
Proteases and HPV-Induced Carcinogenesis

 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. HPV Carcinogenesis
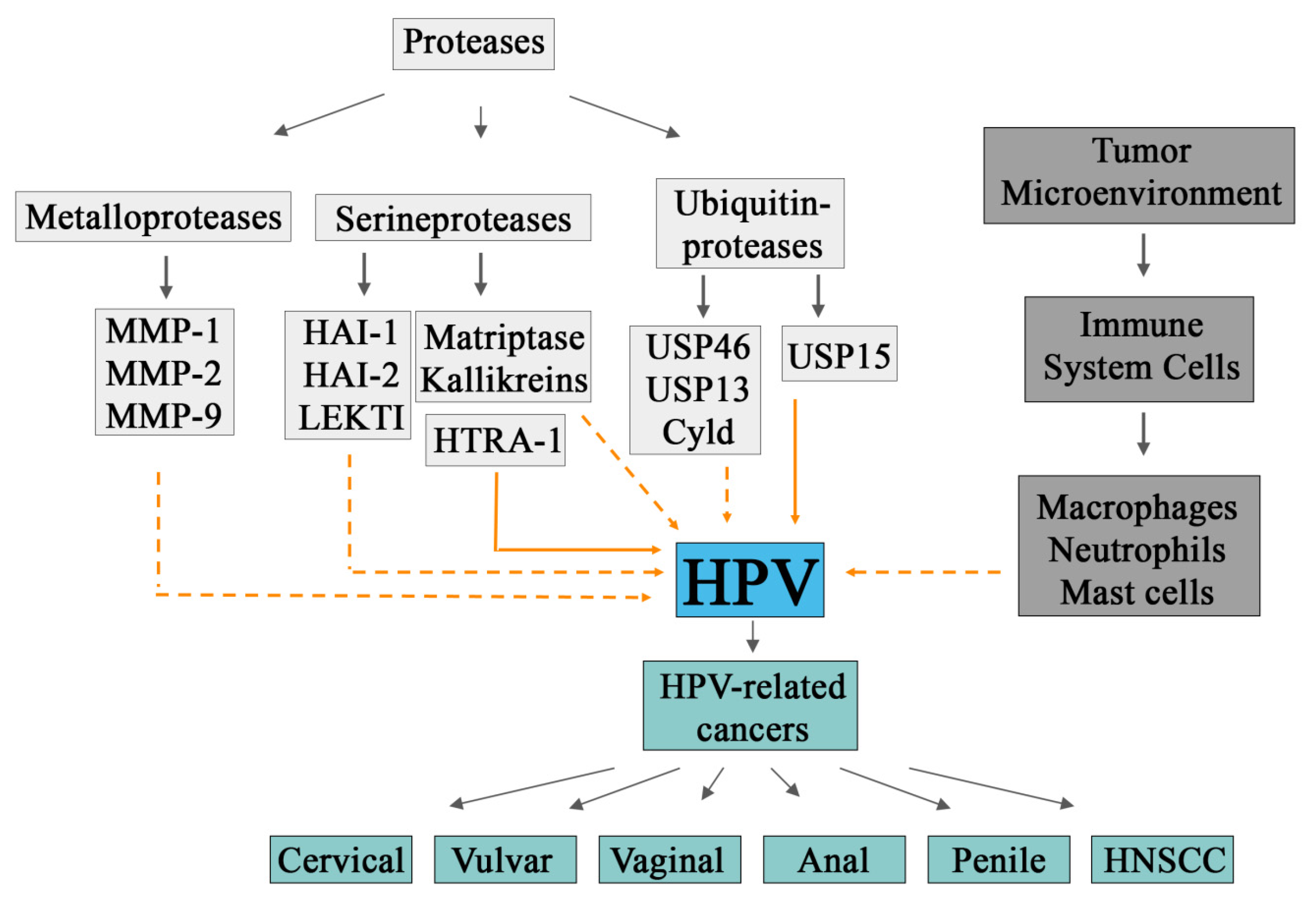
3. Proteases and HPV Carcinogenesis
4. HPV and the Microenvironment
5. HPV and Cervical Intraepithelial Lesions
6. HPV and Cervical Cancer
7. HPV and Vulvar Cancer
8. HPV and Vaginal Cancer
9. HPV and Anal Cancer
10. HPV and Penile Cancer
11. HPV and Head and Neck Squamous Cell Carcinoma (HNSCC)
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Graham, B.S.; Sullivan, N.J. Emerging viral diseases from a vaccinology perspective: Preparing for the next pandemic. Nat. Immunol. 2018, 19, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Fineberg, H.V. Pandemic Preparedness and Response—Lessons from the H1N1 Influenza of 2009. N. Engl. J. Med. 2014, 370, 1335–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, H.F.; Chan, L.W.C.; Cho, W.C.S.; Yu, A.C.S.; Yim, A.K.Y.; Chan, A.K.C.; Ng, L.P.W.; Wong, Y.K.E.; Pei, X.M.; Li, M.J.W.; et al. An update on COVID-19 pandemic: The epidemiology, pathogenesis, prevention and treatment strategies. Expert Rev. Anti Infect. Ther. 2021, 19, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Akram, N.; Imran, M.; Noreen, M.; Ahmed, F.; Atif, M.; Fatima, Z.; Bilal Waqar, A. Oncogenic Role of Tumor Viruses in Humans. Viral Immunol. 2017, 30, 20–27. [Google Scholar] [CrossRef]
- Martin, D.; Gutkind, J.S. Human tumor-associated viruses and new insights into the molecular mechanisms of cancer. Oncogene 2008, 27, S31–S42. [Google Scholar] [CrossRef] [Green Version]
- Pearce, N.; Blair, A.; Vineis, P.; Ahrens, W.; Andersen, A.; Anto, J.M.; Armstrong, B.K.; Baccarelli, A.A.; Beland, F.A.; Berrington, A.; et al. IARC Monographs: 40 Years of Evaluating Carcinogenic Hazards to Humans. Environ. Health Perspect. 2015, 123, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Li, D. Searching for human oncoviruses: Histories, challenges, and opportunities. J. Cell. Biochem. 2018, 119, 4897–4906. [Google Scholar] [CrossRef]
- Chen, C.-J.; Hsu, W.-L.; Yang, H.-I.; Lee, M.-H.; Chen, H.-C.; Chien, Y.-C.; You, S.-L. Epidemiology of Virus Infection and Human Cancer. In Viruses and Human Cancer; Chang, M.H., Jeang, K.-T., Eds.; Recent Results in Cancer Research; Springer: Berlin/Heidelberg, Germany, 2014; Volume 193, pp. 11–32. ISBN 978-3-642-38964-1. [Google Scholar]
- Weinberg, R.A. The Biology of Cancer; W.W. Norton & Company: New York City, NY, USA, 2013; ISBN 978-1-317-96346-2. [Google Scholar]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human Viral Oncogenesis: A Cancer Hallmarks Analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primer 2016, 2, 16086. [Google Scholar] [CrossRef] [PubMed]
- de Sanjosé, S.; Brotons, M.; Pavón, M.A. The natural history of human papillomavirus infection. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Serrano, B.; Brotons, M.; Bosch, F.X.; Bruni, L. Epidemiology and burden of HPV-related disease. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Anna Szymonowicz, K.; Chen, J. Biological and clinical aspects of HPV-related cancers. Cancer Biol. Med. 2020, 17, 864–878. [Google Scholar] [CrossRef]
- Petca, A.; Borislavschi, A.; Zvanca, M.; Petca, R.-C.; Sandru, F.; Dumitrascu, M. Non-sexual HPV transmission and role of vaccination for a better future (Review). Exp. Ther. Med. 2020, 20, 1. [Google Scholar] [CrossRef]
- Rintala, M.A.M.; Grénman, S.E.; Puranen, M.H.; Isolauri, E.; Ekblad, U.; Kero, P.O.; Syrjänen, S.M. Transmission of High-Risk Human Papillomavirus (HPV) between Parents and Infant: A Prospective Study of HPV in Families in Finland. J. Clin. Microbiol. 2005, 43, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.M.; Parker, M.A.; Rubenstein, L.M.; Haugen, T.H.; Hamsikova, E.; Turek, L.P. Evidence for Vertical Transmission of HPV from Mothers to Infants. Infect. Dis. Obstet. Gynecol. 2010, 2010, 326369. [Google Scholar] [CrossRef]
- Dassi, L.; Annunziata, C.; Botti, C.; Micillo, A.; Cerasuolo, A.; Starita, N.; Buonaguro, F.M.; Tornesello, M.L. Detection of Human Papillomaviruses in the Nasopharynx of Breastfed Infants: New Findings and Meta-Analysis. Viruses 2020, 12, 1119. [Google Scholar] [CrossRef]
- Sinal, S.H.; Woods, C.R. Human Papillomavirus Infections of the Genital and Respiratory Tracts in Young Children. Semin. Pediatr. Infect. Dis. 2005, 16, 306–316. [Google Scholar] [CrossRef]
- Bussen, S.; Sütterlin, M.; Schmidt, U.; Bussen, D. Anogenital Warts in Childhood—Always a Marker for Sexual Abuse? Geburtshilfe Frauenheilkd. 2012, 72, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, C.D.; Merjanian, L.; Pierre, J.; Balica, A. A Discussion of High-Risk HPV in a 6-Year-Old Female Survivor of Child Sexual Abuse. Case Rep. Obstet. Gynecol. 2017, 2017, 6014026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betz, S.J. HPV-Related Papillary Lesions of the Oral Mucosa: A Review. Head Neck Pathol. 2019, 13, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The Biology and Life-Cycle of Human Papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef]
- Vonsky, M.; Shabaeva, M.; Runov, A.; Lebedeva, N.; Chowdhury, S.; Palefsky, J.M.; Isaguliants, M. Carcinogenesis Associated with Human Papillomavirus Infection. Mechanisms and Potential for Immunotherapy. Biochem. Mosc. 2019, 84, 782–799. [Google Scholar] [CrossRef]
- Haedicke, J.; Iftner, T. Human papillomaviruses and cancer. Radiother. Oncol. 2013, 108, 397–402. [Google Scholar] [CrossRef]
- Gheit, T. Mucosal and Cutaneous Human Papillomavirus Infections and Cancer Biology. Front. Oncol. 2019, 9, 355. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, Á.M.; Ruiz, J.E.; Gavilanes, A.V.; Eriksson, T.; Lehtinen, M.; Pérez, G.; Sings, H.L.; James, M.K.; Haupt, R.M. Proximity of First Sexual Intercourse to Menarche and Risk of High-Grade Cervical Disease. J. Infect. Dis. 2012, 206, 1887–1896. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.A.; Costa, M.C.; Alves, R.R.F.; Villa, L.L.; Saddi, V.A.; dos Santos Carneiro, M.A.; Zeferino, L.C.; Rabelo-Santos, S.H. HPV infection and cervical neoplasia: Associated risk factors. Infect. Agent. Cancer 2015, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Bruni, L.; Albero, G.; Serrano, B.; Mena, M.; Collado, J.; Gómez, D.; Muñoz, J.; Bosch, F.; de Sanjosé, S. Human Papillomavirus and Related Diseases in the World Summary Report. Available online: Chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://hpvcentre.net/statistics/reports/XWX.pdf (accessed on 1 May 2022).
