
Monitoring of Measurable Residual Disease Using Circulating DNA after Allogeneic Hematopoietic Cell Transplantation

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
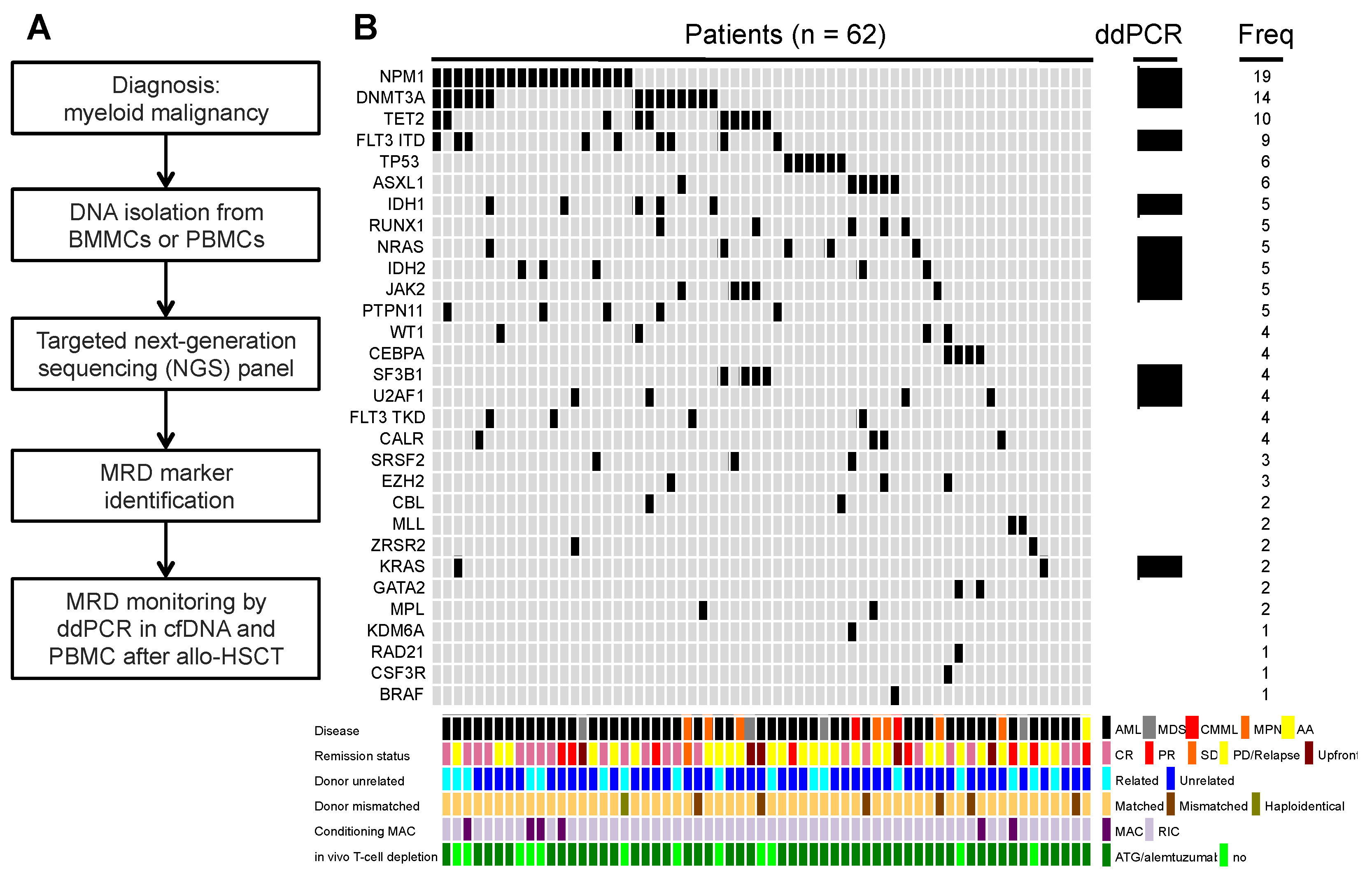
2. Materials and Methods
2.1. Patient Samples
2.2. DNA Isolation
2.3. Next-Generation Sequencing
2.4. Chimerism Testing and Mutational Analysis
2.5. Statistical Analysis
3. Results
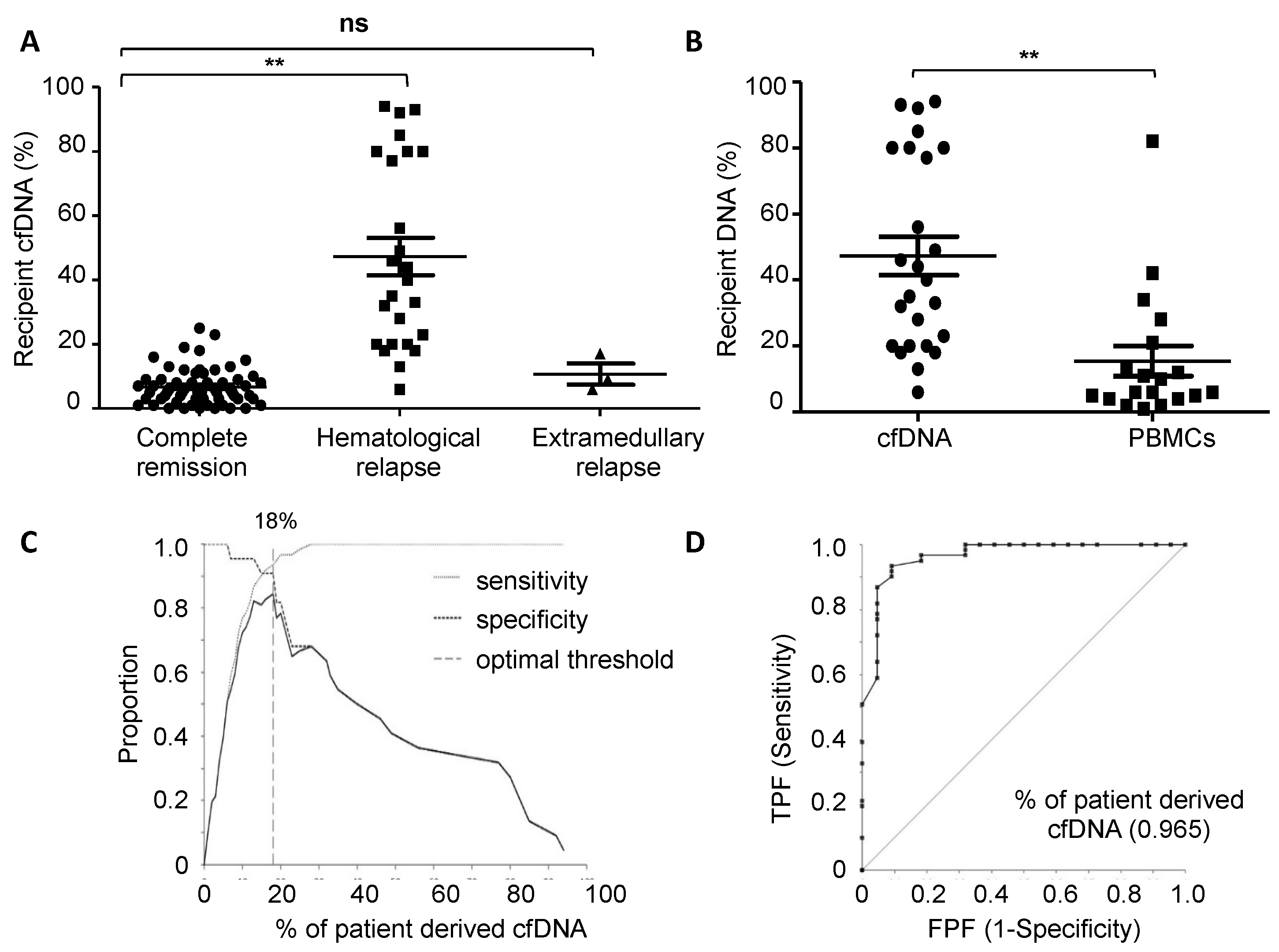
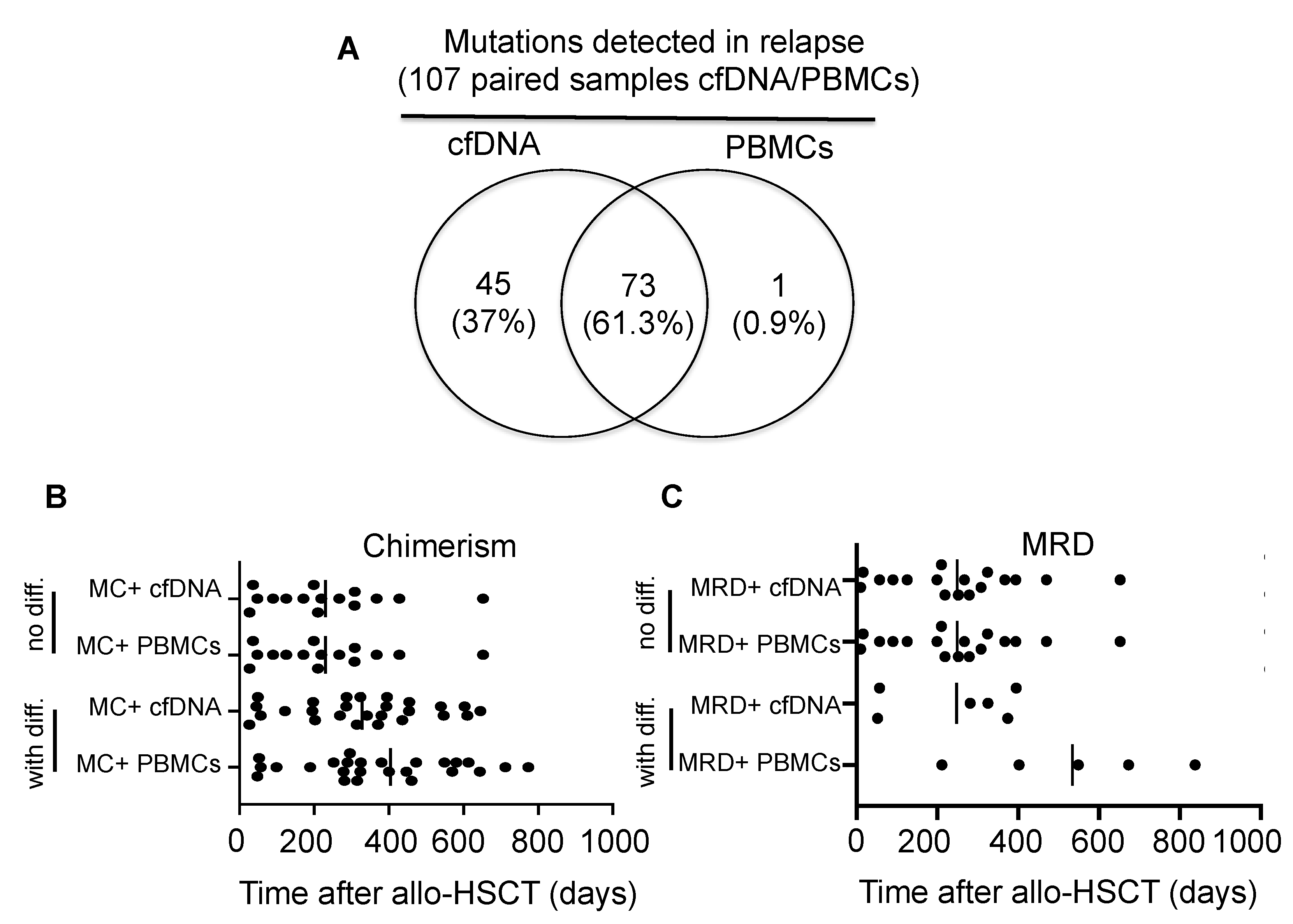
3.1. Chimerism Analysis of cfDNA at Relapse
3.2. MRD Monitoring in cfDNA and PBMCs in Patients Relapsing after Allo-HSCT
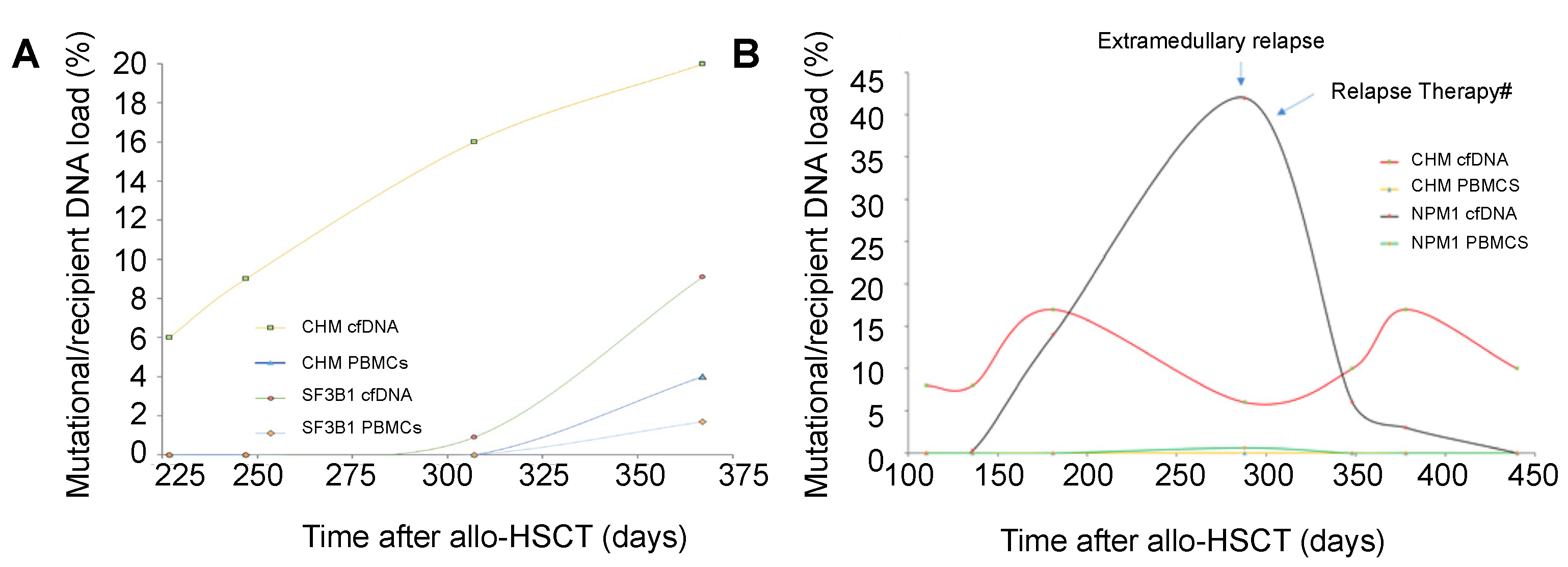
3.3. Chimerism and Mutation Kinetics in cfDNA after Transplantation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barrett, A.J.; Battiwalla, M. Relapse after allogeneic stem cell transplantation. Expert Rev. Hematol. 2010, 3, 429–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rhee, F.; Lin, F.; Cullis, J.O.; Spencer, A.; Cross, N.C.; Chase, A.; Garicochea, B.; Bungey, J.; Barrett, J.; Goldman, J.M. Relapse of chronic myeloid leukemia after allogeneic bone marrow transplant: The case for giving donor leukocyte transfusions before onset of hematologic relapse. Blood 1994, 83, 3377–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiser, R.; Beelen, D.W.; Bethge, W.; Bornhäuser, M.; Bug, G.; Burchert, A.; Christopeit, M.; Duyster, J.; Finke, J.; Gerbitz, A.; et al. Biology-Driven Approaches to Prevent and Treat Relapse of Myeloid Neoplasia after Allogeneic Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, e128–e140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, W.; Voskova, D.; Schoch, C.; Hiddemann, W.; Schnittger, S.; Haferlach, T. Determination of relapse risk based on assessment of minimal residual disease during complete remission by multiparameter flow cytometry in unselected patients with acute myeloid leukemia. Blood 2004, 104, 3078–3085. [Google Scholar] [CrossRef] [Green Version]
