
Novel Approaches in Molecular Characterization of Classical Hodgkin Lymphoma

 ,
,
Abstract
:Simple Summary
Abstract
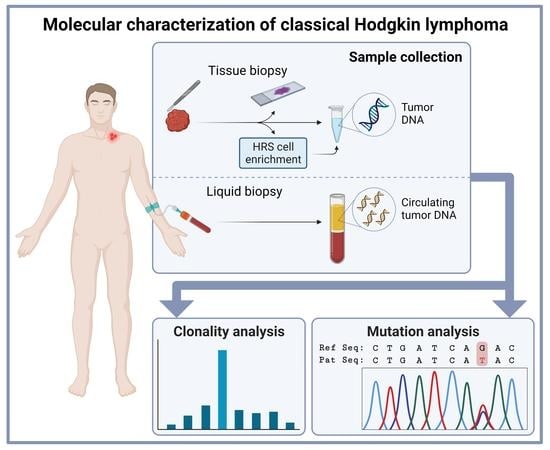
1. Introduction
2. Clonality Assessment in Classical Hodgkin Lymphoma Tissue
3. New Developments in Molecular Testing of Classical Hodgkin Lymphoma with cfDNA
4. Molecular Pathogenesis of Classical Hodgkin Lymphoma
4.1. Mutational Landscape of Classical Hodgkin Lymphoma
4.2. The Role of Epstein–Barr Virus Infection in Classical Hodgkin Lymphoma Pathogenesis
4.3. T-Cell Immune Microenvironment in Classical Hodgkin Lymphoma
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Glaser, S.L.; Clarke, C.A.; Keegan, T.H.; Chang, E.T.; Weisenburger, D.D. Time Trends in Rates of Hodgkin Lymphoma Histologic Subtypes: True Incidence Changes or Evolving Diagnostic Practice? Cancer Epidemiol. Biomark. Prev. 2015, 24, 1474–1488. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Pileri, S.A. Myeloproliferative neoplasms. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Aldinucci, D.; Gloghini, A.; Pinto, A.; De Filippi, R.; Carbone, A. The classical Hodgkin’s lymphoma microenvironment and its role in promoting tumour growth and immune escape. J. Pathol. 2010, 221, 248–263. [Google Scholar] [CrossRef]
- Campo, E.; Harris, N.L.; Pileri, S.A.; Jaffe, E.S.; Stein, H.; Thiele, J. Mature B-cell neoplasms. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Kuppers, R.; Engert, A.; Hansmann, M.L. Hodgkin lymphoma. J. Clin. Investig. 2012, 122, 3439–3447. [Google Scholar] [CrossRef]
- Parente, P.; Zanelli, M.; Sanguedolce, F.; Mastracci, L.; Graziano, P. Hodgkin Reed-Sternberg-Like Cells in Non-Hodgkin Lymphoma. Diagnostics 2020, 10, 1019. [Google Scholar] [CrossRef]
- Agbay, R.L.; Loghavi, S.; Medeiros, L.J.; Khoury, J.D. High-grade Transformation of Low-grade B-cell Lymphoma: Pathology and Molecular Pathogenesis. Am. J. Surg. Pathol. 2016, 40, e1–e16. [Google Scholar] [CrossRef]
- Montoto, S.; Fitzgibbon, J. Transformation of indolent B-cell lymphomas. J. Clin. Oncol. 2011, 29, 1827–1834. [Google Scholar] [CrossRef] [Green Version]
- Montanari, F.; Diefenbach, C. Relapsed Hodgkin lymphoma: Management strategies. Curr. Hematol. Malig. Rep. 2014, 9, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Shanbhag, S.; Ambinder, R.F. Hodgkin lymphoma: A review and update on recent progress. CA Cancer J. Clin. 2018, 68, 116–132. [Google Scholar] [CrossRef]
- Brune, M.M.; Juskevicius, D.; Haslbauer, J.; Dirnhofer, S.; Tzankov, A. Genomic Landscape of Hodgkin Lymphoma. Cancers 2021, 13, 682. [Google Scholar] [CrossRef]
- Cuceu, C.; Hempel, W.M.; Sabatier, L.; Bosq, J.; Carde, P.; M’Kacher, R. Chromosomal Instability in Hodgkin Lymphoma: An In-Depth Review and Perspectives. Cancers 2018, 10, 91. [Google Scholar] [CrossRef] [Green Version]
- Weniger, M.A.; Küppers, R. Molecular biology of Hodgkin lymphoma. Leukemia 2021, 35, 968–981. [Google Scholar] [CrossRef]
- Camus, V.; Jardin, F. Cell-Free DNA for the Management of Classical Hodgkin Lymphoma. Pharmaceuticals 2021, 14, 207. [Google Scholar] [CrossRef]
- Kuppers, R.; Klein, U.; Hansmann, M.L.; Rajewsky, K. Cellular origin of human B-cell lymphomas. N. Engl. J. Med. 1999, 341, 1520–1529. [Google Scholar] [CrossRef]
- Neuberger, M.S.; Milstein, C. Somatic hypermutation. Curr. Opin. Immunol. 1995, 7, 248–254. [Google Scholar] [CrossRef]
- Marafioti, T.; Hummel, M.; Foss, H.D.; Laumen, H.; Korbjuhn, P.; Anagnostopoulos, I.; Lammert, H.; Demel, G.; Theil, J.; Wirth, T.; et al. Hodgkin and reed-sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood 2000, 95, 1443–1450. [Google Scholar] [CrossRef]
- Kuppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc. Natl. Acad. Sci. USA 1994, 91, 10962–10966. [Google Scholar] [CrossRef] [Green Version]
- Brinker, M.G.; Poppema, S.; Buys, C.H.; Timens, W.; Osinga, J.; Visser, L. Clonal immunoglobulin gene rearrangements in tissues involved by Hodgkin’s disease. Blood 1987, 70, 186–191. [Google Scholar] [CrossRef]
- Daus, H.; Schwarze, G.; Pees, H.; Radtke, H.; Kumel, G.; Scheurlen, P.G. Immunoglobulin and T cell receptor gene rearrangements in lymphoproliferative disorders. Exp. Cell Biol. 1989, 57, 177–184. [Google Scholar] [CrossRef]
- Griesser, H.; Feller, A.C.; Mak, T.W.; Lennert, K. Clonal rearrangements of T-cell receptor and immunoglobulin genes and immunophenotypic antigen expression in different subclasses of Hodgkin’s disease. Int. J. Cancer 1987, 40, 157–160. [Google Scholar] [CrossRef]