- Estêvão, D.; Costa, N.R.; Gil da Costa, R.M.; Medeiros, R. Hallmarks of HPV carcinogenesis: The role of E6, E7 and E5 oncoproteins in cellular malignancy. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2019, 1862, 153–162. [Google Scholar] [CrossRef]
- Münger, K.; Howley, P.M. Human papillomavirus immortalization and transformation functions. Virus Res. 2002, 89, 213–228. [Google Scholar] [CrossRef]
- Shai, A.; Brake, T.; Somoza, C.; Lambert, P.F. The Human Papillomavirus E6 Oncogene Dysregulates the Cell Cycle and Contributes to Cervical Carcinogenesis through Two Independent Activities. Cancer Res. 2007, 67, 1626–1635. [Google Scholar] [CrossRef] [Green Version]
- Danos, O.; Katinka, M.; Yaniv, M. Human papillomavirus 1a complete DNA sequence: A novel type of genome organization among papovaviridae. EMBO J. 1982, 1, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.-P.; Ando, T.; Fankhauser, R.L.; Beard, R.S.; Glass, R.I.; Monroe, S.S. Norovirus classification and proposed strain nomenclature. Virology 2006, 346, 312–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, C.B.; Cheng, N.; Thompson, C.D.; Lowy, D.R.; Steven, A.C.; Schiller, J.T.; Trus, B.L. Arrangement of L2 within the Papillomavirus Capsid. J. Virol. 2008, 82, 5190–5197. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Mahata, S.; Shishodia, G.; Pande, S.; Verma, G.; Hedau, S.; Bhambhani, S.; Kumari, A.; Batra, S.; Basir, S.F.; et al. Physical state & copy number of high risk human papillomavirus type 16 DNA in progression of cervical cancer. Indian J. Med. Res. 2014, 139, 531. [Google Scholar]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [CrossRef] [Green Version]
- Peitsaro, P.; Johansson, B.; Syrjänen, S. Integrated Human Papillomavirus Type 16 Is Frequently Found in Cervical Cancer Precursors as Demonstrated by a Novel Quantitative Real-Time PCR Technique. J. Clin. Microbiol. 2002, 40, 886–891. [Google Scholar] [CrossRef] [Green Version]
- Kalof, A.N.; Cooper, K. Our approach to squamous intraepithelial lesions of the uterine cervix. J. Clin. Pathol. 2006, 60, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Mello, V.; Sundstrom, R.K. Cervical Intraepithelial Neoplasia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK544371/ (accessed on 2 May 2022).
- Chellappan, S.; Kraus, V.B.; Kroger, B.; Munger, K.; Howley, P.M.; Phelps, W.C.; Nevins, J.R. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. USA 1992, 89, 4549–4553. [Google Scholar] [CrossRef] [Green Version]
- Helin, K.; Harlow, E.; Fattaey, A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol. Cell. Biol. 1993, 13, 6501–6508. [Google Scholar] [CrossRef] [PubMed]
- Zerfass-Thome, K.; Zwerschke, W.; Mannhardt, B.; Tindle, R.; Botz, J.W.; Jansen-Dürr, P. Inactivation of the CDK Inhibitor p27KIP1 by the Human Papillomavirus Type 16 E7 Oncoprotein—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/8957073/ (accessed on 3 May 2022).
- Jones, D.L.; Alani, R.M.; Münger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 1997, 11, 2101–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demers, G.W.; Halbert, C.L.; Galloway, D.A. Elevated Wild-Type p53 Protein Levels in Human Epithelial Cell Lines Immortalized by the Human Papillomavirus Type 16 E7 Gene. Virology 1994, 198, 169–174. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Scheffner, M.; Whitaker, N.J. Human papillomavirus-induced carcinogenesis and the ubiquitin–proteasome system. Semin. Cancer Biol. 2003, 13, 59–67. [Google Scholar] [CrossRef]
- Martínez-Bailón, C.; Mantilla-Morales, A.; Méndez-Matías, G.; Alvarado-Cabrero, I.; Maldonado-Rodríguez, R.; Quintero-Becerra, J.; Arias-Flores, R.; Piña-Sánchez, P. Human papillomavirus genotypes and P16INK4A expression in squamous penile carcinoma in Mexican patients. BMC Infect. Dis. 2019, 19, 1068. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dakic, A.; Zhang, Y.; Dai, Y.; Chen, R.; Schlegel, R. HPV E6 protein interacts physically and functionally with the cellular telomerase complex. Proc. Natl. Acad. Sci. USA 2009, 106, 18780–18785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzenellenbogen, R.A. Activation of telomerase by HPVs. Virus Res. 2017, 231, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Kim, E.-J.; Kwon, H.-J.; Hwang, E.-S.; Namkoong, S.-E.; Um, S.-J. Inactivation of Interferon Regulatory Factor-1 Tumor Suppressor Protein by HPV E7 Oncoprotein. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [Green Version]
- Beglin, M.; Melar-New, M.; Laimins, L. Human Papillomaviruses and the Interferon Response. J. Interferon Cytokine Res. 2009, 29, 629–635. [Google Scholar] [CrossRef]
- Abraham, A.G.; D’Souza, G.; Jing, Y.; Gange, S.J.; Sterling, T.R.; Silverberg, M.J.; Saag, M.S.; Rourke, S.B.; Rachlis, A.; Napravnik, S.; et al. Invasive Cervical Cancer Risk Among HIV-Infected Women: A North American Multicohort Collaboration Prospective Study. JAIDS J. Acquir. Immune Defic. Syndr. 2013, 62, 405–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Martel, C.; Shiels, M.S.; Franceschi, S.; Simard, E.P.; Vignat, J.; Hall, H.I.; Engels, E.A.; Plummer, M. Cancers attributable to infections among adults with HIV in the United States. AIDS Lond. Engl. 2015, 29, 2173–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention (CDC) Revised surveillance case definition for HIV infection--United States, 2014. MMWR Recomm. Rep. Morb. Mortal. Wkly. Rep. Recomm. Rep. 2014, 63, 1–10.
