldasici, O.; Balacescu, O.; Berindan-Neagoe, I. The dual role of tumor necrosis fac-tor-alpha (TNF-α) in breast cancer: Molecular insights and therapeutic approaches. Cell. Oncol. 2020, 43, 1–18. [Google Scholar] [CrossRef]
- Tulotta, C.; Ottewell, P. The role of IL-1B in breast cancer bone metastasis. Endocr.-Relat. Cancer 2018, 25, R421–R434. [Google Scholar] [CrossRef] [Green Version]
- Soria, G.; Ofri-Shahak, M.; Haas, I.; Yaal-Hahoshen, N.; Leider-Trejo, L.; Leibovich-Rivkin, T.; Weitzenfeld, P.; Meshel, T.; Shabtai, E.; Gutman, M.; et al. Inflammatory mediators in breast cancer: Coordinated ex-pression of TNFalpha & IL-1beta with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer 2011, 11, 130. [Google Scholar]
- Malkova, A.M.; Gubal, A.R.; Petrova, A.L.; Voronov, E.; Apte, R.N.; Semenov, K.N.; Sharoyko, V.V. Pathoge-netic role and clinical significance of interleukin-1β in cancer. Immunology 2022. [Google Scholar] [CrossRef] [PubMed]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Korner, C.; Ben-Baruch, A. Tumor-Stroma-Inflammation Networks Promote Pro-metastatic Chemokines and Aggressiveness Characteristics in Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.L.; Fan, W.; Yang, G.; Yu, M.X. The clinical significance of PR, ER, NF- kappa B, and TNF- alpha in breast cancer. Dis. Markers 2014, 2014, 494581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Yuan, R.Q.; Fuchs, A.; Yao, Y.; Joseph, A.; Schwall, R.; Schnitt, S.J.; Guida, A.; Hastings, H.M.; Andres, J.; et al. Expression of interleukin-1beta in human breast carcinoma. Cancer 1997, 80, 421–434. [Google Scholar] [CrossRef]
- Semesiuk, N.I.; Zhylchuk, A.; Bezdenezhnykh, N.; Lykhova, A.; Vorontsova, A.L.; Zhylchuk, V.E.; Kudrya-vets, Y.I. Disseminated tumor cells and enhanced level of some cytokines in bone marrow and pe-ripheral blood of breast cancer patients as predictive factors of tumor progression. Exp. Oncol. 2013, 35, 295–302. [Google Scholar] [PubMed]
- Abrahamsson, A.; Morad, V.; Saarinen, N.M.; Dabrosin, C. Estradiol, tamoxifen, and flaxseed alter IL-1beta and IL-1Ra levels in normal human breast tissue in vivo. J. Clin. Endocrinol. Metab. 2012, 97, E2044–E2054. [Google Scholar] [CrossRef] [Green Version]
- Perrier, S.; Caldefie-Chezet, F.; Vasson, M.P. IL-1 family in breast cancer: Potential interplay with leptin and other adipocytokines. FEBS Lett. 2009, 583, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Reza, I.; Diaz, L.; Garcia-Becerra, R. Preclinical and clinical aspects of TNF-α and its re-ceptors TNFR1 and TNFR2 in breast cancer. J. Biomed. Sci. 2017, 24, 90. [Google Scholar] [CrossRef] [Green Version]
- Salamanna, F.; Borsari, V.; Contartese, D.; Costa, V.; Giavaresi, G.; Fini, M. What Is the Role of Interleukins in Breast Cancer Bone Metastases? A Systematic Review of Preclinical and Clinical Evidence. Cancers 2019, 11, 2018. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.N.; Voronov, E. Immunotherapeutic approaches of IL-1 neutralization in the tumor micro-environment. J. Leukoc. Biol. 2017, 102, 293–306. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Peng, Z.; Li, X.; Yan, Z.; Yang, Y.; Qiao, Z.; Liu, Y. Neutralization of TNFalpha in tumor with a novel nanobody potentiates paclitaxel-therapy and inhibits metastasis in breast cancer. Cancer Lett. 2017, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Voigt, C.; May, P.; Gottschlich, A.; Markota, A.; Wenk, D.; Gerlach, I.; Voigt, S.; Stathopoulos, G.T.; Arendt, K.A.M.; Heise, C.; et al. Cancer cells induce interleukin-22 production from memory CD4+ T cells via interleukin-1 to promote tumor growth. Proc. Natl. Acad. Sci. USA 2017, 114, 12994–12999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kak, G.; Raza, M.; Tiwari, B.K. Interferon-γ (IFN-γ): Exploring its implications in infectious diseases. Biomol. Concepts 2018, 9, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Alspach, E.; Lussier, D.M.; Schreiber, R.D. Interferon γ and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harb. Perspect. Biol. 2019, 11, a028480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Γ at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozman, P.; Svajger, U. The tolerogenic role of IFN-γ. Cytokine Growth Factor Rev. 2018, 41, 40–53. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regres-sion: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, S.; Srivastava, R.K.; Nandi, A.; Thacker, G.; Murali, H.; Kim, S.; Baldeon, M.; Tobias, J.; Blanco, M.A.; et al. Loss of ELF5-FBXW7 stabilizes IFNGR1 to promote the growth and metastasis of tri-ple-negative breast cancer through interferon-γ signalling. Nat. Cell Biol. 2020, 22, 591–602. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, Y.; Qu, Q.; Zhu, J.; Liu, Z.; Ning, W.; Zeng, H.; Zhang, N.; Du, W.; Chen, C.; et al. PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int. J. Clin. Oncol. 2017, 22, 1026–1033. [Google Scholar] [CrossRef]
- Zaidi, M.R. The Interferon-gamma Paradox in Cancer. J. Interferon Cytokine Res. 2019, 39, 30–38. [Google Scholar] [CrossRef]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. TNFalpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017, 8, 2256. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Shen, Q.; Tong, J.; Huang, L.; Cheng, Y.; Zhong, W. Anti-Tumor Necrosis Factor Receptor 2 Anti-body Combined with Anti-PD-L1 Therapy Exerts Robust Antitumor Effects in Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 720472. [Google Scholar] [CrossRef] [PubMed]
- Case, K.; Tran, L.; Yang, M.; Zheng, H.; Kuhtreiber, W.M.; Faustman, D.L. TNFR2 blockade alone or in combination with PD-1 blockade shows therapeutic efficacy in murine cancer models. J. Leukoc. Biol. 2020, 107, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lao, M.; Xu, J.; Duan, Y.; Yang, H.; Li, M.; Ying, H.; He, L.; Sun, K.; Guo, C.; et al. Combination cancer immunotherapy targeting TNFR2 and PD-1/PD-L1 signaling reduces immunosuppressive effects in the microenvironment of pancreatic tumors. J. Immunother. Cancer 2022, 10, e003982. [Google Scholar] [CrossRef] [PubMed]
- Baram, T.; Erlichman, N.; Dadiani, M.; Balint-Lahat, N.; Pavlovski, A.; Meshel, T.; Morzaev-Sulzbach, D.; Gal-Yam, E.N.; Barshack, I.; Ben-Baruch, A. Chemotherapy Shifts the Balance in Favor of CD8+ TNFR2+ TILs in Triple-Negative Breast Tumors. Cells 2021, 10, 1429. [Google Scholar] [CrossRef]
- Dadiani, M.; Necula, D.; Kahana-Edwin, S.; Oren, N.; Baram, T.; Marin, I.; Morzaev-Sulzbach, D.; Pavlovski, A.; Balint-Lahat, N.; Anafi, L.; et al. TNFR2+ TILs are significantly associated with improved survival in triple-negative breast cancer patients. Cancer Immunol. Immunother. 2020, 69, 1315–1326. [Google Scholar] [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and syner-gizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Yati, S.; Silathapanasakul, A.; Thakaeng, C.; Chanasakulniyom, M.; Songtawee, N.; Porntadavity, S.; Pothacharoen, P.; Pruksakorn, D.; Kongtawelert, P.; Yenchitsomanus, P.T.; et al. Extracellular Vesi-cle-Mediated IL-1 Signaling in Response to Doxorubicin Activates PD-L1 Expression in Osteo-sarcoma Models. Cells 2022, 11, 1042. [Google Scholar] [CrossRef]