- Herbst, H.; Tippelmann, G.; Anagnostopoulos, I.; Gerdes, J.; Schwarting, R.; Boehm, T.; Pileri, S.; Jones, D.B.; Stein, H. Immunoglobulin and T-cell receptor gene rearrangements in Hodgkin’s disease and Ki-1-positive anaplastic large cell lymphoma: Dissociation between phenotype and genotype. Leuk. Res. 1989, 13, 103–116. [Google Scholar] [CrossRef]
- Knowles, D.M., 2nd; Neri, A.; Pelicci, P.G.; Burke, J.S.; Wu, A.; Winberg, C.D.; Sheibani, K.; Dalla-Favera, R. Immunoglobulin and T-cell receptor beta-chain gene rearrangement analysis of Hodgkin’s disease: Implications for lineage determination and differential diagnosis. Proc. Natl. Acad. Sci. USA 1986, 83, 7942–7946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavachar, A.; Binder, T.; Bartram, C.R. Immunoglobulin and T-cell receptor gene rearrangements in Hodgkin’s disease. Cancer Res. 1988, 48, 3591–3594. [Google Scholar] [PubMed]
- Roth, M.S.; Schnitzer, B.; Bingham, E.L.; Harnden, C.E.; Hyder, D.M.; Ginsburg, D. Rearrangement of immunoglobulin and T-cell receptor genes in Hodgkin’s disease. Am. J. Pathol. 1988, 131, 331–338. [Google Scholar] [PubMed]
- Sundeen, J.; Lipford, E.; Uppenkamp, M.; Sussman, E.; Wahl, L.; Raffeld, M.; Cossman, J. Rearranged antigen receptor genes in Hodgkin’s disease. Blood 1987, 70, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamel, O.W.; Chang, P.P.; Hsu, F.J.; Dolezal, M.V.; Warnke, R.A.; van de Rijn, M. Clonal VDJ recombination of the immunoglobulin heavy chain gene by PCR in classical Hodgkin’s disease. Am. J. Clin. Pathol. 1995, 104, 419–423. [Google Scholar] [CrossRef]
- Manzanal, A.; Santon, A.; Oliva, H.; Bellas, C. Evaluation of clonal immunoglobulin heavy chain rearrangements in Hodgkin’s disease using the polymerase chain reaction (PCR). Histopathology 1995, 27, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Manzanal, A.I.; Santon, A.; Acevedo, A.; Aguilera, B.; Oliva, H.; Bellas, C. Molecular analysis of the IgH gene in 212 cases of Hodgkin’s disease: Correlation of IgH clonality with the histologic and the immunocytochemical features. Mod. Pathol. 1997, 10, 679–685. [Google Scholar]
- van Dongen, J.J.; Langerak, A.W.; Bruggemann, M.; Evans, P.A.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; Garcia-Sanz, R.; et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef] [Green Version]
- Burack, W.R.; Laughlin, T.S.; Friedberg, J.W.; Spence, J.M.; Rothberg, P.G. PCR assays detect B-lymphocyte clonality in formalin-fixed, paraffin-embedded specimens of classical hodgkin lymphoma without microdissection. Am. J. Clin. Pathol. 2010, 134, 104–111. [Google Scholar] [CrossRef]
- Chute, D.J.; Cousar, J.B.; Mahadevan, M.S.; Siegrist, K.A.; Silverman, L.M.; Stoler, M.H. Detection of immunoglobulin heavy chain gene rearrangements in classic hodgkin lymphoma using commercially available BIOMED-2 primers. Diagn. Mol. Pathol. 2008, 17, 65–72. [Google Scholar] [CrossRef]
- Ghorbian, S.; Jahanzad, I.; Estiar, M.A.; Ziae, J.E.; Asvadi-Kermani, I.; Andalib, S.; Javadi, G.R.; Sakhinia, E. Molecular Analysis of IGH and Incomplete IGH D-J Clonality Gene Rearrangements in Hodgkin Lymphoma Malignancies. Clin. Lab. 2015, 61, 951–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorbian, S.; Jahanzad, I.; Javadi, G.R.; Sakhinia, E. Evaluation of IGK and IGL molecular gene rearrangements according to the BIOMED-2 protocols for clinical diagnosis of Hodgkin lymphoma. Hematology 2016, 21, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Masaki, A.; Sakamoto, Y.; Takino, H.; Murase, T.; Iida, S.; Inagaki, H. Improved clonality detection in Hodgkin lymphoma using a semi-nested modification of the BIOMED-2 PCR assay for IGH and IGK rearrangements: A paraffin-embedded tissue study. Pathol. Int. 2018, 68, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Helling, A.; Doring, C.; Renne, C.; Hansmann, M.L. Clonality testing of malignant lymphomas with the BIOMED-2 primers in a large cohort of 1969 primary and consultant biopsies. Pathol. Res. Pract. 2013, 209, 495–502. [Google Scholar] [CrossRef]
- Hebeda, K.M.; Van Altena, M.C.; Rombout, P.; Van Krieken, J.H.; Groenen, P.J. PCR clonality detection in Hodgkin lymphoma. J. Hematop. 2009, 2, 34–41. [Google Scholar] [CrossRef] [Green Version]
- McClure, R.F.; Kaur, P.; Pagel, E.; Ouillette, P.D.; Holtegaard, C.E.; Treptow, C.L.; Kurtin, P.J. Validation of immunoglobulin gene rearrangement detection by PCR using commercially available BIOMED-2 primers. Leukemia 2006, 20, 176–179. [Google Scholar] [CrossRef]