- Guiguet, M.; Boué, F.; Cadranel, J.; Lang, J.-M.; Rosenthal, E.; Costagliola, D. Effect of immunodeficiency, HIV viral load, and antiretroviral therapy on the risk of individual malignancies (FHDH-ANRS CO4): A prospective cohort study. Lancet Oncol. 2009, 10, 1152–1159. [Google Scholar] [CrossRef]
- Sajid, M.; McKerrow, J.H. Cysteine proteases of parasitic organisms. Mol. Biochem. Parasitol. 2002, 120, 1–21. [Google Scholar] [CrossRef]
- Ivey, M.E.; Little, P.J. Thrombin regulates vascular smooth muscle cell proteoglycan synthesis via PAR-1 and multiple downstream signalling pathways. Thromb. Res. 2008, 123, 288–297. [Google Scholar] [CrossRef]
- Varghese, V.; Shahriar, R.; Rhee, S.-Y.; Liu, T.; Simen, B.B.; Egholm, M.; Hanczaruk, B.; Blake, L.A.; Gharizadeh, B.; Babrzadeh, F.; et al. Minority variants associated with transmitted and acquired HIV-1 nonnucleoside reverse transcriptase inhibitor resistance: Implications for the use of second-generation nonnucleoside reverse transcriptase inhibitors. J. Acquir. Immune Defic. Syndr. 1999 2009, 52, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Wensing, A.M.J.; van Maarseveen, N.M.; Nijhuis, M. Fifteen years of HIV Protease Inhibitors: Raising the barrier to resistance. Antiviral Res. 2010, 85, 59–74. [Google Scholar] [CrossRef]
- Lu, D.; Sham, Y.Y.; Vince, R. Design, asymmetric synthesis, and evaluation of pseudosymmetric sulfoximine inhibitors against HIV-1 protease. Bioorg. Med. Chem. 2010, 18, 2037–2048. [Google Scholar] [CrossRef]
- Adrian Meredith, J.; Wallberg, H.; Vrang, L.; Oscarson, S.; Parkes, K.; Hallberg, A.; Samuelsson, B. Design and synthesis of novel P2 substituents in diol-based HIV protease inhibitors. Eur. J. Med. Chem. 2010, 45, 160–170. [Google Scholar] [CrossRef]
- Perona, J.J.; Craik, C.S. Evolutionary Divergence of Substrate Specificity within the Chymotrypsin-like Serine Protease Fold. J. Biol. Chem. 1997, 272, 29987–29990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillmor, S.A.; Craik, C.S.; Fletterick, R.J. Structural determinants of specificity in the cysteine protease cruzain. Protein Sci. 1997, 6, 1603–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maupin-Furlow, J.A.; Gil, M.A.; Humbard, M.A.; Kirkland, P.A.; Li, W.; Reuter, C.J.; Wright, A.J. Archaeal proteasomes and other regulatory proteases. Curr. Opin. Microbiol. 2005, 8, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Diamond, S.L. Methods for mapping protease specificity. Curr. Opin. Chem. Biol. 2007, 11, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Gurumallesh, P.; Alagu, K.; Ramakrishnan, B.; Muthusamy, S. A systematic reconsideration on proteases. Int. J. Biol. Macromol. 2019, 128, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K. Proteases cathepsins—A view. Biochem. Educ. 1990, 18, 67–72. [Google Scholar] [CrossRef]
- Mótyán, J.; Tóth, F.; Tőzsér, J. Research Applications of Proteolytic Enzymes in Molecular Biology. Biomolecules 2013, 3, 923–942. [Google Scholar] [CrossRef] [Green Version]
- Sanman, L.E.; Bogyo, M. Activity-Based Profiling of Proteases. Annu. Rev. Biochem. 2014, 83, 249–273. [Google Scholar] [CrossRef] [Green Version]
- Bastians, H.; Topper, L.M.; Gorbsky, G.L.; Ruderman, J.V. Cell Cycle–regulated Proteolysis of Mitotic Target Proteins. Mol. Biol. Cell 1999, 10, 3927–3941. [Google Scholar] [CrossRef] [Green Version]
- Paliouras, M.; Borgono, C.; Diamandis, E.P. Human tissue kallikreins: The cancer biomarker family. Cancer Lett. 2007, 249, 61–79. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Szabo, R.; Bugge, T.H. Membrane-Anchored Serine Proteases in Vertebrate Cell and Developmental Biology. Annu. Rev. Cell Dev. Biol. 2011, 27, 213–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.R.; Khazanovich-Bernstein, N.; Bergmann, E.M.; James, M.N.G. Structural aspects of activation pathways of aspartic protease zymogens and viral 3C protease precursors. Proc. Natl. Acad. Sci. USA 1999, 96, 10968–10975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, A.S.; Varela, F.A.; List, K. Type II transmembrane serine proteases as potential targets for cancer therapy. Biol. Chem. 2016, 397, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Di Cera, E. Serine proteases. IUBMB Life 2009, 61, 510–515. [Google Scholar] [CrossRef]
- Puente, X.S.; Sánchez, L.M.; Gutiérrez-Fernández, A.; Velasco, G.; López-Otín, C. A genomic view of the complexity of mammalian proteolytic systems. Biochem. Soc. Trans. 2005, 33, 331–334. [Google Scholar] [CrossRef]
- Szabo, R.; Bugge, T.H. Membrane-anchored serine proteases as regulators of epithelial function. Biochem. Soc. Trans. 2020, 48, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, T.; Shuman, M.A.; Craik, C.S. Reverse biochemistry: Use of macromolecular protease inhibitors to dissect complex biological processes and identify a membrane-type serine protease in epithelial cancer and normal tissue. Proc. Natl. Acad. Sci. USA 1999, 96, 11054–11061. [Google Scholar] [CrossRef] [Green Version]
- Oberst, M.D.; Johnson, M.D.; Dickson, R.B.; Lin, C.-Y.; Singh, B.; Stewart, M.; Williams, A.; al-Nafussi, A.; Smyth, J.F.; Gabra, H.; et al. Expression of the serine protease matriptase and its inhibitor HAI-1 in epithelial ovarian cancer: Correlation with clinical outcome and tumor clinicopathological parameters. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 1101–1107. [Google Scholar]
- Sidenius, N.; Blasi, F. The urokinase plasminogen activator system in cancer: Recent advances and implication for prognosis and therapy. Cancer Metastasis Rev. 2003, 22, 205–222. [Google Scholar] [CrossRef]
- Uhland, K. Matriptase and its putative role in cancer. Cell. Mol. Life Sci. 2006, 63, 2968–2978. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kobayashi, H.; Kanayama, N.; Saga, Y.; Suzuki, M.; Lin, C.-Y.; Dickson, R.B.; Terao, T. Inhibition of Tumor Invasion by Genomic Down-regulation of Matriptase through Suppression of Activation of Receptor-bound Pro-urokinase. J. Biol. Chem. 2004, 279, 14899–14908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Förbs, D.; Thiel, S.; Stella, M.; Stürzebecher, A.; Schweinitz, A.; Steinmetzer, T.; Stürzebecher, J.; Uhland, K. In vitro inhibition of matriptase prevents invasive growth of cell lines of prostate and colon carcinoma. Int. J. Oncol. 2005, 27, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Demerjian, M.; Hachem, J.-P.; Tschachler, E.; Denecker, G.; Declercq, W.; Vandenabeele, P.; Mauro, T.; Hupe, M.; Crumrine, D.; Roelandt, T.; et al. Acute Modulations in Permeability Barrier Function Regulate Epidermal Cornification. Am. J. Pathol. 2008, 172, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Sales, K.U.; Friis, S.; Konkel, J.E.; Godiksen, S.; Hatakeyama, M.; Hansen, K.K.; Rogatto, S.R.; Szabo, R.; Vogel, L.K.; Chen, W.; et al. Non-hematopoietic PAR-2 is essential for matriptase-driven pre-malignant progression and potentiation of ras-mediated squamous cell carcinogenesis. Oncogene 2015, 34, 346–356. [Google Scholar] [CrossRef] [Green Version]
- List, K.; Szabo, R.; Molinolo, A.; Sriuranpong, V.; Redeye, V.; Murdock, T.; Burke, B.; Nielsen, B.S.; Gutkind, J.S.; Bugge, T.H. Deregulated matriptase causes ras -independent multistage carcinogenesis and promotes ras -mediated malignant transformation. Genes Dev. 2005, 19, 1934–1950. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.-F.; Tzao, C.; Tsai, W.-C.; Lee, W.-H.; Chen, A.; Chiang, H.; Sheu, L.-F.; Jin, J.-S. Expression of Emmprin and matriptase in esophageal squamous cell carcinoma: Correlation with clinicopathological parameters. Dis. Esophagus 2006, 19, 482–486. [Google Scholar] [CrossRef]
- Vogel, L.K.; Sæbø, M.; Skjelbred, C.F.; Abell, K.; Pedersen, E.D.; Vogel, U.; Kure, E.H. The ratio of Matriptase/HAI-1mRNA is higher in colorectal cancer adenomas and carcinomas than corresponding tissue from control individuals. BMC Cancer 2006, 6, 176. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.-F.; Huang, M.-S.; Lin, C.-S.; Lin, L.-H.; Lee, H.-S.; Jiang, J.-C.; Hsia, K.-T. Expression of matriptase correlates with tumour progression and clinical prognosis in oral squamous cell carcinoma. Histopathology 2014, 65, 24–34. [Google Scholar] [CrossRef]
- Kanemaru, K.; Nakamura, Y.; Totoki, K.; Fukuyama, T.; Shoji, M.; Kaneko, H.; Shiratori, K.; Yoneda, A.; Inoue, T.; Iwakura, Y.; et al. Phospholipase Cδ1 regulates p38 MAPK activity and skin barrier integrity. Cell Death Differ. 2017, 24, 1079–1090. [Google Scholar] [CrossRef] [Green Version]
- Szabo, R.; Bugge, T. Type II transmembrane serine proteases in development and disease. Int. J. Biochem. Cell Biol. 2008, 40, 1297–1316. [Google Scholar] [CrossRef] [PubMed]
- Nonboe, A.W.; Krigslund, O.; Soendergaard, C.; Skovbjerg, S.; Friis, S.; Andersen, M.N.; Ellis, V.; Kawaguchi, M.; Kataoka, H.; Bugge, T.H.; et al. HAI-2 stabilizes, inhibits and regulates SEA-cleavage-dependent secretory transport of matriptase. Traffic 2017, 18, 378–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura The role of hepatocyte growth factor activator inhibitor-1 (HAI-1) as a prognostic indicator in cervical cancer. Int. J. Oncol. 2009, 35, 239–248. [CrossRef] [Green Version]
