
Epstein-Barr Virus (EBV) Is Mostly Latent and Clonal in Angioimmunoblastic T Cell Lymphoma (AITL)

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Patients
2.3. EBER In Situ Hybridization
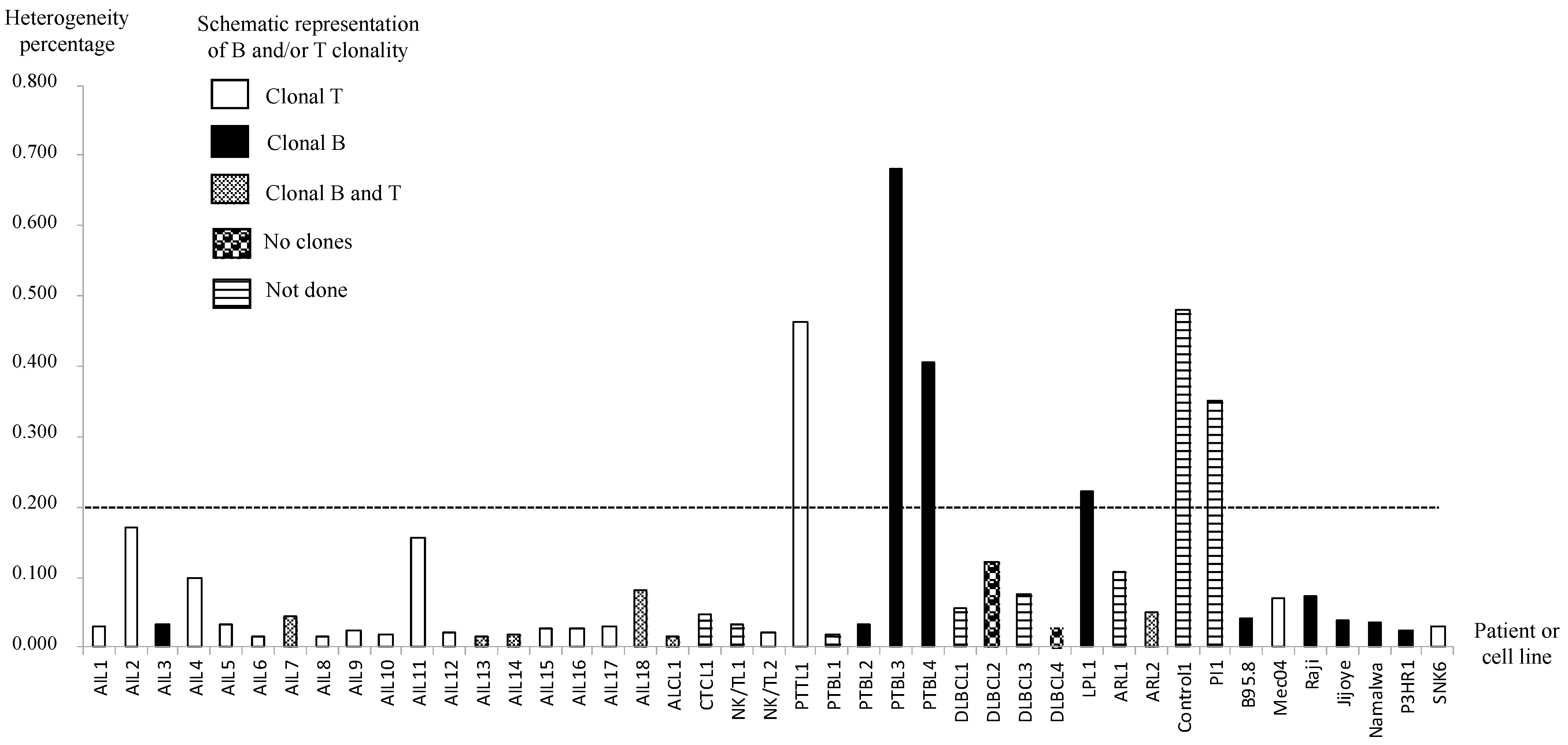
2.4. B and T Clonality Determination
2.5. DNA Extractions and Generic Amplification of Serum DNA