- Tapia, G.; Sanz, C.; Mate, J.L.; Munoz-Marmol, A.M.; Ariza, A. Improved clonality detection in Hodgkin lymphoma using the BIOMED-2-based heavy and kappa chain assay: A paraffin-embedded tissue study. Histopathology 2012, 60, 768–773. [Google Scholar] [CrossRef]
- Evans, P.A.; Pott, C.; Groenen, P.J.; Salles, G.; Davi, F.; Berger, F.; Garcia, J.F.; van Krieken, J.H.; Pals, S.; Kluin, P.; et al. Significantly improved PCR-based clonality testing in B-cell malignancies by use of multiple immunoglobulin gene targets. Report of the BIOMED-2 Concerted Action BHM4-CT98-3936. Leukemia 2007, 21, 207–214. [Google Scholar] [CrossRef]
- Gameiro, P.; Sebastião, M.; Spetalen, S.; da Silva, M.G.; Cabeçadas, J. The added value of immunoglobulin Kappa light chain gene (IGK) rearrangement analysis in suspected B-cell lymphomas: Three illustrative cases. J. Hematop. 2012, 5, 45–56. [Google Scholar] [CrossRef]
- Aguilera, N.S.; Chen, J.; Bijwaard, K.E.; Director-Myska, A.E.; Barekman, C.L.; Millward, C.; Lichy, J.; Abbondanzo, S.L. Gene rearrangement and comparative genomic hybridization studies of classic Hodgkin lymphoma expressing T-cell antigens. Arch. Pathol. Lab. Med. 2006, 130, 1772–1779. [Google Scholar] [CrossRef]
- Deng, F.; Lü, G.; Li, G.; Yang, G. Hodgkin’s disease: Immunoglobulin heavy and light chain gene rearrangements revealed in single Hodgkin/Reed-Sternberg cells. Mol. Pathol. 1999, 52, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummel, M.; Ziemann, K.; Lammert, H.; Pileri, S.; Sabattini, E.; Stein, H. Hodgkin’s disease with monoclonal and polyclonal populations of Reed-Sternberg cells. N. Engl. J. Med. 1995, 333, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Izban, K.F.; Nawrocki, J.F.; Alkan, S.; Hsi, E.D. Monoclonal IgH gene rearrangement in microdissected nodules from nodular sclerosis Hodgkin disease. Am. J. Clin. Pathol. 1998, 110, 599–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, J.; Daus, H.; Trumper, L.; Gause, A.; Salamon-Looijen, M.; Pfreundschuh, M. Detection of immunoglobulin heavy-chain gene rearrangement at the single-cell level in malignant lymphomas: No rearrangement is found in Hodgkin and Reed-Sternberg cells. Int. J. Cancer 1994, 57, 799–804. [Google Scholar] [CrossRef]
- Seitz, V.; Hummel, M.; Marafioti, T.; Anagnostopoulos, I.; Assaf, C.; Stein, H. Detection of clonal T-cell receptor gamma-chain gene rearrangements in Reed-Sternberg cells of classic Hodgkin disease. Blood 2000, 95, 3020–3024. [Google Scholar] [CrossRef]
- Kanzler, H.; Kuppers, R.; Hansmann, M.L.; Rajewsky, K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J. Exp. Med. 1996, 184, 1495–1505. [Google Scholar] [CrossRef]
- Brauninger, A.; Wacker, H.H.; Rajewsky, K.; Kuppers, R.; Hansmann, M.L. Typing the histogenetic origin of the tumor cells of lymphocyte-rich classical Hodgkin’s lymphoma in relation to tumor cells of classical and lymphocyte-predominance Hodgkin’s lymphoma. Cancer Res. 2003, 63, 1644–1651. [Google Scholar]
- Oki, Y.; Neelapu, S.S.; Fanale, M.; Kwak, L.W.; Fayad, L.; Rodriguez, M.A.; Wallace, M.; Klinger, M.; Carlton, V.; Kong, K.; et al. Detection of classical Hodgkin lymphoma specific sequence in peripheral blood using a next-generation sequencing approach. Br. J. Haematol. 2015, 169, 689–693. [Google Scholar] [CrossRef]
- van Bladel, D.A.G.; van den Brand, M.; Rijntjes, J.; Pamidimarri Naga, S.; Haacke, D.; Luijks, J.; Hebeda, K.M.; van Krieken, J.; Groenen, P.; Scheijen, B. Clonality assessment and detection of clonal diversity in classic Hodgkin lymphoma by next-generation sequencing of immunoglobulin gene rearrangements. Mod. Pathol. 2022, 35, 757–766. [Google Scholar] [CrossRef]
- Desch, A.K.; Hartung, K.; Botzen, A.; Brobeil, A.; Rummel, M.; Kurch, L.; Georgi, T.; Jox, T.; Bielack, S.; Burdach, S.; et al. Genotyping circulating tumor DNA of pediatric Hodgkin lymphoma. Leukemia 2020, 34, 151–166. [Google Scholar] [CrossRef]
- Muschen, M.; Rajewsky, K.; Brauninger, A.; Baur, A.S.; Oudejans, J.J.; Roers, A.; Hansmann, M.L.; Kuppers, R. Rare occurrence of classical Hodgkin’s disease as a T cell lymphoma. J. Exp. Med. 2000, 191, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, G.; Song, J.Y.; Tzankov, A.; Dirnhofer, S.; Heinze, G.; Kohl, M.; Traverse-Glehen, A.; Eberle, F.C.; Hanson, J.C.; Raffeld, M.A.; et al. Aberrant T-cell antigen expression in classical Hodgkin lymphoma is associated with decreased event-free survival and overall survival. Blood 2013, 121, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Bruggemann, M.; Kotrova, M.; Knecht, H.; Bartram, J.; Boudjogrha, M.; Bystry, V.; Fazio, G.; Fronkova, E.; Giraud, M.; Grioni, A.; et al. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia 2019, 33, 2241–2253. [Google Scholar] [CrossRef] [Green Version]