- Nakamura, K.; Abarzua, F.; Hongo, A.; Kodama, J.; Nasu, Y.; Kumon, H.; Hiramatsu, Y. Hepatocyte growth factor activator inhibitor-2 (HAI-2) is a favorable prognosis marker and inhibits cell growth through the apoptotic pathway in cervical cancer. Ann. Oncol. 2009, 20, 63–70. [Google Scholar] [CrossRef]
- Sotiropoulou, G.; Pampalakis, G.; Diamandis, E.P. Functional Roles of Human Kallikrein-related Peptidases. J. Biol. Chem. 2009, 284, 32989–32994. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, B.S.; Whittaker, G.R. Cleavage Activation of Human-adapted Influenza Virus Subtypes by Kallikrein-related Peptidases 5 and 12. J. Biol. Chem. 2013, 288, 17399–17407. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.; Greune, L.; Schmidt, M.A.; Schelhaas, M. Extracellular Conformational Changes in the Capsid of Human Papillomaviruses Contribute to Asynchronous Uptake into Host Cells. J. Virol. 2018, 92, e02106-17. [Google Scholar] [CrossRef] [Green Version]
- Giroglou, T.; Florin, L.; Schäfer, F.; Streeck, R.E.; Sapp, M. Human Papillomavirus Infection Requires Cell Surface Heparan Sulfate. J. Virol. 2001, 75, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Culp, T.D.; Budgeon, L.R.; Marinkovich, M.P.; Meneguzzi, G.; Christensen, N.D. Keratinocyte-Secreted Laminin 5 Can Function as a Transient Receptor for Human Papillomaviruses by Binding Virions and Transferring Them to Adjacent Cells. J. Virol. 2006, 80, 8940–8950. [Google Scholar] [CrossRef] [Green Version]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [Green Version]
- Cerqueira, C.; Samperio Ventayol, P.; Vogeley, C.; Schelhaas, M. Kallikrein-8 Proteolytically Processes Human Papillomaviruses in the Extracellular Space to Facilitate Entry into Host Cells. J. Virol. 2015, 89, 7038–7052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Wang, J.; Cheng, J.; Zang, C.; Chen, F.; Wang, W.; Zhao, H.; Wang, Y.; Wang, D. Comprehensive Identification of the Human Secretome as Potential Indicators in Treatment Outcome of HPV-Positive and -Negative Cervical Cancer Patients. Gynecol. Obstet. Investig. 2020, 85, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Brummer, O.; Böhmer, G.; Hollwitz, B.; Flemming, P.; Petry, K.-U.; Kühnle, H. MMP-1 and MMP-2 in the Cervix Uteri in Different Steps of Malignant Transformation—An Immunohistochemical Study. Gynecol. Oncol. 2002, 84, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Woessner, J.F. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991, 5, 2145–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolmatov, I.Y.; Nizhnichenko, V.A.; Dolmatova, L.S. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Echinoderms: Structure and Possible Functions. Cells 2021, 10, 2331. [Google Scholar] [CrossRef] [PubMed]
- Sekton, B. Matrix metalloproteinases—An overview. Res. Rep. Biol. 2010, 1, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Solovyeva, N.I.; Timoshenko, O.S.; Kugaevskaya, E.V.; Gureeva, T.A. Interstitial collagenase MMP-1 and EMMPRIN in cell lines and in clinical specimens of cervical squamous cell carcinoma. Mol. Biol. Rep. 2021, 48, 6879–6886. [Google Scholar] [CrossRef]
- Davidson, B.; Goldberg, I.; Gotlieb, W.H.; Lerner-Geva, L.; Ben-Baruch, G.; Agulansky, L.; Novikov, I.; Kopolovic, J. Macrophage infiltration and angiogenesis in cervical squamous cell carcinomaclinicopathologic correlation. Acta Obstet. Gynecol. Scand. 1999, 78, 240–244. [Google Scholar] [CrossRef]
- Lee, S.S.; Weiss, R.S.; Javier, R.T. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6670–6675. [Google Scholar] [CrossRef] [Green Version]
- Kiyono, T.; Hiraiwa, A.; Fujita, M.; Hayashi, Y.; Akiyama, T.; Ishibashi, M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 11612–11616. [Google Scholar] [CrossRef] [Green Version]
- Grau, S.; Richards, P.J.; Kerr, B.; Hughes, C.; Caterson, B.; Williams, A.S.; Junker, U.; Jones, S.A.; Clausen, T.; Ehrmann, M. The Role of Human HtrA1 in Arthritic Disease. J. Biol. Chem. 2006, 281, 6124–6129. [Google Scholar] [CrossRef] [Green Version]
- Zurawa-Janicka, D.; Skorko-Glonek, J.; Lipinska, B. HtrA proteins as targets in therapy of cancer and other diseases. Expert Opin. Ther. Targets 2010, 14, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Vierkotten, S.; Muether, P.S.; Fauser, S. Overexpression of HTRA1 Leads to Ultrastructural Changes in the Elastic Layer of Bruch’s Membrane via Cleavage of Extracellular Matrix Components. PLoS ONE 2011, 6, e22959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Khurana, A.; Maguire, J.L.; Chien, J.; Shridhar, V. HtrA1 sensitizes ovarian cancer cells to cisplatin-induced cytotoxicity by targeting XIAP for degradation. Int. J. Cancer 2012, 130, 1029–1035. [Google Scholar] [CrossRef] [Green Version]
- Stuqui, B.; Conceição, A.L.G.; Termini, L.; Sichero, L.; Villa, L.L.; Rahal, P.; Calmon, M. de F. The differential role of HTRA1 in HPV-positive and HPV-negative cervical cell line proliferation. BMC Cancer 2016, 16, 840. [Google Scholar] [CrossRef] [Green Version]
- Kiran, S.; Dar, A.; Singh, S.K.; Lee, K.Y.; Dutta, A. The Deubiquitinase USP46 Is Essential for Proliferation and Tumor Growth of HPV-Transformed Cancers. Mol. Cell 2018, 72, 823–835.e5. [Google Scholar] [CrossRef] [Green Version]
- Morgan, E.L.; Patterson, M.R.; Barba-Moreno, D.; Scarth, J.A.; Wilson, A.; Macdonald, A. The deubiquitinase (DUB) USP13 promotes Mcl-1 stabilisation in cervical cancer. Oncogene 2021, 40, 2112–2129. [Google Scholar] [CrossRef]
- Yaginuma, Y.; Yoshimoto, M.; Tokuda, A. USP15 inhibits HPV16 E6 degradation and catalytically inactive USP15 has reduced inhibitory activity. Acta Virol. 2018, 62, 147–156. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Mo, D.; Liu, H.; Veena, M.S.; Srivatsan, E.S.; Massoumi, R.; Rettig, M.B. Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-kappaB activation. Cancer Cell 2008, 14, 394–407. [Google Scholar] [CrossRef] [Green Version]
- van der Burg, S.H.; Piersma, S.J.; de Jong, A.; van der Hulst, J.M.; Kwappenberg, K.M.C.; van den Hende, M.; Welters, M.J.P.; Van Rood, J.J.; Fleuren, G.J.; Melief, C.J.M.; et al. Association of cervical cancer with the presence of CD4+ regulatory T cells specific for human papillomavirus antigens. Proc. Natl. Acad. Sci. USA 2007, 104, 12087–12092. [Google Scholar] [CrossRef] [Green Version]
- Hammes, L.; Tekmal, R.; Naud, P.; Edelweiss, M.; Kirma, N.; Valente, P.; Syrjanen, K.; Cunhafilho, J. Macrophages, inflammation and risk of cervical intraepithelial neoplasia (CIN) progression—Clinicopathological correlation. Gynecol. Oncol. 2007, 105, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Weinberg, V.; Darragh, T.; Smith-McCune, K. Evolving immunosuppressive microenvironment during human cervical carcinogenesis. Mucosal Immunol. 2008, 1, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Lepique, A.P.; Daghastanli, K.R.P.; Cuccovia, I.M.; Villa, L.L. HPV16 Tumor Associated Macrophages Suppress Antitumor T Cell Responses. Clin. Cancer Res. 2009, 15, 4391–4400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Investig. 2004, 114, 623–633. [Google Scholar] [CrossRef]
- Pahler, J.C.; Tazzyman, S.; Erez, N.; Chen, Y.-Y.; Murdoch, C.; Nozawa, H.; Lewis, C.E.; Hanahan, D. Plasticity in Tumor-Promoting Inflammation: Impairment of Macrophage Recruitment Evokes a Compensatory Neutrophil Response. Neoplasia 2008, 10, 329-IN2. [Google Scholar] [CrossRef] [Green Version]
- Benítez–Bribiesca, L.; Wong, A.; Utrera, D.; Castellanos, E. The Role of Mast Cell Tryptase in Neoangiogenesis of Premalignant and Malignant Lesions of the Uterine Cervix. J. Histochem. Cytochem. 2001, 49, 1061–1062. [Google Scholar] [CrossRef] [Green Version]
- Kalyani, R.; Rajeshwari, G. Significance of mast cells in non-neoplastic and neoplastic lesions of uterine cervix. Biomed. Res. Ther. 2016, 3, 3. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1—NFκB—IL-6 signalling axis. PLoS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef] [Green Version]
- Kawano, M.; Mabuchi, S.; Matsumoto, Y.; Sasano, T.; Takahashi, R.; Kuroda, H.; Kozasa, K.; Hashimoto, K.; Isobe, A.; Sawada, K.; et al. The significance of G-CSF expression and myeloid-derived suppressor cells in the chemoresistance of uterine cervical cancer. Sci. Rep. 2015, 5, 18217. [Google Scholar] [CrossRef]
- Mabuchi, S.; Matsumoto, Y.; Kawano, M.; Minami, K.; Seo, Y.; Sasano, T.; Takahashi, R.; Kuroda, H.; Hisamatsu, T.; Kakigano, A.; et al. Uterine Cervical Cancer Displaying Tumor-Related Leukocytosis: A Distinct Clinical Entity with Radioresistant Feature. JNCI J. Natl. Cancer Inst. 2014, 106, dju147. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, H.; Auer, R.; Hörmann, R.; Pechriggl, E.; Regauer, S.; Reich, O. The development of the human vaginal fornix and the portio cervicis. Clin. Anat. 2021, 34, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Prendiville, W.; Sankaranarayanan, R. Colposcopy and Treatment of Cervical Precancer; IARC Technical Publications; International Agency for Research on Cancer: Lyon, FR, USA, 2017; ISBN 978-92-832-2458-7. Available online: http://www.ncbi.nlm.nih.gov/books/NBK568370/ (accessed on 3 May 2022).