- Zong, Z.; Zou, J.; Mao, R.; Ma, C.; Li, N.; Wang, J.; Wang, X.; Zhou, H.; Zhang, L.; Shi, Y. M1 Macrophages Induce PD-L1 Expression in Hepatocellular Carcinoma Cells Through IL-1beta Signaling. Front. Immunol. 2019, 10, 1643. [Google Scholar] [CrossRef] [Green Version]
- Baptista, M.Z.; Sarian, L.O.; Derchain, S.F.; Pinto, G.A.; Vassallo, J. Prognostic significance of PD-L1 and PD-L2 in breast cancer. Hum. Pathol. 2016, 47, 78–84. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, X.; Sun, L.; Mao, Y. Correlation Between PD-L2 Expression and Clinical Outcome in Solid Cancer Patients: A Meta-Analysis. Front. Oncol. 2019, 9, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, M.T.; Anderson, K.S.; Lenkiewicz, E.; Andreozzi, M.; Cunliffe, H.E.; Klassen, C.L.; Dueck, A.C.; McCullough, A.E.; Reddy, S.K.; Ramanathan, R.K.; et al. Genomic amplification of 9p24.1 targeting JAK2, PD-L1, and PD-L2 is enriched in high-risk triple negative breast cancer. Oncotarget 2015, 6, 26483–26493. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.; Lee, N.; Park, M.J.; Jeon, K.; Song, W. Currently Used Laboratory Methodologies for Assays Detecting PD-1, PD-L1, PD-L2 and Soluble PD-L1 in Patients with Metastatic Breast Cancer. Cancers 2021, 13, 5225. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Aiello, M.; Rozali, E.; Lambertini, M.; Willard-Gallo, K.; Migliori, E. Programmed cell death-ligand 2: A neglected but important target in the immune response to cancer? Transl. Oncol. 2020, 13, 100811. [Google Scholar] [CrossRef]
- Van Zee, K.J.; Kohno, T.; Fischer, E.; Rock, C.S.; Moldawer, L.L.; Lowry, S.F. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor alpha in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 4845–4849. [Google Scholar] [CrossRef] [Green Version]
- van Mierlo, G.J.; Scherer, H.U.; Hameetman, M.; Morgan, M.E.; Flierman, R.; Huizinga, T.W.; Toes, R.E. Cutting edge: TNFR-shedding by CD4+CD25+ regulatory T cells inhibits the induction of inflammatory mediators. J. Immunol. 2008, 180, 2747–2751. [Google Scholar] [CrossRef] [Green Version]
- DeBerge, M.P.; Ely, K.H.; Wright, P.F.; Thorp, E.B.; Enelow, R.I. Shedding of TNF receptor 2 by effector CD8+ T cells by ADAM17 is important for regulating TNF-α availability during influenza infection. J. Leukoc. Biol. 2015, 98, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rego, S.L.; Swamydas, M.; Kidiyoor, A.; Helms, R.; De Piante, A.; Lance, A.L.; Mukherjee, P.; Dreau, D. Soluble tumor necrosis factor receptors shed by breast tumor cells inhibit macrophage chemotaxis. J. Interferon Cytokine Res. 2013, 33, 672–681. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Mac-rophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Shichkin, V.P.; Lon, A.D.; Yugrinova, L.G.; Grinevich, Y.A.; Belova, O.B.; Berezhnaya, N.M.; Akalovich, S.; Pashkova, O.; Voitenok, N.N. TNF receptor p55 and IL-8(72) and IL-8(77) isoforms: Blood and urine levels in breast cancer patients. J. Immunotoxicol. 2009, 6, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.K.; Wong, A.L.; Wong, F.L.; Breen, E.C.; Hurria, A.; Smith, M.; Kinjo, C.; Paz, I.B.; Kruper, L.; Somlo, G.; et al. Inflammatory Biomarkers, Comorbidity, and Neurocognition in Women with Newly Diagnosed Breast Cancer. J. Natl. Cancer Inst. 2015, 107, djv131. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, J.; Zemmler, M.; Abrahamsson, A.; Lundberg, P.; Forsgren, M.; Dabrosin, C. Breast Density and Estradiol Are Major Determinants for Soluble TNF-TNF-R Proteins in vivo in Human Breast Tis-sue. Front. Immunol. 2022, 13, 850240. [Google Scholar] [CrossRef] [PubMed]
