
HBV preS Mutations Promote Hepatocarcinogenesis by Inducing Endoplasmic Reticulum Stress and Upregulating Inflammatory Signaling

Abstract
:Simple Summary
Abstract
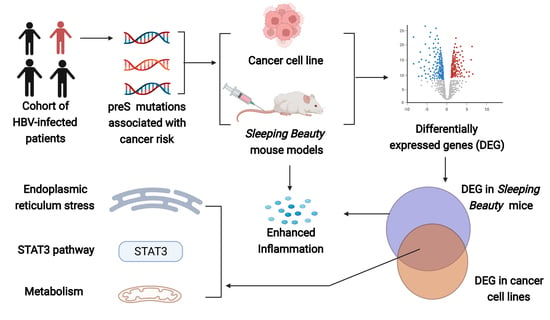
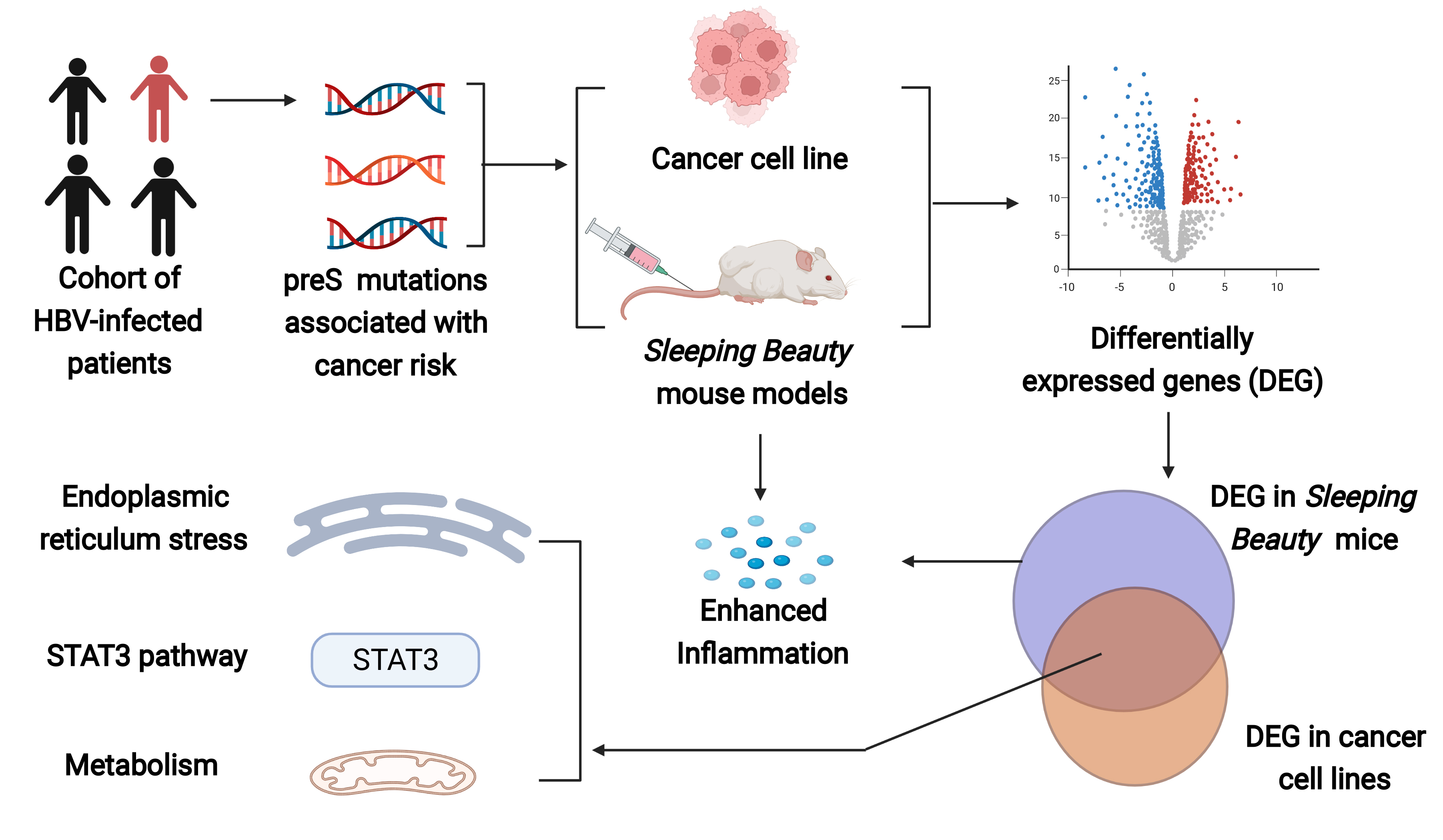
1. Background
2. Methods
2.1. Study Population Involved in the Prospective Cohort Study
2.2. Antiviral Treatment and Follow-Up
2.3. Serum HBV Genotyping and Viral Mutation Analysis
2.4. Plasmid Construction
2.5. Cell Experiments
2.6. Immunofluorescence Staining
2.7. Mouse Models
2.8. HBV-Capture Sequencing
2.9. Gene Expression Profiling Analysis
2.10. Statistical Analysis
3. Result
3.1. Effects of HBV preS Mutations on the Risk of HCC Occurrence
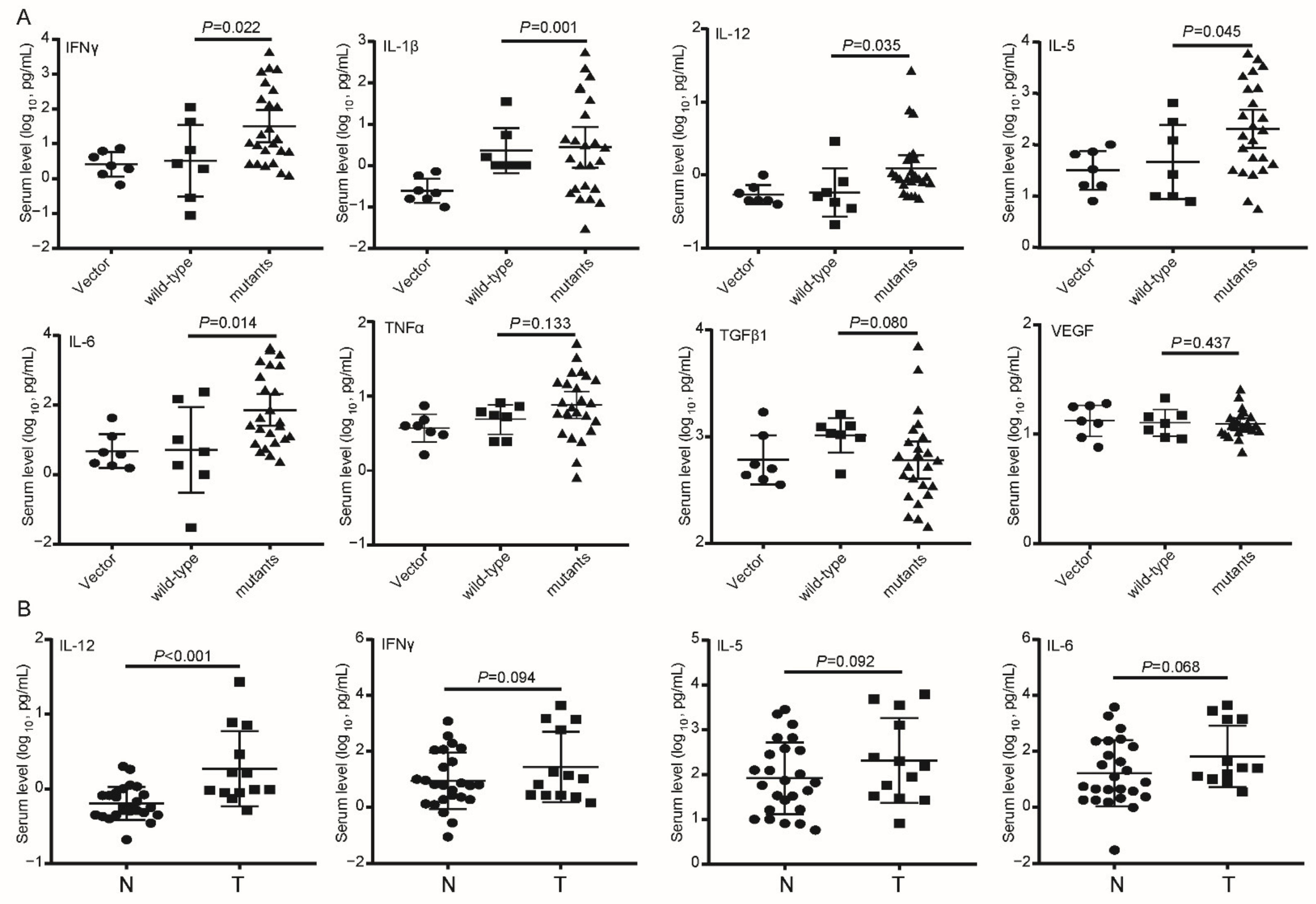
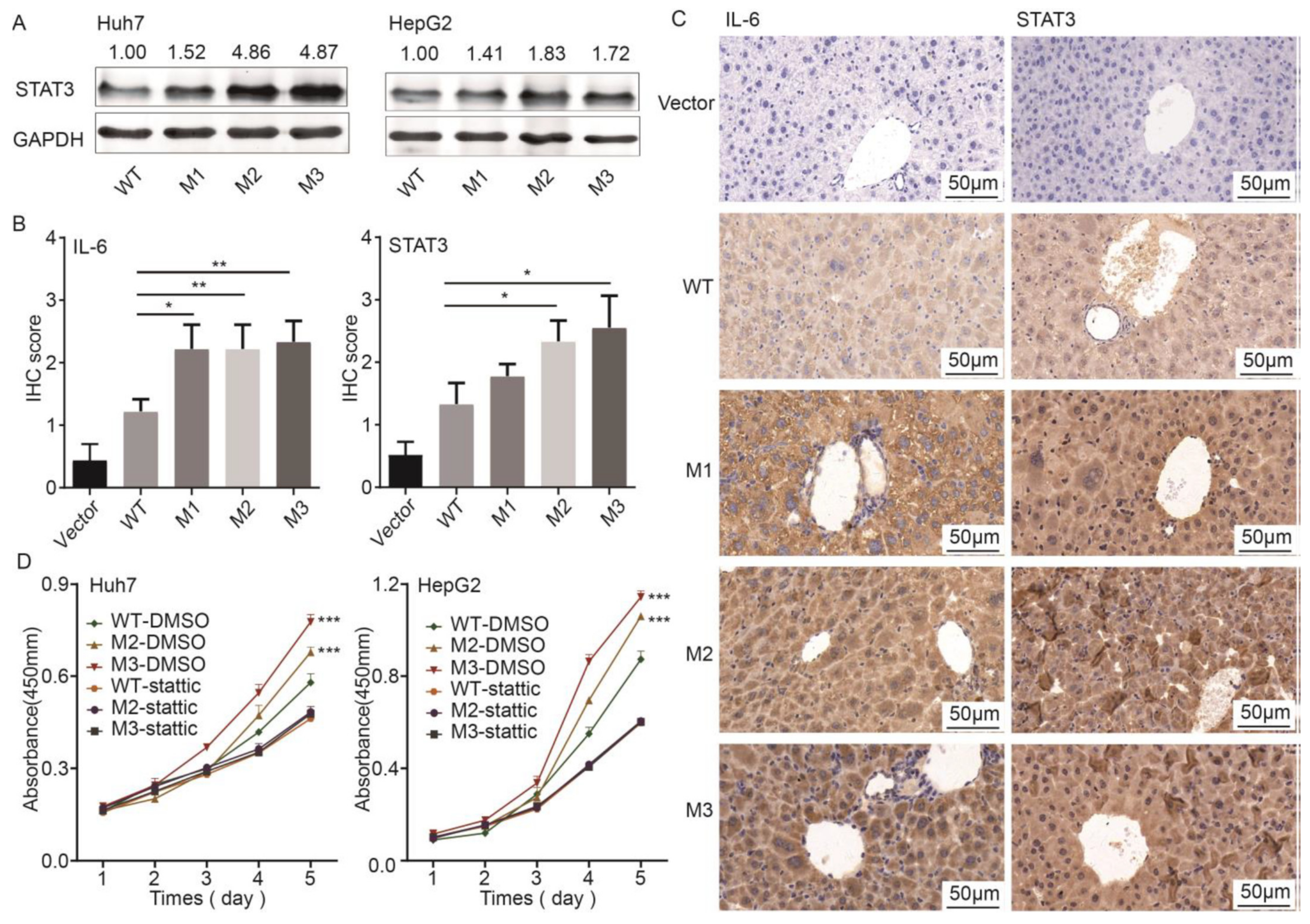
3.2. Effects of Wild-Type and Mutant preS1/preS2/S on Hepatocarcinogenesis and Inflammation in SB Mice
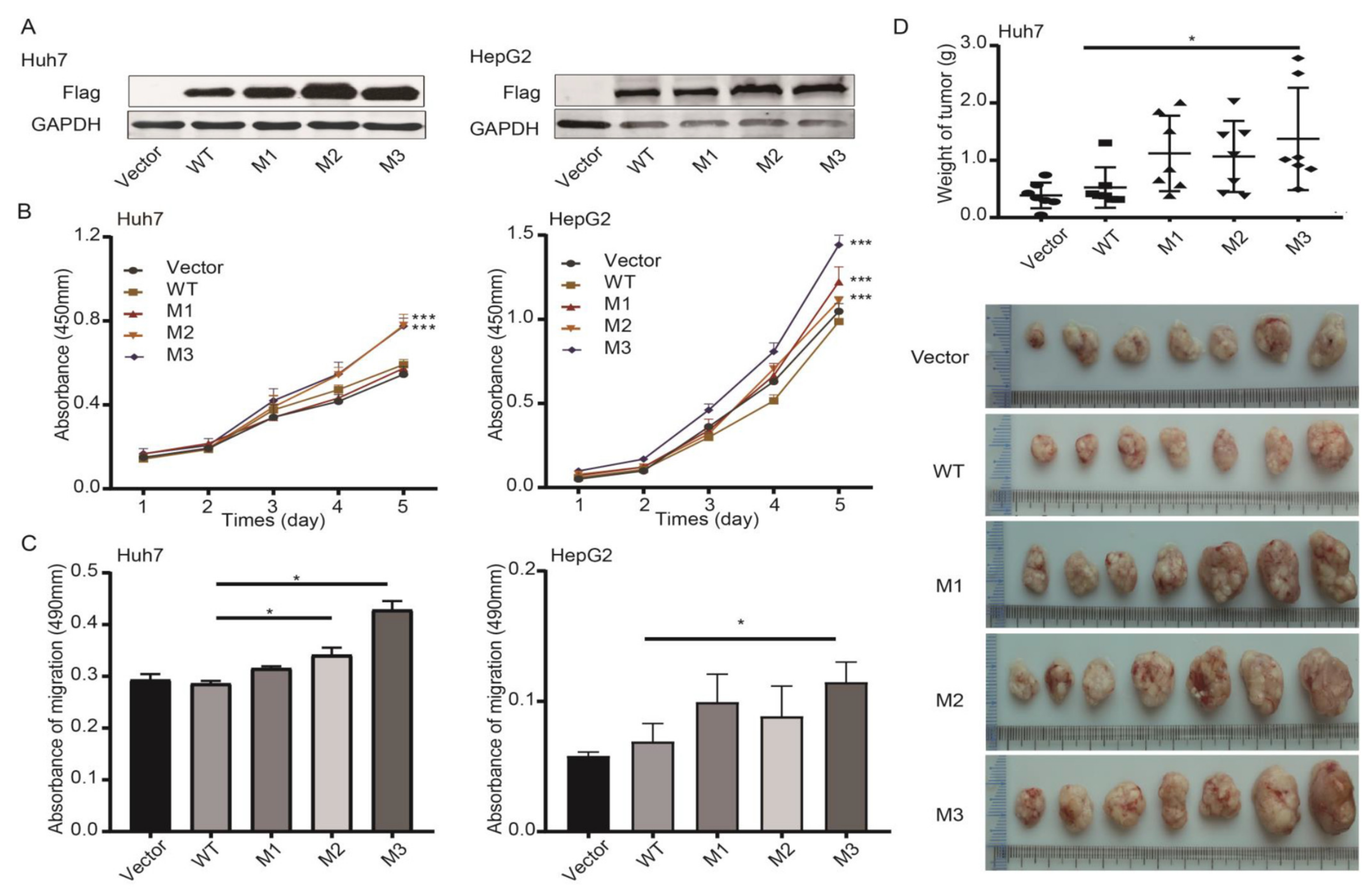
3.3. Effects of HBV preS1/preS2/S Mutants on Malignant Phenotypes of Cancer Cells
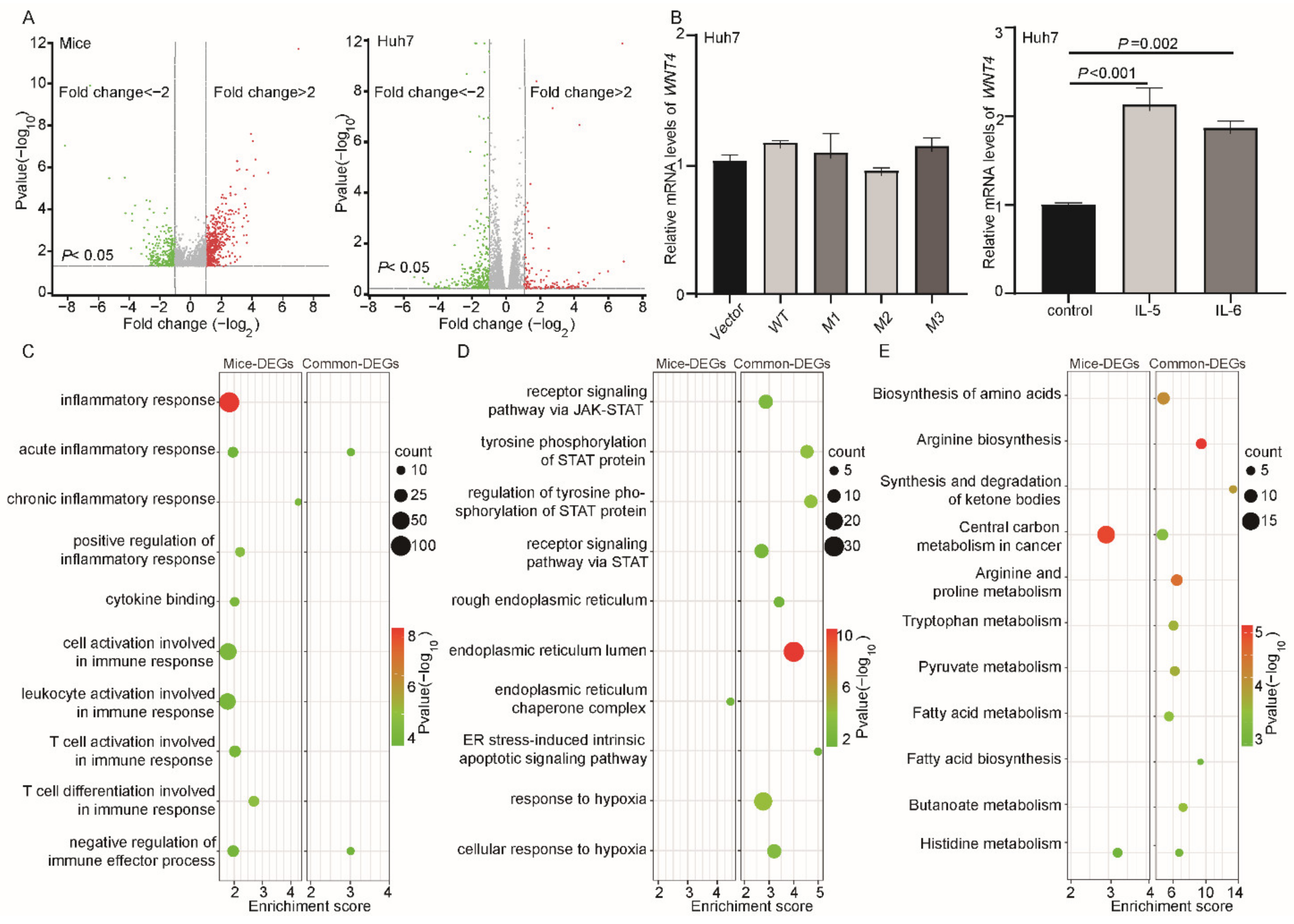
3.4. Potential Function of HBV preS Mutations
3.5. STAT3 Participated in the preS1/preS2/S Mutant-Induced Hepatocarcinogenesis
3.6. HBV preS1/preS2/S Mutants Enhanced ER Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.M.; Zhang, L.J.; Liu, W.B.; Ding, Y.B.; Yin, J.H.; Ren, R.B.; Li, Q.; Chen, Y.F.; Shen, J.Y.; Tan, X.J.; et al. Trends in cancer mortality in China from 2004 to 2018: A nationwide longitudinal study. Cancer Commun. 2021, 41, 1024–1036. [Google Scholar] [CrossRef]
- Rumgay, H.; Ferlay, J.; de Martel, C.; Georges, D.; Ibrahim, A.S.; Zheng, R.; Wei, W.; Lemmens, V.; Soerjomataram, I. Global, regional and national burden of primary liver cancer by subtype. Eur. J. Cancer 2022, 161, 108–118. [Google Scholar] [CrossRef]
- Lin, J.S.; Zhang, H.W.; Yu, H.P.; Bi, X.Y.; Zhang, W.L.; Yin, J.H.; Zhao, P.; Liang, X.M.; Qu, C.F.; Wang, M.J.; et al. Epidemiological characteristics of primary liver cancer in mainland China during 2003 and 2020: A representative multicenter study. Front. Oncol. 2022, 12, 906778. [Google Scholar] [CrossRef]