- Scheijen, B.; Meijers, R.W.J.; Rijntjes, J.; van der Klift, M.Y.; Möbs, M.; Steinhilber, J.; Reigl, T.; van den Brand, M.; Kotrová, M.; Ritter, J.M.; et al. Next-generation sequencing of immunoglobulin gene rearrangements for clonality assessment: A technical feasibility study by EuroClonality-NGS. Leukemia 2019, 33, 2227–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.; Warnke, R.A.; Seo, K.; Rosenberg, S.A.; Arber, D.A. Rare presentation of classical Hodgkin lymphoma with a clonal T-cell receptor gene rearrangement in the tissue. Leuk. Lymphoma 2010, 51, 1356–1359. [Google Scholar] [CrossRef] [PubMed]
- Willenbrock, K.; Ichinohasama, R.; Kadin, M.E.; Miura, I.; Terui, T.; Meguro, K.; Fukuhara, O.; DeCoteau, J.F.; Hansmann, M.L. T-cell variant of classical Hodgkin’s lymphoma with nodal and cutaneous manifestations demonstrated by single-cell polymerase chain reaction. Lab. Investig. 2002, 82, 1103–1109. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Jaffe, E.S. How I Diagnose Angioimmunoblastic T-Cell Lymphoma. Am. J. Clin. Pathol. 2021, 156, 1–14. [Google Scholar] [CrossRef]
- Moroch, J.; Copie-Bergman, C.; de Leval, L.; Plonquet, A.; Martin-Garcia, N.; Delfau-Larue, M.H.; Molinier-Frenkel, V.; Belhadj, K.; Haioun, C.; Audouin, J.; et al. Follicular peripheral T-cell lymphoma expands the spectrum of classical Hodgkin lymphoma mimics. Am. J. Surg. Pathol. 2012, 36, 1636–1646. [Google Scholar] [CrossRef]
- Obermann, E.C.; Mueller, N.; Rufle, A.; Menter, T.; Mueller-Garamvoelgyi, E.; Cathomas, G.; Dirnhofer, S.; Tzankov, A. Clonal relationship of classical hodgkin lymphoma and its recurrences. Clin. Cancer Res. 2011, 17, 5268–5274. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Alcoceba, M.; García-Álvarez, M.; Chillón, M.C.; Jiménez, C.; Medina, A.; Antón, A.; Blanco, O.; Díaz, L.G.; Tamayo, P.; González-Calle, V.; et al. Liquid biopsy: A non-invasive approach for Hodgkin lymphoma genotyping. Br. J. Haematol. 2021, 195, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Bessi, L.; Viailly, P.J.; Bohers, E.; Ruminy, P.; Maingonnat, C.; Bertrand, P.; Vasseur, N.; Beaussire, L.; Cornic, M.; Etancelin, P.; et al. Somatic mutations of cell-free circulating DNA detected by targeted next-generation sequencing and digital droplet PCR in classical Hodgkin lymphoma. Leuk. Lymphoma 2019, 60, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Stamatoullas, A.; Mareschal, S.; Viailly, P.J.; Sarafan-Vasseur, N.; Bohers, E.; Dubois, S.; Picquenot, J.M.; Ruminy, P.; Maingonnat, C.; et al. Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell-free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica 2016, 101, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Viennot, M.; Lequesne, J.; Viailly, P.J.; Bohers, E.; Bessi, L.; Marcq, B.; Etancelin, P.; Dubois, S.; Picquenot, J.M.; et al. Targeted genotyping of circulating tumor DNA for classical Hodgkin lymphoma monitoring: A prospective study. Haematologica 2021, 106, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef] [Green Version]
- Eijkelenboom, A.; Kamping, E.J.; Kastner-van Raaij, A.W.; Hendriks-Cornelissen, S.J.; Neveling, K.; Kuiper, R.P.; Hoischen, A.; Nelen, M.R.; Ligtenberg, M.J.; Tops, B.B. Reliable Next-Generation Sequencing of Formalin-Fixed, Paraffin-Embedded Tissue Using Single Molecule Tags. J. Mol. Diagn. 2016, 18, 851–863. [Google Scholar] [CrossRef] [Green Version]
- Kou, R.; Lam, H.; Duan, H.; Ye, L.; Jongkam, N.; Chen, W.; Zhang, S.; Li, S. Benefits and Challenges with Applying Unique Molecular Identifiers in Next Generation Sequencing to Detect Low Frequency Mutations. PLoS ONE 2016, 11, e0146638. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Liu, X.; Zheng, B.; Ke, R.; Tzeng, C.M. Liquid Biopsy, ctDNA Diagnosis through NGS. Life 2021, 11, 890. [Google Scholar] [CrossRef]
- Lui, Y.Y.; Chik, K.W.; Chiu, R.W.; Ho, C.Y.; Lam, C.W.; Lo, Y.M. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.T.; Chin, Y.M.; Nakamura, Y.; Low, S.K. Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications. Cancers 2020, 12, 2277. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Venanzi, A.; Marra, A.; Schiavoni, G.; Milner, S.G.; Limongello, R.; Santi, A.; Pettirossi, V.; Ultimo, S.; Tasselli, L.; Pucciarini, A.; et al. Dissecting Clonal Hematopoiesis in Tissues of Classical Hodgkin Lymphoma Patients. Blood Cancer Discov. 2021, 2, 216–225. [Google Scholar] [CrossRef]