- Schnittger, S.; Weisser, M.; Schoch, C.; Hiddemann, W.; Haferlach, T.; Kern, W. New score predicting for prognosis in PML-RARA+, AML1-ETO+, or CBFBMYH11+ acute myeloid leukemia based on quantification of fusion transcripts. Blood 2003, 102, 2746–2755. [Google Scholar] [CrossRef] [Green Version]
- Weisser, M.; Kern, W.V.; Rauhut, S.; Schoch, C.L.; Hiddemann, W.; Haferlach, T.; Schnittger, S. Prognostic impact of RT-PCR-based quantification of WT1 gene expression during MRD monitoring of acute myeloid leukemia. Leukemia 2005, 19, 1416–1423. [Google Scholar] [CrossRef]
- Waterhouse, M.; Pfeifer, D.; Follo, M.; Duyster, J.; Schäfer, H.; Bertz, H.; Finke, J. Early mixed hematopoietic chimerism detection by digital droplet PCR in patients undergoing gender-mismatched hematopoietic stem cell transplantation. Clin. Chem. Lab. Med. (CCLM) 2017, 55, 1115–1121. [Google Scholar] [CrossRef]
- Waterhouse, M.; Pfeifer, D.; Duque-Afonso, J.; Follo, M.; Duyster, J.; Depner, M.; Bertz, H.; Finke, J. Droplet digital PCR for the simultaneous analysis of minimal residual disease and hematopoietic chimerism after allogeneic cell transplantation. Clin. Chem. Lab. Med. (CCLM) 2018, 57, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Shumilov, E.; Wiedemann, G.; Porret, N.; Shakhanova, I.; Bürki, S.; Legros, M.; Joncourt, R.; Pabst, T.; Bacher, U. Clinical potential of introducing next-generation sequencing in patients at relapse of acute myeloid leukemia. Hematol. Oncol. 2020, 38, 425–431. [Google Scholar] [CrossRef]