- Tesarova, P.; Kvasnicka, J.; Umlaufova, A.; Homolkova, H.; Jirsa, M.; Tesar, V. Soluble TNF and IL-2 re-ceptors in patients with breast cancer. Med. Sci. Monit. 2000, 6, 661–667. [Google Scholar]
- Fuksiewicz, M.; Kowalska, M.; Kotowicz, B.; Rubach, M.; Chechlinska, M.; Pienkowski, T.; Kaminska, J. Serum soluble tumour necrosis factor receptor type I concentrations independently predict prog-nosis in patients with breast cancer. Clin. Chem. Lab. Med. 2010, 48, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Hayslip, J.; Dressler, E.V.; Weiss, H.; Taylor, T.J.; Chambers, M.; Noel, T.; Miriyala, S.; Keeney, J.T.; Ren, X.; Sul-tana, R.; et al. Plasma TNF-α and Soluble TNF Receptor Levels after Doxorubicin with or without Co-Administration of Mesna-A Randomized, Cross-Over Clinical Study. PLoS ONE 2015, 10, e0124988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlichman, N.; Baram, T.; Meshel, T.; Morein, D.; Da’adoosh, B.; Ben-Baruch, A. Tumor Cell-Autonomous Pro-Metastatic Activities of PD-L1 in Human Breast Cancer Are Mediated by PD-L1-S283 and Chemokine Axes. Cancers 2022, 14, 1042. [Google Scholar] [CrossRef] [PubMed]
- Anaya-Eugenio, G.D.; Eggers, N.A.; Ren, Y.; Rivera-ChAvez, J.; Kinghorn, A.D.; Carcache, D.E.B.E.J. Apoptosis Induced by (+)-Betulin Through NF-kappaB Inhibition in MDA-MB-231 Breast Cancer Cells. Anticancer. Res. 2020, 40, 6637–6647. [Google Scholar] [CrossRef]
- Cai, Z.; Korner, M.; Tarantino, N.; Chouaib, S. IkaBα overexpression in human breast carcinoma MCF7 cells inhibits nuclear factor-kappaB activation but not tumor necrosis factor-alpha-induced apoptosis. J. Biol. Chem. 1997, 272, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mountziaris, P.M.; Tzouanas, S.N.; Mikos, A.G. Dose effect of tumor necrosis factor-alpha on in vitro osteogenic differentiation of mesenchymal stem cells on biodegradable polymeric microfiber scaffolds. Biomaterials 2010, 31, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- Naldini, A.; Filippi, I.; Miglietta, D.; Moschetta, M.; Giavazzi, R.; Carraro, F. Interleukin-1β regulates the migratory potential of MDAMB231 breast cancer cells through the hypoxia-inducible factor-1α. Eur. J. Cancer 2010, 46, 3400–3408. [Google Scholar] [CrossRef]
- Rossi, M.; Sharkey, A.M.; Vigano, P.; Fiore, G.; Furlong, R.; Florio, P.; Ambrosini, G.; Smith, S.K.; Petraglia, F. Identification of genes regulated by interleukin-1beta in human endometrial stromal cells. Reproduction 2005, 130, 721–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulter, K.R.; Wewers, M.D.; Lowe, M.P.; Knoell, D.L. Extracellular regulation of interleukin (IL)-1β through lung epithelial cells and defective IL-1 type II receptor expression. Am. J. Respir. Cell Mol. Biol. 1999, 20, 964–975. [Google Scholar] [CrossRef] [Green Version]
- Weidanz, J.A.; Nguyen, T.; Woodburn, T.; Neethling, F.A.; Chiriva-Internati, M.; Hildebrand, W.H.; Lust-garten, J. Levels of specific peptide-HLA class I complex predicts tumor cell susceptibility to CTL killing. J. Immunol. 2006, 177, 5088–5097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genin, M.; Clement, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 15, 577. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Danziger, O.; Pupko, T.; Bacharach, E.; Ehrlich, M. Interleukin-6 and Interferon-alpha Signaling via JAK1-STAT Differentially Regulate Oncolytic versus Cytoprotective Antiviral States. Front. Immunol. 2018, 9, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimentel, F.F.; Chagas, E.R.; Tiezzi, M.G.; de Andrade, J.M.; Tiezzi, D.G. 171P-Tumour-Immune System Interactions in Breast Cancer: The Role of Pd-L1, Pd-L2 and Cd86 Expression. Ann. Oncol. 2014, 25, iv59. Available online: https://www.sciencedirect.com/science/article/pii/S0923753419516416 (accessed on 26 May 2022). [CrossRef]