- Vandenberghe, P.; Wlodarska, I.; Tousseyn, T.; Dehaspe, L.; Dierickx, D.; Verheecke, M.; Uyttebroeck, A.; Bechter, O.; Delforge, M.; Vandecaveye, V.; et al. Non-invasive detection of genomic imbalances in Hodgkin/Reed-Sternberg cells in early and advanced stage Hodgkin’s lymphoma by sequencing of circulating cell-free DNA: A technical proof-of-principle study. Lancet Haematol. 2015, 2, e55–e65. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.S.; Vergilio, J.A.; Salhia, B.; Huang, H.J.; Oki, Y.; Garrido-Laguna, I.; Park, H.; Westin, J.R.; Meric-Bernstam, F.; Fabrizio, D.; et al. Comprehensive Genomic Profiling of Hodgkin Lymphoma Reveals Recurrently Mutated Genes and Increased Mutation Burden. Oncologist 2019, 24, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Hussaini, M.O.; Srivastava, J.; Lee, L.W.; Nishihori, T.; Shah, B.D.; Alsina, M.; Pinilla-Ibarz, J.; Shain, K.H. Assessment of Clonotypic Rearrangements and Minimal Residual Disease in Lymphoid Malignancies: A Large Cancer Center Experience Using clonoSEQ. Arch. Pathol. Lab. Med. 2021, 146, 485–493. [Google Scholar] [CrossRef]
- Monter, A.; Nomdedeu, J.F. ClonoSEQ assay for the detection of lymphoid malignancies. Expert Rev. Mol. Diagn. 2019, 19, 571–578. [Google Scholar] [CrossRef]
- Sobesky, S.; Mammadova, L.; Cirillo, M.; Drees, E.E.E.; Mattlener, J.; Dörr, H.; Altmüller, J.; Shi, Z.; Bröckelmann, P.J.; Weiss, J.; et al. In-depth cell-free DNA sequencing reveals genomic landscape of Hodgkin’s lymphoma and facilitates ultrasensitive residual disease detection. Med (N. Y.) 2021, 2, 1171–1193. [Google Scholar] [CrossRef]
- Mussolin, L.; Burnelli, R.; Pillon, M.; Carraro, E.; Farruggia, P.; Todesco, A.; Mascarin, M.; Rosolen, A. Plasma cell-free DNA in paediatric lymphomas. J. Cancer 2013, 4, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Primerano, S.; Burnelli, R.; Carraro, E.; Pillon, M.; Elia, C.; Farruggia, P.; Sala, A.; Vinti, L.; Buffardi, S.; Basso, G.; et al. Kinetics of Circulating Plasma Cell-Free DNA in Paediatric Classical Hodgkin Lymphoma. J. Cancer 2016, 7, 364–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Su, H.; Song, Y.; Jiang, W.; Sun, X.; Qian, W.; Zhang, W.; Gao, Y.; Jin, Z.; Zhou, J.; et al. Circulating tumor DNA predicts response in Chinese patients with relapsed or refractory classical hodgkin lymphoma treated with sintilimab. EBioMedicine 2020, 54, 102731. [Google Scholar] [CrossRef] [PubMed]
- Faham, M.; Zheng, J.; Moorhead, M.; Carlton, V.E.; Stow, P.; Coustan-Smith, E.; Pui, C.H.; Campana, D. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood 2012, 120, 5173–5180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Kurtz, D.M. Bringing circulating tumor DNA to the clinic in Hodgkin lymphoma. Haematologica 2021, 106, 5–6. [Google Scholar] [CrossRef]
- Stewart, J.P.; Gazdova, J.; Darzentas, N.; Wren, D.; Proszek, P.; Fazio, G.; Songia, S.; Alcoceba, M.; Sarasquete, M.E.; Villarese, P.; et al. Validation of the EuroClonality-NGS DNA capture panel as an integrated genomic tool for lymphoproliferative disorders. Blood Adv. 2021, 5, 3188–3198. [Google Scholar] [CrossRef]
- Murray, P.G.; Young, L.S. An etiological role for the Epstein-Barr virus in the pathogenesis of classical Hodgkin lymphoma. Blood 2019, 134, 591–596. [Google Scholar] [CrossRef]
- Reichel, J.; Chadburn, A.; Rubinstein, P.G.; Giulino-Roth, L.; Tam, W.; Liu, Y.; Gaiolla, R.; Eng, K.; Brody, J.; Inghirami, G.; et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood 2015, 125, 1061–1072. [Google Scholar] [CrossRef] [Green Version]
- Tiacci, E.; Ladewig, E.; Schiavoni, G.; Penson, A.; Fortini, E.; Pettirossi, V.; Wang, Y.; Rosseto, A.; Venanzi, A.; Vlasevska, S.; et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 2018, 131, 2454–2465. [Google Scholar] [CrossRef] [Green Version]
- Wienand, K.; Chapuy, B.; Stewart, C.; Dunford, A.J.; Wu, D.; Kim, J.; Kamburov, A.; Wood, T.R.; Cader, F.Z.; Ducar, M.D.; et al. Genomic analyses of flow-sorted Hodgkin Reed-Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv. 2019, 3, 4065–4080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilly, H.; Bastard, C.; Delastre, T.; Duval, C.; Bizet, M.; Lenormand, B.; Daucé, J.P.; Monconduit, M.; Piguet, H. Cytogenetic studies in untreated Hodgkin’s disease. Blood 1991, 77, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Martin-Subero, J.I.; Gesk, S.; Hüsken, J.; Giefing, M.; Nagel, I.; Riemke, J.; Chott, A.; Klapper, W.; Parrens, M.; et al. Detection of genomic imbalances in microdissected Hodgkin and Reed-Sternberg cells of classical Hodgkin’s lymphoma by array-based comparative genomic hybridization. Haematologica 2008, 93, 1318–1326. [Google Scholar] [CrossRef]