- Reich, O.; Regauer, S.; McCluggage, W.G.; Bergeron, C.; Redman, C. Defining the Cervical Transformation Zone and Squamocolumnar Junction: Can We Reach a Common Colposcopic and Histologic Definition? Int. J. Gynecol. Pathol. 2017, 36, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Tzafetas, M.; Lyons, D.; Fotopoulou, C.; Paraskevaidis, E.; Kyrgiou, M. Cervical intraepithelial neoplasia: Screening and management. Br. J. Hosp. Med. 2016, 77, C118–C123. [Google Scholar] [CrossRef]
- Darragh, T.M.; Colgan, T.J.; Cox, J.T.; Heller, D.S.; Henry, M.R.; Luff, R.D.; McCalmont, T.; Nayar, R.; Palefsky, J.M.; Stoler, M.H.; et al. The Lower Anogenital Squamous Terminology Standardization Project for HPV-Associated Lesions: Background and Consensus Recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Arch. Pathol. Lab. Med. 2012, 136, 1266–1297. [Google Scholar] [CrossRef]
- Solomon, D. The 2001 Bethesda SystemTerminology for Reporting Results of Cervical Cytology. JAMA 2002, 287, 2114. [Google Scholar] [CrossRef]
- Loopik, D.L.; Bentley, H.A.; Eijgenraam, M.N.; IntHout, J.; Bekkers, R.L.M.; Bentley, J.R. The Natural History of Cervical Intraepithelial Neoplasia Grades 1, 2, and 3: A Systematic Review and Meta-analysis. J. Low. Genit. Tract Dis. 2021, 25, 221–231. [Google Scholar] [CrossRef]
- Zielinski, G.D.; Snijders, P.J.F.; Rozendaal, L.; Voorhorst, F.J.; van der Linden, H.C.; Runsink, A.P.; de Schipper, F.A.; Meijer, C.J.L.M. HPV presence precedes abnormal cytology in women developing cervical cancer and signals false negative smears. Br. J. Cancer 2001, 85, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Snijders, P.J.; Steenbergen, R.D.; Heideman, D.A.; Meijer, C.J. HPV-mediated cervical carcinogenesis: Concepts and clinical implications. J. Pathol. 2006, 208, 152–164. [Google Scholar] [CrossRef]
- Kudela, E.; Holubekova, V.; Farkasova, A.; Danko, J. Determination of malignant potential of cervical intraepithelial neoplasia. Tumor Biol. 2016, 37, 1521–1525. [Google Scholar] [CrossRef]
- Matheus, E.R.; Zonta, M.A.; Discacciati, M.G.; Paruci, P.; Velame, F.; Cardeal, L.B.S.; Barros, S.B.M.; Pignatari, A.C.; Maria-Engler, S.S. MMP-9 expression increases according to the grade of squamous intraepithelial lesion in cervical smears. Diagn. Cytopathol. 2014, 42, 827–833. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Okunade, K.S. Human papillomavirus and cervical cancer. J. Obstet. Gynaecol. 2020, 40, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, F.; Björkholm, E.; Näslund, I. Evaluation of Screening for Cervical Cancer in Sweden: Trends in Incidence and Mortality 1958–1980. Int. J. Epidemiol. 1985, 14, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.; Babb, P.; Jones, J.; Allen, E. Effect of screening on incidence of and mortality from cancer of cervix in England: Evaluation based on routinely collected statistics. BMJ 1999, 318, 904. [Google Scholar] [CrossRef] [Green Version]
- Willoughby, B.J.; Faulkner, K.; Stamp, E.C.; Whitaker, C.J. A descriptive study of the decline in cervical screening coverage rates in the North East and Yorkshire and the Humber Regions of the UK from 1995 to 2005. J. Public Health 2006, 28, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Patel, C.; Brotherton, J.M.; Pillsbury, A.; Jayasinghe, S.; Donovan, B.; Macartney, K.; Marshall, H. The impact of 10 years of human papillomavirus (HPV) vaccination in Australia: What additional disease burden will a nonavalent vaccine prevent? Eurosurveillance 2018, 23, 1700737. [Google Scholar] [CrossRef]
- Williams, N.L.; Werner, T.L.; Jarboe, E.A.; Gaffney, D.K. Adenocarcinoma of the Cervix: Should We Treat It Differently? Curr. Oncol. Rep. 2015, 17, 17. [Google Scholar] [CrossRef]
- Karlsson, R.; Jonsson, M.; Edlund, K.; Evander, M.; Gustavsson, Å.; Bodén, E.; Rylander, E.; Wadell, G. Lifetime Number of Partners As the Only Independent Risk Factor for Human Papillomavirus Infection: A Population-Based Study. Sex. Transm. Dis. 1995, 22, 119–127. [Google Scholar] [CrossRef]
- Burk, R.D.; Ho, G.Y.F.; Beardsley, L.; Lempa, M.; Peters, M.; Bierman, R. Sexual Behavior and Partner Characteristics Are the Predominant Risk Factors for Genital Human Papillomavirus Infection in Young Women. J. Infect. Dis. 1996, 174, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Winer, R.L. Genital Human Papillomavirus Infection: Incidence and Risk Factors in a Cohort of Female University Students. Am. J. Epidemiol. 2003, 157, 218–226. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.R.; Shew, M.L.; Qadadri, B.; Neptune, N.; Vargas, M.; Tu, W.; Juliar, B.E.; Breen, T.E.; Fortenberry, J.D. A Longitudinal Study of Genital Human Papillomavirus Infection in a Cohort of Closely Followed Adolescent Women. J. Infect. Dis. 2005, 191, 182–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelimo, C.; Wouldes, T.A.; Cameron, L.D.; Elwood, J.M. Risk factors for and prevention of human papillomaviruses (HPV), genital warts and cervical cancer. J. Infect. 2013, 66, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.A.; James, D.; Marzan, A.; Armaos, M. Cervical Cancer: An Overview of Pathophysiology and Management. Semin. Oncol. Nurs. 2019, 35, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Vesco, K.K.; Whitlock, E.P.; Eder, M.; Burda, B.U.; Senger, C.A.; Lutz, K. Risk Factors and Other Epidemiologic Considerations for Cervical Cancer Screening: A Narrative Review for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2011, 155, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.; Kahesa, C.; Mwaiselage, J.; West, J.T.; Wood, C.; Angeletti, P.C. How the Cervical Microbiota Contributes to Cervical Cancer Risk in Sub-Saharan Africa. Front. Cell. Infect. Microbiol. 2020, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.; Cerqueira, F.; Medeiros, R. Chlamydia trachomatis infection: Implications for HPV status and cervical cancer. Arch. Gynecol. Obstet. 2014, 289, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shen, Z.; Luo, H.; Zhang, W.; Zhu, X. Chlamydia Trachomatis Infection-Associated Risk of Cervical Cancer: A Meta-Analysis. Medicine 2016, 95, e3077. [Google Scholar] [CrossRef]
- Karim, S.; Souho, T.; Benlemlih, M.; Bennani, B. Cervical Cancer Induction Enhancement Potential of Chlamydia Trachomatis: A Systematic Review. Curr. Microbiol. 2018, 75, 1667–1674. [Google Scholar] [CrossRef]
- Khan, A.A.; Abuderman, A.A.; Ashraf, M.T.; Khan, Z. Protein-protein interactions of HPV-Chlamydia trachomatis-human and their potential in cervical cancer. Future Microbiol. 2020, 15, 509–520. [Google Scholar] [CrossRef]
- The International Collaboration of Epidemiological Studies of Cervical Cancer Comparison of risk factors for invasive squamous cell carcinoma and adenocarcinoma of the cervix: Collaborative reanalysis of individual data on 8097 women with squamous cell carcinoma and 1374 women with adenocarcinoma from 12 epidemiological studies: Squamous Cell Carcinoma and Adenocarcinoma of the Cervix. Int. J. Cancer 2007, 120, 885–891. [CrossRef]
- Collins, S.; Rollason, T.P.; Young, L.S.; Woodman, C.B.J. Cigarette smoking is an independent risk factor for cervical intraepithelial neoplasia in young women: A longitudinal study. Eur. J. Cancer 2010, 46, 405–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinyemiju, T.F. Socio-economic and health access determinants of breast and cervical cancer screening in low-income countries: Analysis of the World Health Survey. PLoS ONE 2012, 7, e48834. [Google Scholar] [CrossRef] [PubMed]
- Chidyaonga-Maseko, F.; Chirwa, M.L.; Muula, A.S. Underutilization of cervical cancer prevention services in low and middle income countries: A review of contributing factors. Pan Afr. Med. J. 2015, 21, 231. [Google Scholar] [CrossRef] [PubMed]