- Taniguchi, K.; Karin, M. NF-kβ, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kβ signaling in inflammation. Signal Transduct. Target 2017, 2, 102660. [Google Scholar]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) pathway. Sci. Signal. 2010, 3, cm1. [Google Scholar] [CrossRef]
- Gregory, A.P.; Dendrou, C.A.; Attfield, K.E.; Haghikia, A.; Xifara, D.K.; Butter, F.; Poschmann, G.; Kaur, G.; Lambert, L.; Leach, O.A.; et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 2012, 488, 508–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lainez, B.; Fernandez-Real, J.M.; Romero, X.; Esplugues, E.; Canete, J.D.; Ricart, W.; Engel, P. Identification and characterization of a novel spliced variant that encodes human soluble tumor necrosis factor receptor 2. Int. Immunol. 2004, 16, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.; Broder, C.; Helmstetter, A.; Schmidt, S.; Yan, I.; Muller, M.; Schmidt-Arras, D.; Becker-Pauly, C.; Koch-Nolte, F.; Mittrucker, H.W.; et al. Short-term TNFalpha shedding is independent of cytoplasmic phosphorylation or furin cleavage of ADAM17. Biochim. Biophys. Acta 2013, 1833, 3355–3367. [Google Scholar] [CrossRef] [Green Version]
- Cook, E.B.; Stahl, J.L.; Graziano, F.M.; Barney, N.P. Regulation of the receptor for TNFalpha, TNFR1, in human conjunctival epithelial cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3992–3998. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.S.; McCann, P.P. Matrix metalloproteinase inhibition as a novel anticancer strategy: A review with special focus on batimastat and marimastat. Pharmacol. Ther. 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Lorenzen, I.; Lokau, J.; Korpys, Y.; Oldefest, M.; Flynn, C.M.; Kunzel, U.; Garbers, C.; Freeman, M.; Grotzinger, J.; Dusterhoft, S. Control of ADAM17 activity by regulation of its cellular localisation. Sci. Rep. 2016, 6, 35067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, P.M.; Baggiolini, M.; Charo, I.F.; Hebert, C.A.; Horuk, R.; Matsushima, K.; Miller, L.H.; Oppenheim, J.J.; Power, C.A. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 2000, 52, 145–176. [Google Scholar]
- Borsig, L.; Wolf, M.J.; Roblek, M.; Lorentzen, A.; Heikenwalder, M. Inflammatory chemokines and me-tastasis-tracing the accessory. Oncogene 2013, 33, 3217–3224. [Google Scholar] [CrossRef] [Green Version]
- Morein, D.; Erlichman, N.; Ben-Baruch, A. Beyond Cell Motility: The Expanding Roles of Chemokines and Their Receptors in Malignancy. Front. Immunol. 2020, 11, 952. [Google Scholar] [CrossRef]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [Green Version]
- Svensson, S.; Abrahamsson, A.; Rodriguez, G.V.; Olsson, A.K.; Jensen, L.; Cao, Y.; Dabrosin, C. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin. Cancer Res. 2015, 21, 3794–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, X.; Nawab, O.; Patel, T.; Kossenkov, A.V.; Halama, N.; Jaeger, D.; Pestell, R.G. Recent Advances Tar-geting CCR5 for Cancer and Its Role in Immuno-Oncology. Cancer Res. 2019, 79, 4801–4807. [Google Scholar] [CrossRef] [Green Version]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, G.; Ben-Baruch, A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008, 267, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Baram, T.; Rubinstein-Achiasaf, L.; Ben-Yaakov, H.; Ben-Baruch, A. Inflammation-Driven Breast Tumor Cell Plasticity: Stemness/EMT, Therapy Resistance and Dormancy. Front. Oncol. 2020, 10, 614468. [Google Scholar] [CrossRef]