- Liu, W.B.; Wu, J.F.; Yang, F.; Ma, L.T.; Ni, C.; Hou, X.M.; Wang, L.; Xu, A.; Song, J.H.; Deng, Y.; et al. Genetic Polymorphisms Predisposing the Interleukin 6-Induced APOBEC3B-UNG Imbalance Increase HCC Risk via Promoting the Generation of APOBEC-Signature HBV Mutations. Clin. Cancer Res. 2019, 25, 5525–5536. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.H.; Chen, X.; Li, N.; Han, X.; Liu, W.B.; Pu, R.; Wu, T.; Ding, Y.B.; Zhang, H.W.; Zhao, J.; et al. Compartmentalized evolution of hepatitis B virus contributes differently to the prognosis of hepatocellular carcinoma. Carcinogenesis 2021, 42, 461–470. [Google Scholar] [CrossRef]
- Yin, J.H.; Zhang, H.W.; He, Y.C.; Xie, J.X.; Liu, S.J.; Chang, W.J.; Tan, X.J.; Gu, C.Y.; Lu, W.; Wang, H.Y.; et al. Distribution and hepatocellular carcinoma-related viral properties of hepatitis B virus genotypes in Mainland China: A community-based study. Cancer Epidemiol. Biomark. Prev. 2010, 19, 777–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mowafy, M.; Elgaml, A.; El-Mesery, M.; Elegezy, M. Molecular analysis of Hepatitis B virus sub-genotypes and incidence of preS1/preS2 region mutations in HBV-infected Egyptian patients from Mansoura. J. Med. Virol. 2017, 89, 1559–1566. [Google Scholar] [CrossRef]
- Yin, J.H.; Xie, J.X.; Zhang, H.W.; Shen, Q.X.; Han, L.; Lu, W.Y.; Han, Y.F.; Li, C.Z.; Ni, W.; Wang, H.Y.; et al. Significant association of different preS mutations with hepatitis B-related cirrhosis or hepatocellular carcinoma. J. Gastroenterol. 2010, 45, 1063–1071. [Google Scholar] [CrossRef]
- Chen, C.H.; Hung, C.H.; Lee, C.M.; Hu, T.H.; Wang, J.H.; Wang, J.C.; Lu, S.N.; Changchien, C.S. Pre-S deletion and complex mutations of hepatitis B virus related to advanced liver disease in HBeAg-negative patients. Gastroenterology 2007, 133, 1466–1474. [Google Scholar] [CrossRef]
- Huy, T.T.; Ushijima, H.; Win, K.M.; Luengrojanakul, P.; Shrestha, P.K.; Zhong, Z.H.; Smirnov, A.V.; Taltavull, T.C.; Sata, T.; Abe, K. High prevalence of hepatitis B virus pre-s mutant in countries where it is endemic and its relationship with genotype and chronicity. J. Clin. Microbiol. 2003, 41, 5449–5455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, M.F.; Li, N.; Pu, R.; Wu, T.; Ding, Y.B.; Cai, P.; Zhang, H.W.; Zhao, J.; Yin, J.H.; et al. Nucleotide variants in hepatitis B virus preS region predict the recurrence of hepatocellular carcinoma. Aging 2021, 13, 22256–22275. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.L.; Hung, J.H.; Huang, W. Association of the Hepatitis B Virus Large Surface Protein with Viral Infectivity and Endoplasmic Reticulum Stress-mediated Liver Carcinogenesis. Cells 2020, 9, 2052. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.H.; Su, I.J.; Lei, H.Y.; Wang, H.C.; Lin, W.C.; Chang, W.T.; Huang, W.; Chang, W.C.; Chang, Y.S.; Chen, C.C.; et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J. Biol. Chem. 2004, 279, 46384–46392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Xu, J.; Zhou, L.; Yun, X.; Chen, L.; Wang, S.; Sun, L.; Wen, Y.; Gu, J. Hepatitis B virus large surface antigen promotes liver carcinogenesis by activating the Src/PI3K/Akt pathway. Cancer Res. 2011, 71, 7547–7557. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.J.; Teng, W.; Chen, C.L.; Sun, C.P.; Teng, R.D.; Huang, Y.H.; Liang, K.H.; Chen, Y.W.; Lin, C.C.; Su, C.W.; et al. Clinical Implications of HBV PreS/S Mutations and the Effects of PreS2 Deletion on Mitochondria, Liver Fibrosis, and Cancer Development. Hepatology 2021, 74, 641–655. [Google Scholar] [CrossRef]
- Yin, J.H.; Li, N.; Han, Y.F.; Xue, J.; Deng, Y.; Shi, J.; Guo, W.X.; Zhang, H.W.; Wang, H.Y.; Cheng, S.Q.; et al. Effect of antiviral treatment with nucleotide/nucleoside analogs on postoperative prognosis of hepatitis B virus-related hepatocellular carcinoma: A two-stage longitudinal clinical study. J. Clin. Oncol. 2013, 31, 3647–3655. [Google Scholar] [CrossRef]
- Yin, J.H.; Wang, J.X.; Pu, R.; Xin, H.G.; Li, Z.X.; Han, X.; Ding, Y.B.; Du, Y.; Liu, W.B.; Deng, Y.; et al. Hepatitis B Virus Combo Mutations Improve the Prediction and Active Prophylaxis of Hepatocellular Carcinoma: A Clinic-Based Cohort Study. Cancer Prev. Res. 2015, 8, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Pu, R.; Liu, W.B.; Zhou, X.Y.; Chen, X.; Hou, X.M.; Cai, S.L.; Chen, L.P.; Wu, J.F.; Yang, F.; Tan, X.J.; et al. The Effects and Underlying Mechanisms of Hepatitis B Virus X Gene Mutants on the Development of Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 836517. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, J.; Lin, C.; Liu, W.; Huo, Y.; Yang, M.; Jiang, S.H.; Sun, Y.; Hua, R. Endoplasmic Reticulum stress-dependent expression of ERO1L promotes aerobic glycolysis in Pancreatic Cancer. Theranostics 2020, 10, 8400–8414. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Teng, C.F.; Wu, H.C.; Tsai, H.W.; Chuang, H.C.; Tsai, T.F.; Hsu, Y.H.; Huang, W.; Wu, L.W.; Su, I.J. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology 2009, 49, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Wang, Z.; Chowdhury, S.; Simadu, M.; Koura, M.; Muramatsu, M. Uracil DNA glycosylase counteracts APOBEC3G-induced hypermutation of hepatitis B viral genomes: Excision repair of covalently closed circular DNA. PLoS Pathog. 2013, 9, e1003361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.J.; Chen, E.Q.; Yang, J.H.; Zhou, T.Y.; Liu, L.; Tang, H. A mutation in the interferon regulatory element of HBV may influence the response of interferon treatment in chronic hepatitis B patients. Virol. J. 2012, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.C.; Neo, J.C.; Wu, J.C.; Chen, Y.F.; Kao, C.H.; Tsai, T.F. Expression of a hepatitis B virus pre-S2 deletion mutant in the liver results in hepatomegaly and hepatocellular carcinoma in mice. J. Pathol. 2017, 241, 463–474. [Google Scholar] [CrossRef]
- Sun, H.; Chang, L.; Yan, Y.; Wang, L. Hepatitis B virus pre-S region: Clinical implications and applications. Rev. Med. Virol. 2020, 31, e2201. [Google Scholar] [CrossRef]
- Yin, F.; Xie, Y.; Fan, H.; Zhang, J.; Guo, Z. Mutations in hepatitis B virus polymerase are associated with the postoperative survival of hepatocellular carcinoma patients. PLoS ONE 2017, 12, e0189730. [Google Scholar] [CrossRef] [Green Version]
- Teng, C.F.; Li, T.C.; Wang, T.; Liao, D.C.; Wen, Y.H.; Wu, T.H.; Wang, J.; Wu, H.C.; Shyu, W.C.; Su, I.J.; et al. Increased infiltration of regulatory T cells in hepatocellular carcinoma of patients with hepatitis B virus pre-S2 mutant. Sci. Rep. 2021, 11, 1136. [Google Scholar] [CrossRef]
- Teng, C.F.; Li, T.C.; Wang, T.; Wu, T.H.; Wang, J.; Wu, H.C.; Shyu, W.C.; Su, I.J.; Jeng, L.B. Increased Expression of Programmed Death Ligand 1 in Hepatocellular Carcinoma of Patients with Hepatitis B Virus Pre-S2 Mutant. J. Hepatocell. Carcinoma 2020, 7, 385–401. [Google Scholar] [CrossRef]
- Fu, J.; Xu, D.; Liu, Z.; Shi, M.; Zhao, P.; Fu, B.; Zhang, Z.; Yang, H.; Zhang, H.; Zhou, C.; et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 2007, 132, 2328–2339. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, X.Y.; Qiu, S.J.; Yamato, I.; Sho, M.; Nakajima, Y.; Zhou, J.; Li, B.Z.; Shi, Y.H.; Xiao, Y.S.; et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin. Cancer Res. 2009, 15, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploeger, C.; Schreck, J.; Huth, T.; Fraas, A.; Albrecht, T.; Charbel, A.; Ji, J.; Singer, S.; Breuhahn, K.; Pusch, S.; et al. STAT1 and STAT3 Exhibit a Crosstalk and Are Associated with Increased Inflammation in Hepatocellular Carcinoma. Cancers 2022, 14, 1154. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.; Kurt, R.; Song, K.; Lin, D.; Osman, H.; Youngquist, B.; Scott, J.W.; Shores, N.J.; Thevenot, P.; Cohen, A.; et al. Hepatic Stress Response in HCV Infection Promotes STAT3-Mediated Inhibition of HNF4A-miR-122 Feedback Loop in Liver Fibrosis and Cancer Progression. Cancers 2019, 11, 1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonnella, R.; Gilardini Montani, M.S.; Guttieri, L.; Romeo, M.A.; Santarelli, R.; Cirone, M. IRE1 Alpha/XBP1 Axis Sustains Primary Effusion Lymphoma Cell Survival by Promoting Cytokine Release and STAT3 Activation. Biomedicines 2021, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.X.; Sokol, E.S.; Del Vecchio, C.A.; Sanduja, S.; Claessen, J.H.; Proia, T.A.; Jin, D.X.; Reinhardt, F.; Ploegh, H.L.; Wang, Q.; et al. Epithelial-to-mesenchymal transition activates PERK-eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 2014, 4, 702–715. [Google Scholar] [CrossRef] [Green Version]
- Gonen, N.; Meller, A.; Sabath, N.; Shalgi, R. Amino Acid Biosynthesis Regulation during Endoplasmic Reticulum Stress Is Coupled to Protein Expression Demands. iScience 2019, 19, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P.; et al. Hypoxic activation of the PERK/eIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 2013, 19, 6126–6137. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Zang, R.; Lu, Z.; Zhang, G.; Huang, J.; Liu, C.; Wang, Z.; Mao, S.; Che, Y.; Wang, X.; et al. ERO1L promotes IL6/sIL6R signaling and regulates MUC16 expression to promote CA125 secretion and the metastasis of lung cancer cells. Cell Death Dis. 2020, 11, 853. [Google Scholar] [CrossRef]
- Fang, P.; Xiang, L.; Huang, S.; Jin, L.; Zhou, G.; Zhuge, L.; Li, J.; Fan, H.; Zhou, L.; Pan, C.; et al. IRE1α-XBP1 signaling pathway regulates IL-6 expression and promotes progression of hepatocellular carcinoma. Oncol. Lett. 2018, 16, 4729–4736. [Google Scholar] [CrossRef] [Green Version]