- Re, D.; Zander, T.; Diehl, V.; Wolf, J. Genetic instability in Hodgkin’s lymphoma. Ann. Oncol. 2002, 13 (Suppl. 1), 19–22. [Google Scholar] [CrossRef]
- Martín-Subero, J.I.; Klapper, W.; Sotnikova, A.; Callet-Bauchu, E.; Harder, L.; Bastard, C.; Schmitz, R.; Grohmann, S.; Höppner, J.; Riemke, J.; et al. Chromosomal breakpoints affecting immunoglobulin loci are recurrent in Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma. Cancer Res. 2006, 66, 10332–10338. [Google Scholar] [CrossRef] [Green Version]
- Chui, D.T.; Hammond, D.; Baird, M.; Shield, L.; Jackson, R.; Jarrett, R.F. Classical Hodgkin lymphoma is associated with frequent gains of 17q. Genes Chromosomes Cancer 2003, 38, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.; Menz, C.K.; Wrobel, G.; Siebert, R.; Gesk, S.; Ohl, S.; Mechtersheimer, G.; Trümper, L.; Möller, P.; Lichter, P.; et al. Classical Hodgkin lymphoma is characterized by recurrent copy number gains of the short arm of chromosome 2. Blood 2002, 99, 1381–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steidl, C.; Telenius, A.; Shah, S.P.; Farinha, P.; Barclay, L.; Boyle, M.; Connors, J.M.; Horsman, D.E.; Gascoyne, R.D. Genome-wide copy number analysis of Hodgkin Reed-Sternberg cells identifies recurrent imbalances with correlations to treatment outcome. Blood 2010, 116, 418–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [Green Version]
- Buedts, L.; Wlodarska, I.; Finalet-Ferreiro, J.; Gheysens, O.; Dehaspe, L.; Tousseyn, T.; Fornecker, L.M.; Lazarovici, J.; Casasnovas, R.O.; Gac, A.C.; et al. The landscape of copy number variations in classical Hodgkin lymphoma: A joint KU Leuven and LYSA study on cell-free DNA. Blood Adv. 2021, 5, 1991–2002. [Google Scholar] [CrossRef]
- Healy, J.A.; Nugent, A.; Rempel, R.E.; Moffitt, A.B.; Davis, N.S.; Jiang, X.; Shingleton, J.R.; Zhang, J.; Love, C.; Datta, J.; et al. GNA13 loss in germinal center B cells leads to impaired apoptosis and promotes lymphoma in vivo. Blood 2016, 127, 2723–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hayre, M.; Inoue, A.; Kufareva, I.; Wang, Z.; Mikelis, C.M.; Drummond, R.A.; Avino, S.; Finkel, K.; Kalim, K.W.; DiPasquale, G.; et al. Inactivating mutations in GNA13 and RHOA in Burkitt’s lymphoma and diffuse large B-cell lymphoma: A tumor suppressor function for the Gα13/RhoA axis in B cells. Oncogene 2016, 35, 3771–3780. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Zhang, X.; Liu, P.; Zhang, R.; Huang, Z.; Li, D.; Xiao, X.; Wu, M.; Ning, N.; Zhang, Q.; et al. GNA13 regulates BCL2 expression and the sensitivity of GCB-DLBCL cells to BCL2 inhibitors in a palmitoylation-dependent manner. Cell Death Dis. 2021, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Maggio, E.M.; Stekelenburg, E.; Van den Berg, A.; Poppema, S. TP53 gene mutations in Hodgkin lymphoma are infrequent and not associated with absence of Epstein-Barr virus. Int. J. Cancer 2001, 94, 60–66. [Google Scholar] [CrossRef]
- Chen, W.G.; Chen, Y.Y.; Kamel, O.W.; Koo, C.H.; Weiss, L.M. p53 mutations in Hodgkin’s disease. Lab. Investig. 1996, 75, 519–527. [Google Scholar]
- Mata, E.; Fernández, S.; Astudillo, A.; Fernández, R.; García-Cosío, M.; Sánchez-Beato, M.; Provencio, M.; Estévez, M.; Montalbán, C.; Piris, M.A.; et al. Genomic analyses of microdissected Hodgkin and Reed-Sternberg cells: Mutations in epigenetic regulators and p53 are frequent in refractory classic Hodgkin lymphoma. Blood Cancer J. 2019, 9, 34. [Google Scholar] [CrossRef]
- Elenitoba-Johnson, K.S.; Medeiros, L.J.; Khorsand, J.; King, T.C. P53 expression in Reed-Sternberg cells does not correlate with gene mutations in Hodgkin’s disease. Am. J. Clin. Pathol. 1996, 106, 728–738. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sattarzadeh, A.; Diepstra, A.; Visser, L.; van den Berg, A. The microenvironment in classical Hodgkin lymphoma: An actively shaped and essential tumor component. Semin. Cancer Biol. 2014, 24, 15–22. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus-associated lymphomas. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef]
- Vrzalikova, K.; Pugh, M.; Mundo, L.; Murray, P. The contribution of ebv to the pathogenesis of classical hodgkin lymphoma. Ann. Lymphoma 2021, 5, 30. [Google Scholar] [CrossRef]