- Rosso, A.; Juliusson, G.; Lorenz, F.; Lehmann, S.; Derolf, Å.; Deneberg, S.; Jädersten, M.; Antunovic, P.; Cammenga, J.; Möllgård, L.; et al. Is there an impact of measurable residual disease as assessed by multiparameter flow cytometry on survival of AML patients treated in clinical practice? A population-based study. Leuk. Lymphoma 2021, 62, 1973–1981. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthias, C.; Dignan, F.L.; Morilla, R.; Morilla, A.; Ethell, M.E.; Potter, M.N.; Shaw, B.E. Pre-transplant MRD predicts outcome following reduced-intensity and myeloablative allogeneic hemopoietic SCT in AML. Bone Marrow Transplant. 2014, 49, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, R.; Hills, R.; Freeman, S.; Potter, N.; Jovanovic, J.; Ivey, A.; Kanda, A.S.; Runglall, M.; Foot, N.; Valganon, M.; et al. Molecular MRD status and outcome after transplantation in NPM1-mutated AML. Blood 2020, 135, 680–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, R.B.; Gooley, T.A.; Wood, B.L.; Milano, F.; Fang, M.; Sorror, M.L.; Estey, E.H.; Salter, A.I.; Lansverk, E.; Chien, J.W.; et al. Impact of pretransplantation minimal residual disease, as detected by multiparametric flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 1190–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, M.; McGowan, K.S.; Lu, K.; Jain, N.; Candia, J.; Hensel, N.F.; Tang, J.; Calvo, K.R.; Battiwalla, M.; Barrett, A.J.; et al. A multigene array for measurable residual disease detection in AML patients undergoing SCT. Bone Marrow Transplant. 2015, 50, 642–651. [Google Scholar] [CrossRef] [Green Version]