- Todorovic-Rakovic, N.; Milovanovic, J. Interleukin-8 in breast cancer progression. J. Interferon Cytokine Res. 2013, 33, 563–570. [Google Scholar] [CrossRef]
- Kondo, A.; Yamashita, T.; Tamura, H.; Zhao, W.; Tsuji, T.; Shimizu, M.; Shinya, E.; Takahashi, H.; Tamada, K.; Chen, L.; et al. Interferon-γ and tumor necrosis factor-alpha induce an immunoinhibitory molecule, B7-H1, via nuclear factor-kappaB activation in blasts in myelodysplastic syndromes. Blood 2010, 116, 1124–1131. [Google Scholar] [CrossRef] [Green Version]
- Gowrishankar, K.; Gunatilake, D.; Gallagher, S.J.; Tiffen, J.; Rizos, H.; Hersey, P. Inducible but not consti-tutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kβ. PLoS ONE 2015, 10, e0123410. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Szondy, Z.; Pallai, A. Transmembrane TNF-α reverse signaling leading to TGF-beta production is selectively activated by TNF targeting molecules: Therapeutic implications. Pharmacol. Res. 2017, 115, 124–132. [Google Scholar] [CrossRef]
- Qu, Y.; Zhao, G.; Li, H. Forward and Reverse Signaling Mediated by Transmembrane Tumor Necrosis Factor-Alpha and TNF Receptor 2: Potential Roles in an Immunosuppressive Tumor Microenvi-ronment. Front. Immunol. 2017, 8, 1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Yan, D.; Shi, X.; Liang, H.; Pang, Y.; Qin, N.; Chen, H.; Wang, J.; Yin, B.; Jiang, X.; et al. Transmem-brane TNF-α mediates “forward” and “reverse” signaling, inducing cell death or survival via the NF-kβ pathway in Raji Burkitt lymphoma cells. J. Leukoc. Biol. 2008, 84, 789–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willrich, M.A.; Murray, D.L.; Snyder, M.R. Tumor necrosis factor inhibitors: Clinical utility in autoim-mune diseases. Transl. Res. 2015, 165, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Billmeier, U.; Dieterich, W.; Neurath, M.F.; Atreya, R. Molecular mechanism of action of anti-tumor ne-crosis factor antibodies in inflammatory bowel diseases. World J. Gastroenterol. 2016, 22, 9300–9313. [Google Scholar] [CrossRef] [PubMed]
- Montfort, A.; Filleron, T.; Virazels, M.; Dufau, C.; Milhes, J.; Pages, C.; Olivier, P.; Ayyoub, M.; Mounier, M.; Lusque, A.; et al. Combining Nivolumab and Ipilimumab with Infliximab or Certolizumab in Pa-tients with Advanced Melanoma: First Results of a Phase Ib Clinical Trial. Clin. Cancer Res. 2021, 27, 1037–1047. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baram, T.; Oren, N.; Erlichman, N.; Meshel, T.; Ben-Baruch, A. Inflammation-Driven Regulation of PD-L1 and PD-L2, and Their Cross-Interactions with Protective Soluble TNFα Receptors in Human Triple-Negative Breast Cancer. Cancers 2022, 14, 3513. https://doi.org/10.3390/cancers14143513
Baram T, Oren N, Erlichman N, Meshel T, Ben-Baruch A. Inflammation-Driven Regulation of PD-L1 and PD-L2, and Their Cross-Interactions with Protective Soluble TNFα Receptors in Human Triple-Negative Breast Cancer. Cancers. 2022; 14(14):3513. https://doi.org/10.3390/cancers14143513
Chicago/Turabian StyleBaram, Tamir, Nino Oren, Nofar Erlichman, Tsipi Meshel, and Adit Ben-Baruch. 2022. "Inflammation-Driven Regulation of PD-L1 and PD-L2, and Their Cross-Interactions with Protective Soluble TNFα Receptors in Human Triple-Negative Breast Cancer" Cancers 14, no. 14: 3513. https://doi.org/10.3390/cancers14143513