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med. Microbiol. Immunol. 2019, 208, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.Y.; Lu, J.; Shih, Y.C.; Tsai, C.H. Epstein-Barr virus latent membrane protein 2A regulates c-Jun protein through extracellular signal-regulated kinase. J. Virol. 2002, 76, 9556–9561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portis, T.; Longnecker, R. Epstein-Barr virus (EBV) LMP2A alters normal transcriptional regulation following B-cell receptor activation. Virology 2004, 318, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Liu, Y.; Wang, C.; Gan, R. Signaling pathways of EBV-induced oncogenesis. Cancer Cell Int. 2021, 21, 93. [Google Scholar] [CrossRef]
- Vrzalikova, K.; Sunmonu, T.; Reynolds, G.; Murray, P. Contribution of Epstein(-)Barr Virus Latent Proteins to the Pathogenesis of Classical Hodgkin Lymphoma. Pathogens 2018, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.Y.; Wu, S.J.; Huang, M.H.; Lo, F.Y.; Tsai, M.H.; Tsai, C.H.; Hsu, S.M.; Lin, C.W. EBV-positive Hodgkin lymphoma is associated with suppression of p21cip1/waf1 and a worse prognosis. Mol. Cancer 2010, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Iwakiri, D.; Takada, K. Role of EBERs in the pathogenesis of EBV infection. Adv. Cancer Res. 2010, 107, 119–136. [Google Scholar] [CrossRef]
- De Re, V.; Caggiari, L.; De Zorzi, M.; Fanotto, V.; Miolo, G.; Puglisi, F.; Cannizzaro, R.; Canzonieri, V.; Steffan, A.; Farruggia, P.; et al. Epstein-Barr virus BART microRNAs in EBV- associated Hodgkin lymphoma and gastric cancer. Infect. Agent Cancer 2020, 15, 42. [Google Scholar] [CrossRef]
- Lake, A.; Shield, L.A.; Cordano, P.; Chui, D.T.; Osborne, J.; Crae, S.; Wilson, K.S.; Tosi, S.; Knight, S.J.; Gesk, S.; et al. Mutations of NFKBIA, encoding IkappaB alpha, are a recurrent finding in classical Hodgkin lymphoma but are not a unifying feature of non-EBV-associated cases. Int. J. Cancer 2009, 125, 1334–1342. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, N.D.; Coward, W.B.t.; Johnson, S.; Yuan, J.; Gulley, M.L.; Mathews, S.P.; Kaiser-Rogers, K.; Rao, K.W.; Sanger, W.G.; Sanmann, J.N.; et al. Karyotypic abnormalities associated with Epstein-Barr virus status in classical Hodgkin lymphoma. Cancer Genet. 2016, 209, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Stanelle, J.; Hansmann, M.L.; Küppers, R. Pathogenesis of classical and lymphocyte-predominant Hodgkin lymphoma. Annu. Rev. Pathol. 2009, 4, 151–174. [Google Scholar] [CrossRef] [PubMed]
- Renné, C.; Hinsch, N.; Willenbrock, K.; Fuchs, M.; Klapper, W.; Engert, A.; Küppers, R.; Hansmann, M.L.; Bräuninger, A. The aberrant coexpression of several receptor tyrosine kinases is largely restricted to EBV-negative cases of classical Hodgkin’s lymphoma. Int. J. Cancer 2007, 120, 2504–2509. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Hansmann, M.L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S.; et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Celegato, M.; Casagrande, N. Microenvironmental interactions in classical Hodgkin lymphoma and their role in promoting tumor growth, immune escape and drug resistance. Cancer Lett. 2016, 380, 243–252. [Google Scholar] [CrossRef]
- Re, D.; Kuppers, R.; Diehl, V. Molecular pathogenesis of Hodgkin’s lymphoma. J. Clin. Oncol. 2005, 23, 6379–6386. [Google Scholar] [CrossRef] [Green Version]
- Wein, F.; Küppers, R. The role of T cells in the microenvironment of Hodgkin lymphoma. J. Leukoc. Biol. 2016, 99, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T.; et al. Single-Cell Transcriptome Analysis Reveals Disease-Defining T-cell Subsets in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Marshall, A.; Chavez, E.A.; Miyata-Takata, T.; Ben-Neriah, S.; Unrau, D.; Telenius, A.; et al. Single-cell profiling reveals the importance of CXCL13/CXCR5 axis biology in lymphocyte-rich classic Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2105822118. [Google Scholar] [CrossRef]
- Cader, F.Z.; Schackmann, R.C.J.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; et al. Mass cytometry of Hodgkin lymphoma reveals a CD4(+) regulatory T-cell-rich and exhausted T-effector microenvironment. Blood 2018, 132, 825–836. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Gruss, H.J.; Pinto, A. CD40 ligand is constitutively expressed in a subset of T cell lymphomas and on the microenvironmental reactive T cells of follicular lymphomas and Hodgkin’s disease. Am. J. Pathol. 1995, 147, 912–922. [Google Scholar] [PubMed]