- Akinyemiju, T.; Ogunsina, K.; Sakhuja, S.; Ogbhodo, V.; Braithwaite, D. Life-course socioeconomic status and breast and cervical cancer screening: Analysis of the WHO’s Study on Global Ageing and Adult Health (SAGE). BMJ Open. 2016, 6, e012753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adegoke, O.; Kulasingam, S.; Virnig, B. Cervical Cancer Trends in the United States: A 35-Year Population-Based Analysis. J. Womens Health 2012, 21, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Franceschi, S.; Howell-Jones, R.; Snijders, P.J.F.; Clifford, G.M. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: Variation by geographical region, histological type and year of publication. Int. J. Cancer 2011, 128, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Landolfi, S.; Olivella, A.; Lloveras, B.; Klaustermeier, J.; Suárez, H.; Alòs, L.; Puig-Tintoré, L.M.; Campo, E.; Ordi, J. p16 Overexpression Identifies HPV-positive Vulvar Squamous Cell Carcinomas. Am. J. Surg. Pathol. 2006, 30, 1347–1356. [Google Scholar] [CrossRef]
- Cheng, A.S.; Karnezis, A.N.; Jordan, S.; Singh, N.; McAlpine, J.N.; Gilks, C.B. p16 Immunostaining Allows for Accurate Subclassification of Vulvar Squamous Cell Carcinoma Into HPV-Associated and HPV-Independent Cases. Int. J. Gynecol. Pathol. 2016, 35, 385–393. [Google Scholar] [CrossRef]
- Kortekaas, K.E.; Bastiaannet, E.; van Doorn, H.C.; de Vos van Steenwijk, P.J.; Ewing-Graham, P.C.; Creutzberg, C.L.; Akdeniz, K.; Nooij, L.S.; van der Burg, S.H.; Bosse, T.; et al. Vulvar cancer subclassification by HPV and p53 status results in three clinically distinct subtypes. Gynecol. Oncol. 2020, 159, 649–656. [Google Scholar] [CrossRef]
- Singh, N.; Gilks, C.B. Vulval squamous cell carcinoma and its precursors. Histopathology 2020, 76, 128–138. [Google Scholar] [CrossRef]
- Tian, R.; Li, X.; Gao, Y.; Li, Y.; Yang, P.; Wang, K. Identification and validation of the role of matrix metalloproteinase-1 in cervical cancer. Int. J. Oncol. 2018, 52, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.S.; Kim, N.; Kim, J.Y.; Do, S.I.; Cho, Y.; Kim, H.S.; Kim, Y.B. Kallikrein 5 overexpression is associated with poor prognosis in uterine cervical cancer. J. Gynecol. Oncol. 2020, 31, e78. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.-J.; Greer, B.E.; Abu-Rustum, N.R.; Campos, S.M.; Cho, K.R.; Chon, H.S.; Chu, C.; Cohn, D.; Crispens, M.A.; Dizon, D.S.; et al. Vulvar Cancer, Version 1.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2017, 15, 92–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pils, S.; Gensthaler, L.; Alemany, L.; Horvat, R.; de Sanjosé, S.; Joura, E.A. HPV prevalence in vulvar cancer in Austria. Wien. Klin. Wochenschr. 2017, 129, 805–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakislova, N.; Saco, A.; Sierra, A.; del Pino, M.; Ordi, J. Role of Human Papillomavirus in Vulvar Cancer. Adv. Anat. Pathol. 2017, 24, 201–214. [Google Scholar] [CrossRef] [PubMed]
- del Pino, M.; Rodriguez-Carunchio, L.; Ordi, J. Pathways of vulvar intraepithelial neoplasia and squamous cell carcinoma. Histopathology 2013, 62, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Preti, M.; Scurry, J.; Marchitelli, C.E.; Micheletti, L. Vulvar intraepithelial neoplasia. Best Pract. Res. Clin. Obstet. Gynaecol. 2014, 28, 1051–1062. [Google Scholar] [CrossRef]
- Bornstein, J.; Bogliatto, F.; Haefner, H.K.; Stockdale, C.K.; Preti, M.; Bohl, T.G.; Reutter, J. The 2015 International Society for the Study of Vulvovaginal Disease (ISSVD) Terminology of Vulvar Squamous Intraepithelial Lesions. Obstet. Gynecol. 2016, 127, 264–268. [Google Scholar] [CrossRef]
- Hampl, M.; Deckers-Figiel, S.; Hampl, J.A.; Rein, D.; Bender, H.G. New aspects of vulvar cancer: Changes in localization and age of onset. Gynecol. Oncol. 2008, 109, 340–345. [Google Scholar] [CrossRef]
- de Bie, R.P.; van de Nieuwenhof, H.P.; Bekkers, R.L.M.; Melchers, W.J.G.; Siebers, A.G.; Bulten, J.; Massuger, L.F.A.G.; de Hullu, J.A. Patients with usual vulvar intraepithelial neoplasia-related vulvar cancer have an increased risk of cervical abnormalities. Br. J. Cancer 2009, 101, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E. Technical Review: In Situ Hybridization: AR Insights. Anat. Rec. 2014, 297, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Määttä, M.; Santala, M.; Soini, Y.; Turpeenniemi-Hujanen, T.; Talvensaari-Mattila, A. Increased matrix metalloproteinases 2 and 9 and tissue inhibitor of matrix metalloproteinase 2 expression is associated with progression from vulvar intraepithelial neoplasia to invasive carcinoma. Acta Obstet. Gynecol. Scand. 2010, 89, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, V.; Bellati, F.; Fischetti, M.; Plotti, F.; Perniola, G.; Panici, P.B. Vaginal cancer. Crit. Rev. Oncol. Hematol. 2012, 81, 286–295. [Google Scholar] [CrossRef]
- Gadducci, A.; Fabrini, M.G.; Lanfredini, N.; Sergiampietri, C. Squamous cell carcinoma of the vagina: Natural history, treatment modalities and prognostic factors. Crit. Rev. Oncol. Hematol. 2015, 93, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.S.; Rogers, L.J.; Cuello, M.A. Cancer of the vagina: 2021 update. Int. J. Gynecol. Obstet. 2021, 155, 19–27. [Google Scholar] [CrossRef]
- Rasmussen, C.L.; Bertoli, H.K.; Sand, F.L.; Kjær, A.K.; Thomsen, L.T.; Kjær, S.K. The prognostic significance of HPV, p16, and p53 protein expression in vaginal cancer: A systematic review. Acta Obstet. Gynecol. Scand. 2021, 100, 2144–2156. [Google Scholar] [CrossRef]
- Daling, J.R.; Madeleine, M.M.; Schwartz, S.M.; Shera, K.A.; Carter, J.J.; McKnight, B.; Porter, P.L.; Galloway, D.A.; McDougall, J.K.; Tamimi, H. A population-based study of squamous cell vaginal cancer: HPV and cofactors. Gynecol. Oncol. 2002, 84, 263–270. [Google Scholar] [CrossRef]
- Rajaram, S.; Maheshwari, A.; Srivastava, A. Staging for vaginal cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2015, 29, 822–832. [Google Scholar] [CrossRef]
- Way, S. Vaginal metastases of carcinoma of the body of the uterus. J. Obstet. Gynaecol. Br. Emp. 1951, 58, 558–572. [Google Scholar] [CrossRef]
- Nerdrum, T.A. Vaginal metastasis of hypernephroma. Report of three cases. Acta Obstet. Gynecol. Scand. 1966, 45, 515–524. [Google Scholar] [CrossRef]
- de Mûelenaere, G.F. Vaginal metastases in endometrial carcinoma. Am. J. Obstet. Gynecol. 1974, 118, 168–172. [Google Scholar] [CrossRef]
- Giacalone, P.L.; Dumontier, C.; Roger, P.; Laffargue, F.; Baldet, P. Vaginal metastases of breast cancer. J. Gynecol. Obstet. Biol. Reprod. 1998, 27, 714–717. [Google Scholar]
- Ng, Q.J.; Namuduri, R.P.; Yam, K.L.; Lim-Tan, S.K. Vaginal metastasis presenting as postmenopausal bleeding. Singap. Med. J. 2015, 56, e134–e136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pampalakis, G.; Zingkou, E.; Sotiropoulou, G. KLK5, a novel potential suppressor of vaginal carcinogenesis. Biol. Chem. 2018, 399, 1107–1111. [Google Scholar] [CrossRef]
- Grulich, A.E.; Jin, F.; Conway, E.L.; Stein, A.N.; Hocking, J. Cancers attributable to human papillomavirus infection. Sex. Health 2010, 7, 244–252. [Google Scholar] [CrossRef]
- Johnson, L.G.; Madeleine, M.M.; Newcomer, L.M.; Schwartz, S.M.; Daling, J.R. Anal cancer incidence and survival: The surveillance, epidemiology, and end results experience, 1973-2000. Cancer 2004, 101, 281–288. [Google Scholar] [CrossRef]
- Robinson, D.; Coupland, V.; Møller, H. An analysis of temporal and generational trends in the incidence of anal and other HPV-related cancers in Southeast England. Br. J. Cancer 2009, 100, 527–531. [Google Scholar] [CrossRef] [Green Version]
- Islami, F.; Ferlay, J.; Lortet-Tieulent, J.; Bray, F.; Jemal, A. International trends in anal cancer incidence rates. Int. J. Epidemiol. 2016, 46, dyw276. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, A.A.; Suk, R.; Shiels, M.S.; Sonawane, K.; Nyitray, A.G.; Liu, Y.; Gaisa, M.M.; Palefsky, J.M.; Sigel, K. Recent Trends in Squamous Cell Carcinoma of the Anus Incidence and Mortality in the United States, 2001–2015. J. Natl. Cancer Inst. 2020, 112, 829–838. [Google Scholar] [CrossRef]