- Pollicino, T.; Amaddeo, G.; Restuccia, A.; Raffa, G.; Alibrandi, A.; Cutroneo, G.; Favaloro, A.; Maimone, S.; Squadrito, G.; Raimondo, G. Impact of hepatitis B virus (HBV) preS/S genomic variability on HBV surface antigen and HBV DNA serum levels. Hepatology 2012, 56, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Bläckberg, J.; Kidd-Ljunggren, K. Mutations within the hepatitis B virus genome among chronic hepatitis B patients with hepatocellular carcinoma. J. Med. Virol. 2003, 71, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Gil-García, A.I.; Madejón, A.; Francisco-Recuero, I.; López-López, A.; Villafranca, E.; Romero, M.; García, A.; Olveira, A.; Mena, R.; Larrubia, J.R.; et al. Prevalence of hepatocarcinoma-related hepatitis B virus mutants in patients in grey zone of treatment. World J. Gastroenterol. 2019, 25, 5883–5896. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Yin, J.H.; Tan, X.J.; Zhang, H.Q.; Zhang, H.W.; Chen, B.C.; Chang, W.J.; Schaefer, S.; Cao, G.W. Improved multiplex-PCR to identify hepatitis B virus genotypes A–F and subgenotypes B1, B2, C1 and C2. J. Clin. Virol. 2007, 38, 238–243. [Google Scholar] [CrossRef]
- Yin, J.H.; Xie, J.X.; Liu, S.J.; Zhang, H.W.; Han, L.; Lu, W.Y.; Shen, Q.X.; Xu, G.Z.; Dong, H.J.; Shen, J.; et al. Association between the various mutations in viral core promoter region to different stages of hepatitis B, ranging of asymptomatic carrier state to hepatocellular carcinoma. Am. J. Gastroenterol. 2011, 106, 81–92. [Google Scholar] [CrossRef]
- Li, X.P.; Tan, X.J.; Yu, Y.W.; Chen, H.T.; Chang, W.J.; Hou, J.G.; Xu, D.F.; Ma, L.Y.; Cao, G.W. D9S168 microsatellite alteration predicts a poor prognosis in patients with clear cell renal cell carcinoma and correlates with the down-regulation of protein tyrosine phosphatase receptor delta. Cancer 2011, 117, 4201–4211. [Google Scholar] [CrossRef]
- Chang, W.J.; Gao, X.H.; Han, Y.F.; Du, Y.; Liu, Q.Z.; Wang, L.; Tan, X.J.; Zhang, Q.; Liu, Y.; Zhu, Y.; et al. Gene expression profiling-derived immunohistochemistry signature with high prognostic value in colorectal carcinoma. Gut 2014, 63, 1457–1467. [Google Scholar] [CrossRef]