- Thol, F.; Gabdoulline, R.; Liebich, A.; Klement, P.; Schiller, J.; Kandziora, C.; Hambach, L.; Stadler, M.; Koenecke, C.; Flintrop, M.; et al. Measurable residual disease monitoring by NGS before allogeneic hematopoietic cell transplantation in AML. Blood 2018, 132, 1703–1713. [Google Scholar] [CrossRef] [Green Version]
- Aljurf, M.; Abalkhail, H.; Alseraihy, A.; Mohamed, S.Y.; Ayas, M.; Alsharif, F.; Alzahrani, H.; Al-Jefri, A.; Aldawsari, G.; Al-Ahmari, A.; et al. Chimerism Analysis of Cell-Free DNA in Patients Treated with Hematopoietic Stem Cell Transplantation May Predict Early Relapse in Patients with Hematologic Malignancies. Biotechnol. Res. Int. 2016, 2016, 8589270. [Google Scholar] [CrossRef]
- Lui, Y.Y.; Chik, K.W.; Chiu, R.W.; Ho, C.Y.; Lam, C.W.; Lo, Y.M. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Duque-Afonso, J.; Waterhouse, M.; Pfeifer, D.; Follo, M.; Duyster, J.; Bertz, H.; Finke, J. Cell-free DNA characteristics and chimerism analysis in patients after allogeneic cell transplantation. Clin. Biochem. 2018, 52, 137–141. [Google Scholar] [CrossRef]
- Nakamura, S.; Yokoyama, K.; Shimizu, E.; Yusa, N.; Kondoh, K.; Ogawa, M.; Takei, T.; Kobayashi, A.; Ito, M.; Isobe, M.; et al. Prognostic impact of circulating tumor DNA status post–allogeneic hematopoietic stem cell transplantation in AML and MDS. Blood 2019, 133, 2682–2695. [Google Scholar] [CrossRef]
- Herrera, A.F.; Kim, H.T.; Kong, K.A.; Faham, M.; Sun, H.; Sohani, A.R.; Alyea, E.P.; Carlton, V.E.; Chen, Y.-B.; Cutler, C.S.; et al. Next-generation sequencing-based detection of circulating tumour DNA After allogeneic stem cell transplantation for lymphoma. Br. J. Haematol. 