- Steidl, C.; Connors, J.M.; Gascoyne, R.D. Molecular pathogenesis of Hodgkin’s lymphoma: Increasing evidence of the importance of the microenvironment. J. Clin. Oncol. 2011, 29, 1812–1826. [Google Scholar] [CrossRef] [PubMed]
- Steinhilber, J.; Mederake, M.; Bonzheim, I.; Serinsöz-Linke, E.; Müller, I.; Fallier-Becker, P.; Lemonnier, F.; Gaulard, P.; Fend, F.; Quintanilla-Martinez, L. The pathological features of angioimmunoblastic T-cell lymphomas with IDH2(R172) mutations. Mod. Pathol. 2019, 32, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Kujala, J.; Hartikainen, J.M.; Tengström, M.; Sironen, R.; Auvinen, P.; Kosma, V.M.; Mannermaa, A. Circulating Cell-Free DNA Reflects the Clonal Evolution of Breast Cancer Tumors. Cancers 2022, 14, 1332. [Google Scholar] [CrossRef]
- Imamura, F.; Uchida, J.; Kukita, Y.; Kumagai, T.; Nishino, K.; Inoue, T.; Kimura, M.; Oba, S.; Kato, K. Monitoring of treatment responses and clonal evolution of tumor cells by circulating tumor DNA of heterogeneous mutant EGFR genes in lung cancer. Lung Cancer 2016, 94, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Perdigones, N.; Murtaza, M. Capturing tumor heterogeneity and clonal evolution in solid cancers using circulating tumor DNA analysis. Pharmacol. Ther. 2017, 174, 22–26. [Google Scholar] [CrossRef]
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Stehr, H.; Craig, A.F.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med. 2016, 8, 364ra155. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C.; et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 2017, 129, 1947–1957. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IG Gene Rearrangements | |||||
| Study Cohorts | Clone Detection (IG) | ||||
| Clonality Assay | Studies (n) | Samples (n) | Mean | Range | References |
| Southern blot * | 8 | 8–39 | 45% | 0–87% | [19,20,21,22,23,24,25,26] |
| PCR-based * | 3 | 32–212 | 30% | 23–44% | [27,28,29] |
| BIOMED-2 * | 8 | 12–58 | 57% | 26–79% | [31,32,33,34,35,36,37,38,39] |
| NGS-based | |||||
| Tissue gDNA * | 2 | 16–17 | 72% | 56–88% | [50,51] |
| cfDNA | 2 | 9–72 | 64% | 38–89% | [50,52] |
| Enriched HRS cells # | 11 | 3–25 | 62% | 0–100% | [17,18,26,42,43,44,45,46,47,48,49] |
| TR Gene Rearrangements | |||||
| Study cohorts | Clone detection (TR) | ||||
| Clonality assay | Studies (n) | Samples (n) | Mean | Range | References |
| Southern blot * | 8 | 8–39 | 15% | 0–68% | [19,20,21,22,23,24,25,26] |
| BIOMED-2 * | 1 | 58 | 17% | NA | [36] |
| Enriched HRS cells # | 4 | 3–19 | 28% | 11–50% | [42,47,53,54] |
| Gene | Genetic Aberration(s) * | Pathways and Biological Processes |
|---|---|---|
| PD-L1 and PD-L2 | CNAs (9p24 gain) | Immune evasion |
| B2M, HLA-A/B | Inactivating mutations | |
| CIITA | Translocations | |
| JAK2 | CNAs (9p24 gain) | JAK/STAT signaling |
| STAT6 | Activating mutations | |
| SOCS1 | Inactivating mutations | |
| CSFR2B | Activating mutations | |
| PTPN1 | Inactivating mutations | |
| TNFAIP3 | Inactivating mutations and deletions | NF-κB pathway |
| IKBKB | Activating mutations | |
| NFKBIA, NFKBIE | Inactivating mutations | |
| BIRC3 | CNAs (11q loss) | |
| REL | CNAs (2p gain) | |
| GNA13 | Inactivating mutations | PI3K/AKT pathway |
| ITPKB | Inactivating mutations | |
| RBM38 | Inactivating mutations | |
| PIK3CA | Activating mutations | |
| XPO1 | Activating mutations | Nuclear–cytoplasmic transport |
| TP53 | Inactivating mutations | Genomic stability |
| ATM | Inactivating mutations | |
| ARID1A | Inactivating mutations | Epigenetic regulation |
| KMT2C | Inactivating mutations | |
| KMT2D | Inactivating mutations |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Bladel, D.A.G.; Stevens, W.B.C.; van den Brand, M.; Kroeze, L.I.; Groenen, P.J.T.A.; van Krieken, J.H.J.M.; Hebeda, K.M.; Scheijen, B. Novel Approaches in Molecular Characterization of Classical Hodgkin Lymphoma. Cancers 2022, 14, 3222. https://doi.org/10.3390/cancers14133222
van Bladel DAG, Stevens WBC, van den Brand M, Kroeze LI, Groenen PJTA, van Krieken JHJM, Hebeda KM, Scheijen B. Novel Approaches in Molecular Characterization of Classical Hodgkin Lymphoma. Cancers. 2022; 14(13):3222. https://doi.org/10.3390/cancers14133222
Chicago/Turabian Stylevan Bladel, Diede A. G., Wendy B. C. Stevens, Michiel van den Brand, Leonie I. Kroeze, Patricia J. T. A. Groenen, J. Han J. M. van Krieken, Konnie M. Hebeda, and Blanca Scheijen. 2022. "Novel Approaches in Molecular Characterization of Classical Hodgkin Lymphoma" Cancers 14, no. 13: 3222. https://doi.org/10.3390/cancers14133222