- Wang, Q.G.; Jia, P.L.; Zhao, Z.M. VERSE: A novel approach to detect virus integration in host genomes through reference genome customization. Genome Med. 2015, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.Y.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.S.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.M.; Li, Q.; Shang, R.D.; Yao, L.W.; Wu, L.L.; Zhang, M.J.; Zhang, L.H.; Xu, M.; Lu, Z.H.; Zhou, J.; et al. WNT4 secreted by tumor tissues promotes tumor progression in colorectal cancer by activation of the Wnt/β-catenin signalling pathway. J. Exp. Clin. Cancer Res. 2020, 39, 251. [Google Scholar] [CrossRef]
- Gu, M.L.; Zhou, X.X.; Ren, M.T.; Shi, K.D.; Yu, M.S.; Jiao, W.R.; Wang, Y.M.; Zhong, W.X.; Ji, F. Blockage of ETS homologous factor inhibits the proliferation and invasion of gastric cancer cells through the c-Met pathway. World J. Gastroenterol. 2020, 26, 7497–7512. [Google Scholar] [CrossRef]
- Chandrika, M.; Chua, P.J.; Muniasamy, U.; Huang, R.Y.J.; Thike, A.A.; Ng, C.T.; Tan, P.H.; Yip, G.W.; Bay, B.H. Prognostic significance of phosphoglycerate dehydrogenase in breast cancer. Breast Cancer Res. Treat. 2021, 186, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cai, J.; Du, S.; Wei, W.; Shen, X. LAMC2 modulates the acidity of microenvironments to promote invasion and migration of pancreatic cancer cells via regulating AKT-dependent NHE1 activity. Exp. Cell Res. 2020, 391, 111984. [Google Scholar] [CrossRef] [PubMed]
- Pei, T.; Meng, F.Z.; Xiao, P.; Han, J.H.; Song, R.P.; Lan, Y.L.; Wang, Y.; Xue, J.L.; Lang, Q.F.; He, Z.F.; et al. MUC13 promotes intrahepatic cholangiocarcinoma progression via EGFR/PI3K/AKT pathways. J. Hepatol. 2020, 72, 761–773. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | No. (%) of Participants (n = 2114) | Person-Years of Follow-Up | No. of HCC (n = 224) | Incidence Rate per 1000 Person-Years | Univariate Analysis HR (95% CI) | p Value | Multivariate Analysis HR (95% CI) | p Value |
|---|---|---|---|---|---|---|---|---|
| Gender | ||||||||
| Female | 490 (23.18) | 5987 | 30 | 5.01 | 1 | 1 | ||
| Male | 1624 (76.82) | 17,858 | 194 | 10.86 | 2.08 (1.41–3.05) | <0.001 | 3.28 (1.78–6.04) | <0.001 |
| Age (years) | ||||||||
| <60 | 1885 (89.17) | 21,363 | 190 | 8.89 | 1 | 1 | ||
| ≥60 | 229 (10.83) | 2482 | 34 | 13.70 | 1.62 (1.12–2.33) | 0.010 | 1.92 (1.15–3.20) | 0.013 |
| Antiviral treatment | ||||||||
| No | 1502 (71.05) | 17,361 | 184 | 10.60 | 1 | 1 | ||
| Yes | 612 (28.95) | 6484 | 40 | 6.17 | 0.54 (0.38–0.76) | <0.001 | 0.61 (0.39–0.96) | 0.031 |
| Cirrhosis | ||||||||