2016, 175, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, M.; Pennisi, S.; Pfeifer, D.; Deuter, M.; von Bubnoff, N.; Scherer, F.; Strüssmann, T.; Wehr, C.; Duyster, J.; Bertz, H.; et al. Colon and liver tissue damage detection using methylated SESN3 and PTK2B genes in circulating cell-free DNA in patients with acute graft-versus-host disease. Bone Marrow Transplant. 2020, 56, 327–333. [Google Scholar] [CrossRef]
- Marks, R.; Potthoff, K.; Hahn, J.; Ihorst, G.; Bertz, H.; Spyridonidis, A.; Holler, E.; Finke, J.M. Reduced-toxicity conditioning with fludarabine, BCNU, and melphalan in allogeneic cell transplantation: Particular activity against advanced hematologic malignancies. Blood 2008, 112, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Jacobsohn, D.A.; Loken, M.R.; Fei, M.; Adams, A.; Brodersen, L.E.; Logan, B.R.; Ahn, K.W.; Shaw, B.E.; Kletzel, M.; Olszewski, M.; et al. Outcomes of Measurable Residual Disease in Pediatric Acute Myeloid Leukemia before and after Hematopoietic Stem Cell Transplant: Validation of Difference from Normal Flow Cytometry with Chimerism Studies and Wilms Tumor 1 Gene Expression. Biol. Blood Marrow Transplant. 2018, 24, 2040–2046. [Google Scholar] [CrossRef] [Green Version]
- Araki, D.; Wood, B.L.; Othus, M.; Radich, J.P.; Halpern, A.B.; Zhou, Y.; Mielcarek, M.; Estey, E.H.; Appelbaum, F.R.; Walter, R.B. Allogeneic Hematopoietic Cell Transplantation for Acute Myeloid Leukemia: Time to Move Toward a Minimal Residual Disease–Based Definition of Complete Remission? J. Clin. Oncol. 2016, 34, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-J.; Cheng, W.-Y.; Lin, X.-J.; Wang, S.-Y.; Jiang, T.-Y.; Ma, T.-T.; Zhu, Y.-M.; Shen, Y. Measurable Residual Disease Detected by Multiparameter Flow Cytometry and Sequencing Improves Prediction of Relapse and Survival in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 677833. [Google Scholar] [CrossRef] [PubMed]