2.6. Probe Design for EBV Sequence Capture
2.7. Illumina Library Construction and Whole EBV Genome Sequencing
2.8. Bioinformatics Analysis
2.8.1. De Novo Assembly
2.8.2. Read Mapping and Variant Calling
2.9. EBV Typing
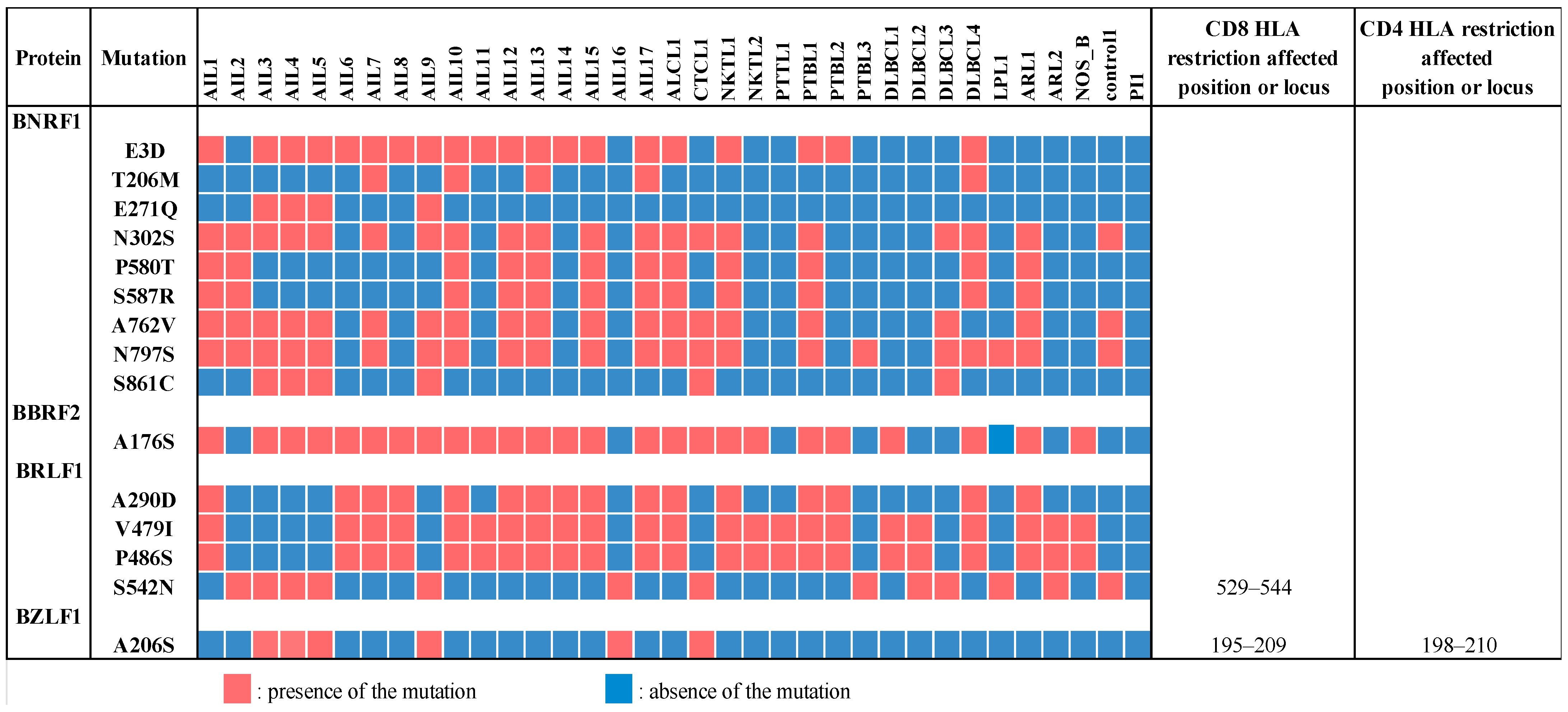