- Clark, M.A.; Hartley, A.; Geh, J.I. Cancer of the anal canal. Lancet Oncol. 2004, 5, 149–157. [Google Scholar] [CrossRef]
- Palefsky, J.M. HPV infection in men. Dis. Markers 2007, 23, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anic, G.M.; Giuliano, A.R. Genital HPV infection and related lesions in men. Prev. Med. 2011, 53 (Suppl. 1), S36–S41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Wieland, U.; Kreuter, A. The importance of HPV16 in anal cancer prevention. Lancet Infect. Dis. 2018, 18, 131–132. [Google Scholar] [CrossRef] [Green Version]
- Krzowska-Firych, J.; Lucas, G.; Lucas, C.; Lucas, N.; Pietrzyk, Ł. An overview of Human Papillomavirus (HPV) as an etiological factor of the anal cancer. J. Infect. Public Health 2019, 12, 1–6. [Google Scholar] [CrossRef]
- Valvo, F.; Ciurlia, E.; Avuzzi, B.; Doci, R.; Ducreux, M.; Roelofsen, F.; Roth, A.; Trama, A.; Wittekind, C.; Bosset, J.-F. Cancer of the anal region. Crit. Rev. Oncol. Hematol. 2019, 135, 115–127. [Google Scholar] [CrossRef]
- Lin, C.; Franceschi, S.; Clifford, G.M. Human papillomavirus types from infection to cancer in the anus, according to sex and HIV status: A systematic review and meta-analysis. Lancet Infect. Dis. 2018, 18, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Clifford, G.M.; Georges, D.; Shiels, M.S.; Engels, E.A.; Albuquerque, A.; Poynten, I.M.; de Pokomandy, A.; Easson, A.M.; Stier, E.A. A meta-analysis of anal cancer incidence by risk group: Toward a unified anal cancer risk scale. Int. J. Cancer 2021, 148, 38–47. [Google Scholar] [CrossRef]
- Svidler López, L.; La Rosa, L. Human Papilloma Virus Infection and Anal Squamous Intraepithelial Lesions. Clin. Colon Rectal Surg. 2019, 32, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Chittleborough, T.; Tapper, R.; Eglinton, T.; Frizelle, F. Anal squamous intraepithelial lesions: An update and proposed management algorithm. Technol. Coloproctol. 2020, 24, 95–103. [Google Scholar] [CrossRef]
- Moscicki, A.-B.; Darragh, T.M.; Berry-Lawhorn, J.M.; Roberts, J.M.; Khan, M.J.; Boardman, L.A.; Chiao, E.; Einstein, M.H.; Goldstone, S.E.; Jay, N.; et al. Screening for Anal Cancer in Women. J. Low. Genit. Tract Dis. 2015, 19, S27–S42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donà, M.G.; Benevolo, M.; Latini, A.; Rollo, F.; Colafigli, M.; Frasca, M.; Zaccarelli, M.; Giglio, A.; Moretto, D.; Pescarmona, E.; et al. Anal cytological lesions and HPV infection in individuals at increased risk for anal cancer: Anal Lesions in HIV-Positive and -Negative MSM. Cancer Cytopathol. 2018, 126, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albuquerque, A. Cytology in Anal Cancer Screening: Practical Review for Clinicians. Acta Cytol. 2020, 64, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Cimic, A.; Saqi, A. Anal cytology and high-risk human papilloma virus testing in atypical squamous categories: Value of concurrent testing in management of high-risk population. Diagn. Cytopathol. 2021, 49, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA. Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Giuliano, A.R.; Lazcano-Ponce, E.; Villa, L.L.; Flores, R.; Salmeron, J.; Lee, J.-H.; Papenfuss, M.R.; Abrahamsen, M.; Jolles, E.; Nielson, C.M.; et al. The Human Papillomavirus Infection in Men Study: Human Papillomavirus Prevalence and Type Distribution among Men Residing in Brazil, Mexico, and the United States. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 2036–2043. [Google Scholar] [CrossRef] [Green Version]
- Maden, C.; Sherman, K.J.; Beckmann, A.M.; Hislop, T.G.; Teh, C.-Z.; Ashley, R.L.; Daling, J.R. History of Circumcision, Medical Conditions, and Sexual Activity and Risk of Penile Cancer. JNCI J. Natl. Cancer Inst. 1993, 85, 19–24. [Google Scholar] [CrossRef]
- Dillner, J.; von Krogh, G.; Horenblas, S.; Meijer, C.J.L.M. Etiology of Squamous Cell Carcinoma of the Penis. Scand. J. Urol. Nephrol. 2000, 34, 189–193. [Google Scholar] [CrossRef]
- Daling, J.R.; Madeleine, M.M.; Johnson, L.G.; Schwartz, S.M.; Shera, K.A.; Wurscher, M.A.; Carter, J.J.; Porter, P.L.; Galloway, D.A.; McDougall, J.K.; et al. Penile cancer: Importance of circumcision, human papillomavirus and smoking inin situ and invasive disease. Int. J. Cancer 2005, 116, 606–616. [Google Scholar] [CrossRef]
- Miralles-Guri, C.; Bruni, L.; Cubilla, A.L.; Castellsagué, X.; Bosch, F.X.; de Sanjosé, S. Human papillomavirus prevalence and type distribution in penile carcinoma. J. Clin. Pathol. 2009, 62, 870–878. [Google Scholar] [CrossRef]
- Alemany, L.; Cubilla, A.; Halec, G.; Kasamatsu, E.; Quirós, B.; Masferrer, E.; Tous, S.; Lloveras, B.; Hernández-Suarez, G.; Lonsdale, R.; et al. Role of Human Papillomavirus in Penile Carcinomas Worldwide. Eur. Urol. 2016, 69, 953–961. [Google Scholar] [CrossRef]
- Viens, L.J.; Henley, S.J.; Watson, M.; Markowitz, L.E.; Thomas, C.C.; Thompson, T.D.; Razzaghi, H.; Saraiya, M. Human Papillomavirus–Associated Cancers—United States, 2008–2012. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Olesen, T.B.; Sand, F.L.; Rasmussen, C.L.; Albieri, V.; Toft, B.G.; Norrild, B.; Munk, C.; Kjær, S.K. Prevalence of human papillomavirus DNA and p16INK4a in penile cancer and penile intraepithelial neoplasia: A systematic review and meta-analysis. Lancet Oncol. 2019, 20, 145–158. [Google Scholar] [CrossRef]
- Harish, K.; Ravi, R. The role of tobacco in penile carcinoma. Br. J. Urol. 1995, 75, 375–377. [Google Scholar] [CrossRef] [PubMed]
- Micali, G. Lichen sclerosus of the glans is significantly associated with penile carcinoma. Sex. Transm. Infect. 2001, 77, 226. [Google Scholar] [CrossRef] [Green Version]
- Poetsch, M.; Hemmerich, M.; Kakies, C.; Kleist, B.; Wolf, E.; vom Dorp, F.; Hakenberg, O.W.; Protzel, C. Alterations in the tumor suppressor gene p16 INK4A are associated with aggressive behavior of penile carcinomas. Virchows Arch. 2011, 458, 221–229. [Google Scholar] [CrossRef]
- Steinestel, J.; Al Ghazal, A.; Arndt, A.; Schnoeller, T.J.; Schrader, A.J.; Moeller, P.; Steinestel, K. The role of histologic subtype, p16INK4a expression, and presence of human papillomavirus DNA in penile squamous cell carcinoma. BMC Cancer 2015, 15, 220. [Google Scholar] [CrossRef] [Green Version]
- Vanthoor, J.; Vos, G.; Albersen, M. Penile cancer: Potential target for immunotherapy? World J. Urol. 2021, 39, 1405–1411. [Google Scholar] [CrossRef]
- Bethune, G.; Campbell, J.; Rocker, A.; Bell, D.; Rendon, R.; Merrimen, J. Clinical and Pathologic Factors of Prognostic Significance in Penile Squamous Cell Carcinoma in a North American Population. Urology 2012, 79, 1092–1097. [Google Scholar] [CrossRef]
- Gunia, S.; Erbersdobler, A.; Hakenberg, O.W.; Koch, S.; May, M. p16INK4a is a Marker of Good Prognosis for Primary Invasive Penile Squamous Cell Carcinoma: A Multi-Institutional Study. J. Urol. 2012, 187, 899–907. [Google Scholar] [CrossRef]
- Djajadiningrat, R.S.; Jordanova, E.S.; Kroon, B.K.; van Werkhoven, E.; de Jong, J.; Pronk, D.T.M.; Snijders, P.J.F.; Horenblas, S.; Heideman, D.A.M. Human Papillomavirus Prevalence in Invasive Penile Cancer and Association with Clinical Outcome. J. Urol. 2015, 193, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primer 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Crooker, K.; Aliani, R.; Ananth, M.; Arnold, L.; Anant, S.; Thomas, S.M. A Review of Promising Natural Chemopreventive Agents for Head and Neck Cancer. Cancer Prev. Res. 2018, 11, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.S.; White, D.K. Human papillomavirus expression in oral mucosa, premalignant conditions, and squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 1996, 82, 57–68. [Google Scholar] [CrossRef]