- Rausch, C.; Rothenberg-Thurley, M.; Buerger, S.A.; Tschuri, S.; Dufour, A.; Neusser, M.; Schneider, S.; Spiekermann, K.; Metzeler, K.H.; Ziemann, F. Double Drop-Off Droplet Digital PCR: A Novel, Versatile Tool for Mutation Screening and Residual Disease Monitoring in Acute Myeloid Leukemia Using Cellular or Cell-Free DNA. J. Mol. Diagn. 2021, 23, 975–985. [Google Scholar] [CrossRef]
- Short, N.J.; Patel, K.P.; Albitar, M.; Franquiz, M.; Luthra, R.; Kanagal-Shamanna, R.; Wang, F.; Assi, R.; Montalban-Bravo, G.; Matthews, J.; et al. Targeted next-generation sequencing of circulating cell-free DNA vs bone marrow in patients with acute myeloid leukemia. Blood Adv. 2020, 4, 1670–1677. [Google Scholar] [CrossRef]
- Gai, W.; Ji, L.; Lam, W.K.J.; Sun, K.; Jiang, P.; Chan, A.W.H.; Wong, J.; Lai, P.B.S.; Ng, S.S.M.; Ma, B.B.Y.; et al. Liver- and Colon-Specific DNA Methylation Markers in Plasma for Investigation of Colorectal Cancers with or without Liver Metastases. Clin. Chem. 2018, 64, 1239–1249. [Google Scholar] [CrossRef]
- Van Ginkel, J.H.; Huibers, M.M.H.; van Es, R.J.J.; de Bree, R.; Willems, S.M. Droplet digital PCR for detection and quantification of circulating tumor DNA in plasma of head and neck cancer patients. BMC Cancer 2017, 17, 428. [Google Scholar] [CrossRef]
- Maier, J.; Lange, T.; Kerle, I.; Specht, K.; Bruegel, M.; Wickenhauser, C.; Jost, P.; Niederwieser, D.; Peschel, C.; Duyster, J.; et al. Detection of mutant free circulating tumor DNA in the plasma of patients with gastrointestinal stromal tumor harboring activating mutations of CKIT