| No | 1624 (76.82) | 19,372 | 126 | 6.50 | 1 | 1 | ||
| Yes | 490 (23.18) | 4473 | 98 | 21.91 | 3.39 (2.60–4.41) | <0.001 | 2.04 (1.25–3.32) | 0.004 |
| C3116T | ||||||||
| C | 755 (35.71) | 8403 | 56 | 6.66 | 1 | 1 | ||
| T | 587 (27.77) | 6765 | 75 | 11.09 | 1.70 (1.20–2.40) | 0.003 | 1.24 (0.55–2.81) | 0.600 |
| T31C | ||||||||
| T | 675 (31.93) | 7605 | 51 | 6.71 | 1 | 1 | ||
| C | 608 (28.76) | 6898 | 73 | 10.58 | 1.60 (1.12–2.28) | 0.011 | 0.97 (0.43–2.22) | 0.95 |
| HBV genotype | ||||||||
| B | 447 (21.14) | 4977 | 27 | 5.42 | 1 | 1 | ||
| C | 1212 (57.33) | 13,887 | 137 | 9.87 | 1.86 (1.23–2.81) | 0.003 | 1.38 (0.79–2.42) | 0.26 |
| Direct bilirubin (mol/L) | ||||||||
| ≤7 | 691 (32.69) | 8370 | 56 | 6.69 | 1 | 1 | ||
| >7 | 1396 (66.04) | 15,157 | 167 | 11.02 | 1.63 (1.21–2.21) | <0.001 | 0.92 (0.58–1.46) | 0.71 |
| Albumin (g/L) | ||||||||
| ≥35 | 1335 (63.15) | 16,255 | 113 | 6.95 | 1 | 1 | ||
| <35 | 772 (36.52) | 7507 | 109 | 14.52 | 2.08 (1.59–2.70) | <0.001 | 1.29 (0.80–2.07) | 0.30 |
| A-fetoprotein (ng/mL) | ||||||||
| ≤20 | 1358 (64.24) | 16,014 | 124 | 7.74 | 1 | 1 | ||
| 20–400 | 752 (35.57) | 7791 | 100 | 12.84 | 1.61 (1.23–2.09) | <0.001 | 1.39 (0.93–2.07) | 0.11 |
| Platelet count (109/L) | ||||||||
| 100–300 | 1004 (47.49) | 12,050 | 68 | 5.64 | 1 | 1 | ||
| <100 | 1110 (52.51) | 11,795 | 156 | 13.23 | 2.35 (1.77–3.12) | <0.001 | 1.22 (0.77–1.91) | 0.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Cai, S.; Pu, R.; Li, Z.; Liu, D.; Zhou, X.; Yin, J.; Chen, X.; Chen, L.; Wu, J.; et al. HBV preS Mutations Promote Hepatocarcinogenesis by Inducing Endoplasmic Reticulum Stress and Upregulating Inflammatory Signaling. Cancers 2022, 14, 3274. https://doi.org/10.3390/cancers14133274
Liu W, Cai S, Pu R, Li Z, Liu D, Zhou X, Yin J, Chen X, Chen L, Wu J, et al. HBV preS Mutations Promote Hepatocarcinogenesis by Inducing Endoplasmic Reticulum Stress and Upregulating Inflammatory Signaling. Cancers. 2022; 14(13):3274. https://doi.org/10.3390/cancers14133274
Chicago/Turabian StyleLiu, Wenbin, Shiliang Cai, Rui Pu, Zixiong Li, Donghong Liu, Xinyu Zhou, Jianhua Yin, Xi Chen, Liping Chen, Jianfeng Wu, and et al. 2022. "HBV preS Mutations Promote Hepatocarcinogenesis by Inducing Endoplasmic Reticulum Stress and Upregulating Inflammatory Signaling" Cancers 14, no. 13: 3274. https://doi.org/10.3390/cancers14133274