- Sugerman, P.B.; Shillitoe, E.J. The high risk human papillomaviruses and oral cancer: Evidence for and against a causal relationship. Oral Dis. 1997, 3, 130–147. [Google Scholar] [CrossRef]
- Neville, B.W.; Day, T.A. Oral cancer and precancerous lesions. CA. Cancer J. Clin. 2002, 52, 195–215. [Google Scholar] [CrossRef]
- Solomon, B.; Young, R.J.; Rischin, D. Head and neck squamous cell carcinoma: Genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin. Cancer Biol. 2018, 52, 228–240. [Google Scholar] [CrossRef]
- Mourad, M.; Jetmore, T.; Jategaonkar, A.A.; Moubayed, S.; Moshier, E.; Urken, M.L. Epidemiological Trends of Head and Neck Cancer in the United States: A SEER Population Study. J. Oral Maxillofac. Surg. Off. J. Am. Assoc. Oral Maxillofac. Surg. 2017, 75, 2562–2572. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [Green Version]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef]
- Forastiere, A.; Koch, W.; Trotti, A.; Sidransky, D. Head and neck cancer. N. Engl. J. Med. 2001, 345, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.D.; Parkinson, E.K.; Harrison, P.R. Profiling early head and neck cancer. Nat. Rev. Cancer 2005, 5, 127–135. [Google Scholar] [CrossRef]
- Vigneswaran, N.; Williams, M.D. Epidemiologic trends in head and neck cancer and aids in diagnosis. Oral Maxillofac. Surg. Clin. N. Am. 2014, 26, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Molinolo, A.A.; Amornphimoltham, P.; Squarize, C.H.; Castilho, R.M.; Patel, V.; Gutkind, J.S. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009, 45, 324–334. [Google Scholar] [CrossRef] [Green Version]
- de Melo Filho, M.R.; Rocha, B.A.; Pires, M.B. de O.; Fonseca, E.S.; Freitas, E.M. de; Martelli Junior, H.; Santos, F.B.G. Quality of life of patients with head and neck cancer. Braz. J. Otorhinolaryngol. 2013, 79, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Costea, D.E.; Tsinkalovsky, O.; Vintermyr, O.K.; Johannessen, A.C.; Mackenzie, I.C. Cancer stem cells—New and potentially important targets for the therapy of oral squamous cell carcinoma. Oral Dis. 2006, 12, 443–454. [Google Scholar] [CrossRef]
- van der Riet, P.; Nawroz, H.; Hruban, R.H.; Corio, R.; Tokino, K.; Koch, W.; Sidransky, D. Frequent loss of chromosome 9p21-22 early in head and neck cancer progression. Cancer Res. 1994, 54, 1156–1158. [Google Scholar]
- González, M.V.; Pello, M.F.; López-Larrea, C.; Suárez, C.; Menéndez, M.J.; Coto, E. Deletion and methylation of the tumour suppressor gene p16/CDKN2 in primary head and neck squamous cell carcinoma. J. Clin. Pathol. 1997, 50, 509–512. [Google Scholar] [CrossRef] [Green Version]
- Mehramiz, M.; Ghasemi, F.; Esmaily, H.; Tayefi, M.; Hassanian, S.M.; Sadeghzade, M.; Sadabadi, F.; Moohebati, M.; Azarpazhooh, M.R.; Parizadeh, S.M.R.; et al. Interaction between a variant of CDKN2A/B-gene with lifestyle factors in determining dyslipidemia and estimated cardiovascular risk: A step toward personalized nutrition. Clin. Nutr. Edinb. Scotl. 2018, 37, 254–261. [Google Scholar] [CrossRef]
- Rivandi, M.; Khorrami, M.-S.; Fiuji, H.; Shahidsales, S.; Hasanzadeh, M.; Jazayeri, M.H.; Hassanian, S.M.; Ferns, G.A.; Saghafi, N.; Avan, A. The 9p21 locus: A potential therapeutic target and prognostic marker in breast cancer. J. Cell. Physiol. 2018, 233, 5170–5179. [Google Scholar] [CrossRef] [PubMed]
- Kamb, A.; Gruis, N.A.; Weaver-Feldhaus, J.; Liu, Q.; Harshman, K.; Tavtigian, S.V.; Stockert, E.; Day, R.S.; Johnson, B.E.; Skolnick, M.H. A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994, 264, 436–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, A.L.; Califano, J.; Cairns, P.; Westra, W.H.; Jones, R.M.; Koch, W.; Ahrendt, S.; Eby, Y.; Sewell, D.; Nawroz, H.; et al. High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res. 1996, 56, 3630–3633. [Google Scholar] [PubMed]
- Ohnishi, T.; Ohnishi, K.; Takahashi, A. Glycerol restores heat-induced p53-dependent apoptosis of human glioblastoma cells bearing mutant p53. BMC Biotechnol. 2002, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Poeta, M.L.; Manola, J.; Goldwasser, M.A.; Forastiere, A.; Benoit, N.; Califano, J.A.; Ridge, J.A.; Goodwin, J.; Kenady, D.; Saunders, J.; et al. TP53 Mutations and Survival in Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2007, 357, 2552–2561. [Google Scholar] [CrossRef] [Green Version]
- Talamini, R.; Bosetti, C.; La Vecchia, C.; Dal Maso, L.; Levi, F.; Bidoli, E.; Negri, E.; Pasche, C.; Vaccarella, S.; Barzan, L.; et al. Combined effect of tobacco and alcohol on laryngeal cancer risk: A case-control study. Cancer Causes Control CCC 2002, 13, 957–964. [Google Scholar] [CrossRef]
- Pai, S.I.; Westra, W.H. Molecular pathology of head and neck cancer: Implications for diagnosis, prognosis, and treatment. Annu. Rev. Pathol. 2009, 4, 49–70. [Google Scholar] [CrossRef] [Green Version]
- Stein, A.P.; Saha, S.; Kraninger, J.L.; Swick, A.D.; Yu, M.; Lambert, P.F.; Kimple, R.J. Prevalence of Human Papillomavirus in Oropharyngeal Cancer: A Systematic Review. Cancer J. Sudbury Mass 2015, 21, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Isayeva, T.; Li, Y.; Maswahu, D.; Brandwein-Gensler, M. Human papillomavirus in non-oropharyngeal head and neck cancers: A systematic literature review. Head Neck Pathol. 2012, 6 (Suppl. 1), S104–S120. [Google Scholar] [CrossRef] [Green Version]
- Michaud, D.S.; Langevin, S.M.; Eliot, M.; Nelson, H.H.; Pawlita, M.; McClean, M.D.; Kelsey, K.T. High-risk HPV types and head and neck cancer. Int. J. Cancer 2014, 135, 1653–1661. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [CrossRef] [PubMed] [Green Version]
- Sacconi, A.; Donzelli, S.; Pulito, C.; Ferrero, S.; Spinella, F.; Morrone, A.; Rigoni, M.; Pimpinelli, F.; Ensoli, F.; Sanguineti, G.; et al. TMPRSS2, a SARS-CoV-2 internalization protease is downregulated in head and neck cancer patients. J. Exp. Clin. Cancer Res. CR 2020, 39, 200. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Li, Y.; Wang, X.; Yang, Y. Secretory leukocyte protease inhibitor suppresses HPV E6-expressing HNSCC progression by mediating NF-κB and Akt pathways. Cancer Cell Int. 2019, 19, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Population | World | |||||||
|---|---|---|---|---|---|---|---|---|
| Women at risk for cervical cancer (Female population aged ≥ 15 years) in millions | 2869.0 | |||||||
| Burden of cervical cancer and other HPV-related cancers | ||||||||
| Annual number of new cervical cancer cases | 604,127 | |||||||
| Annual number of cervical cancer deaths | 341,831 | |||||||
| Standardized incidence rates per 100,000 population: | ||||||||
| Cervical Cancer | Anal Cancer | Vulva Cancer | Vaginal Cancer | Penile Cancer | Oropharyngeal Cancer | Oral cavity Cancer | Laryngeal Cancer | |
| Men | - | 0.49 | - | - | 0.80 | 1.79 | 5.96 | 3.59 |
| Women | 13.3 | 0.58 | 0.85 | 0.36 | - | 0.40 | 2.28 | 0.49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieira, G.V.; Somera dos Santos, F.; Lepique, A.P.; da Fonseca, C.K.; Innocentini, L.M.A.R.; Braz-Silva, P.H.; Quintana, S.M.; Sales, K.U. Proteases and HPV-Induced Carcinogenesis. Cancers 2022, 14, 3038. https://doi.org/10.3390/cancers14133038
Vieira GV, Somera dos Santos F, Lepique AP, da Fonseca CK, Innocentini LMAR, Braz-Silva PH, Quintana SM, Sales KU. Proteases and HPV-Induced Carcinogenesis. Cancers. 2022; 14(13):3038. https://doi.org/10.3390/cancers14133038
Chicago/Turabian StyleVieira, Gabriel Viliod, Fernanda Somera dos Santos, Ana Paula Lepique, Carol Kobori da Fonseca, Lara Maria Alencar Ramos Innocentini, Paulo Henrique Braz-Silva, Silvana Maria Quintana, and Katiuchia Uzzun Sales. 2022. "Proteases and HPV-Induced Carcinogenesis" Cancers 14, no. 13: 3038. https://doi.org/10.3390/cancers14133038