or PDGFRA. Clin. Cancer Res. 2013, 19, 4854–4867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, C.J.; Kennedy, J.A.; Nikiforow, S.; Kuo, F.C.; Alyea, E.P.; Ho, V.; Ritz, J.; Soiffer, R.; Antin, J.H.; Lindsley, R.C. Donor-engrafted CHIP is common among stem cell transplant recipients with unexplained cytopenias. Blood 2017, 130, 91–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean follow-up in days (range) | 827 (52–4363) |
| Gender male/female | 37/25 |
| Mean age at transplant (range) | 57 (21–76) |
| Disease | |
| AML | 48 |
| MDS | 4 |
| MPN | 7 |
| CMML | 2 |
| AA | 1 |
| Karyotype | |
| Normal | 29 |
| Complex | 24 |
| No data | 9 |
| Remission status at allo-HSCT | |
| CR | 21 |
| Non-CR | 41 |
| - Partial remission | 8 |
| - Stable disease | 1 |
| - Progressive disease/relapse | 27 |
| - Upfront | 5 |
| Karnofsky index (%) | |
| 100 | 8 |
| 90 | 21 |
| 80 | 14 |
| ≤70 | 19 |
| Donor female/recipient male | 10 |
| HLA mismatch | |
| Yes | 7 |
| No | 55 |
| Donor type | |
| Related | 17 |
| Non related | 45 |
| Conditioning regimen | |
| Myeloablative | 5 |
| Reduced toxicity | 57 |
| GvHD prophylaxis | |
| CyA/MMF/ATG | 44 |
| CyA/MMF | 10 |
| CyA/MTX | 1 |
| Everolimus/MMF/ATG | 5 |
| CyA/MMF/cyclophosphamide | 1 |
| Mutation | N 1 | Tumor Load PBMCs | Tumor Load cfDNA | p-Value |
|---|---|---|---|---|
| FLT3-ITD | 20 | 0.2 | 0.5 | 0.01 |
| KRAS | 11 | 3.3 | 20 | 0.0014 |
| NPM1 | 28 | 1.5 | 5.4 | 0.002 |
| DNMT3A | 7 | 6.8 | 7.6 | 0.02 |
| SF3B1 | 9 | 3.3 | 4.2 | 0.4 |
| IDH1/2 | 22 | 5.9 | 9.7 | 0.3 |
| JAK2 | 8 | 10 | 17 | 0.2 |
| NRAS | 6 | 10 | 16 | 0.3 |
| U2AF1 | 8 | 3.1 | 15.8 | 0.06 |
| Patient | Age at Allo-HSCT (Years) | Disease | Donor | Conditio-Ning | MRD Pos. in cfDNA (Days) | MRD Pos. in PBMCs (Days) | Hematological Relapse (Days) | Outcome |
|---|---|---|---|---|---|---|---|---|
| #1 | 68 | tAML | Unrelated | Reduced | 325 | 673 | 802 | Death after 2. allo-HSCT |
| #2 | 23 | AML | Unrelated | Reduced | 374 | 402 | 877 (skin) | Alive after 3. allo-HSCT |
| #3 | 59 | AML | Related | Myeloa- blative | 57 | 211 | 1243 | Alive after 2. allo-HSCT |
| #4 | 60 | sAML | Related | Reduced | 281 | 838 | 366 (CNS-relapse) | Death after after 2. allo-HSCT |
| #5 | 52 | OMF | Unrelated | Reduced | 52 | Not detected | n.a. | Non-relapse mortality (pneumonia) |
| #6 | 66 | CMML | Related | Reduced | 395 | 549 | No hematological relapse | Alive at last follow-up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waterhouse, M.; Pennisi, S.; Pfeifer, D.; Scherer, F.; Zeiser, R.; Duyster, J.; Bertz, H.; Finke, J.; Duque-Afonso, J. Monitoring of Measurable Residual Disease Using Circulating DNA after Allogeneic Hematopoietic Cell Transplantation. Cancers 2022, 14, 3307. https://doi.org/10.3390/cancers14143307
Waterhouse M, Pennisi S, Pfeifer D, Scherer F, Zeiser R, Duyster J, Bertz H, Finke J, Duque-Afonso J. Monitoring of Measurable Residual Disease Using Circulating DNA after Allogeneic Hematopoietic Cell Transplantation. Cancers. 2022; 14(14):3307. https://doi.org/10.3390/cancers14143307
Chicago/Turabian StyleWaterhouse, Miguel, Sandra Pennisi, Dietmar Pfeifer, Florian Scherer, Robert Zeiser, Justus Duyster, Hartmut Bertz, Jürgen Finke, and Jesús Duque-Afonso. 2022. "Monitoring of Measurable Residual Disease Using Circulating DNA after Allogeneic Hematopoietic Cell Transplantation" Cancers 14, no. 14: 3307. https://doi.org/10.3390/cancers14143307