2.10. Mutation Analysis and Clonality Assessment
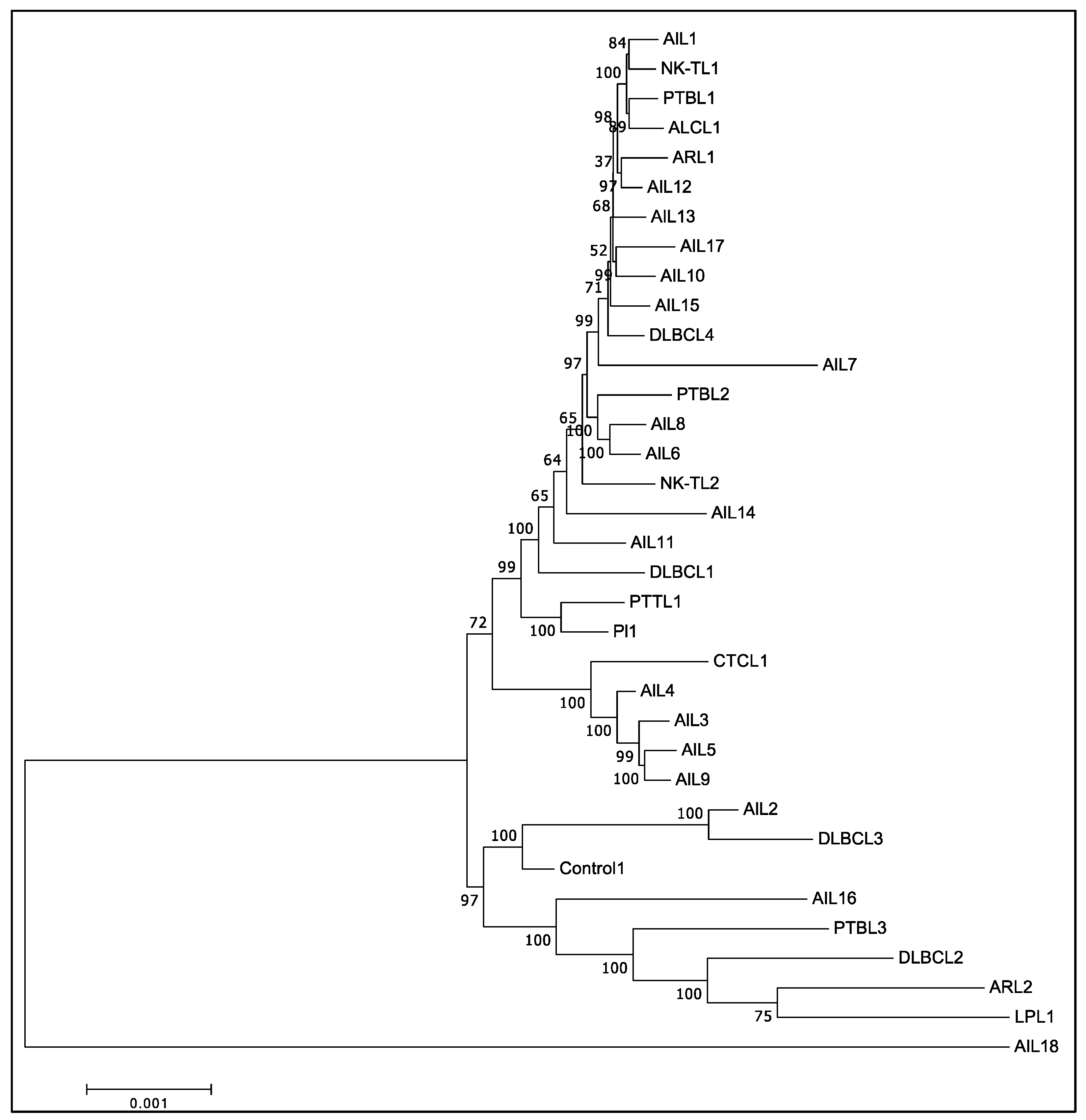
2.11. Phylogenetic Analysis of EBV Genomes
2.12. Quantitative PCRs
3. Results
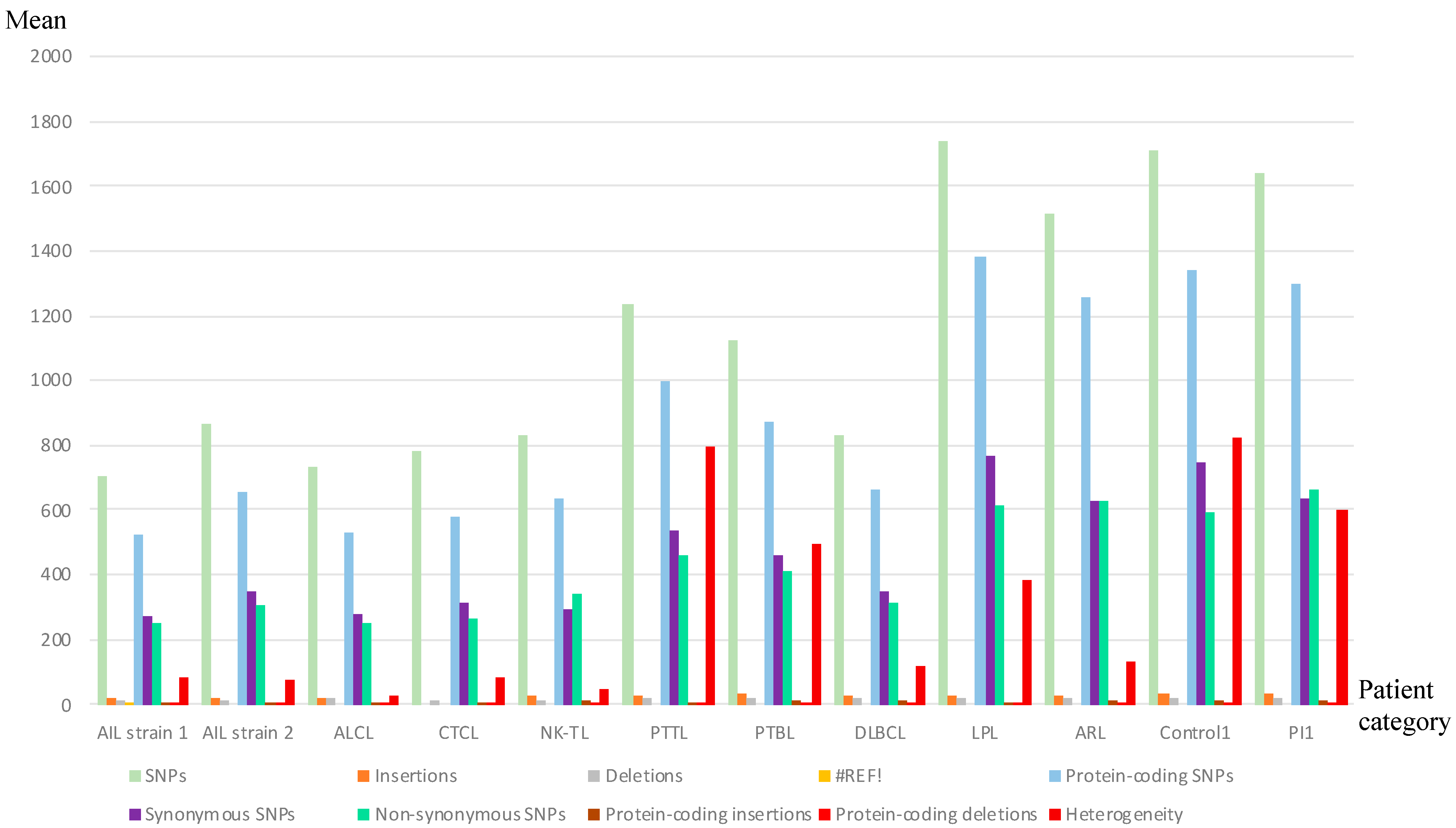
3.1. Analysis of Whole EBV Genome Sequences
3.2. Some AITL Strains Exhibited Similar Distribution Patterns of Mutations
3.3. AITL Biopsies Revealed EBV in a Clonal Form
3.4. EBV Was Almost Always Latent in AITL Biopsies
3.5. Phylogenic Analysis Confirmed the Existence of at Least Two Different Groups of EBV among AITL Biopsies
3.6. Mutations Occurred Mostly in Latency Genes and Secondarily in Tegument Genes
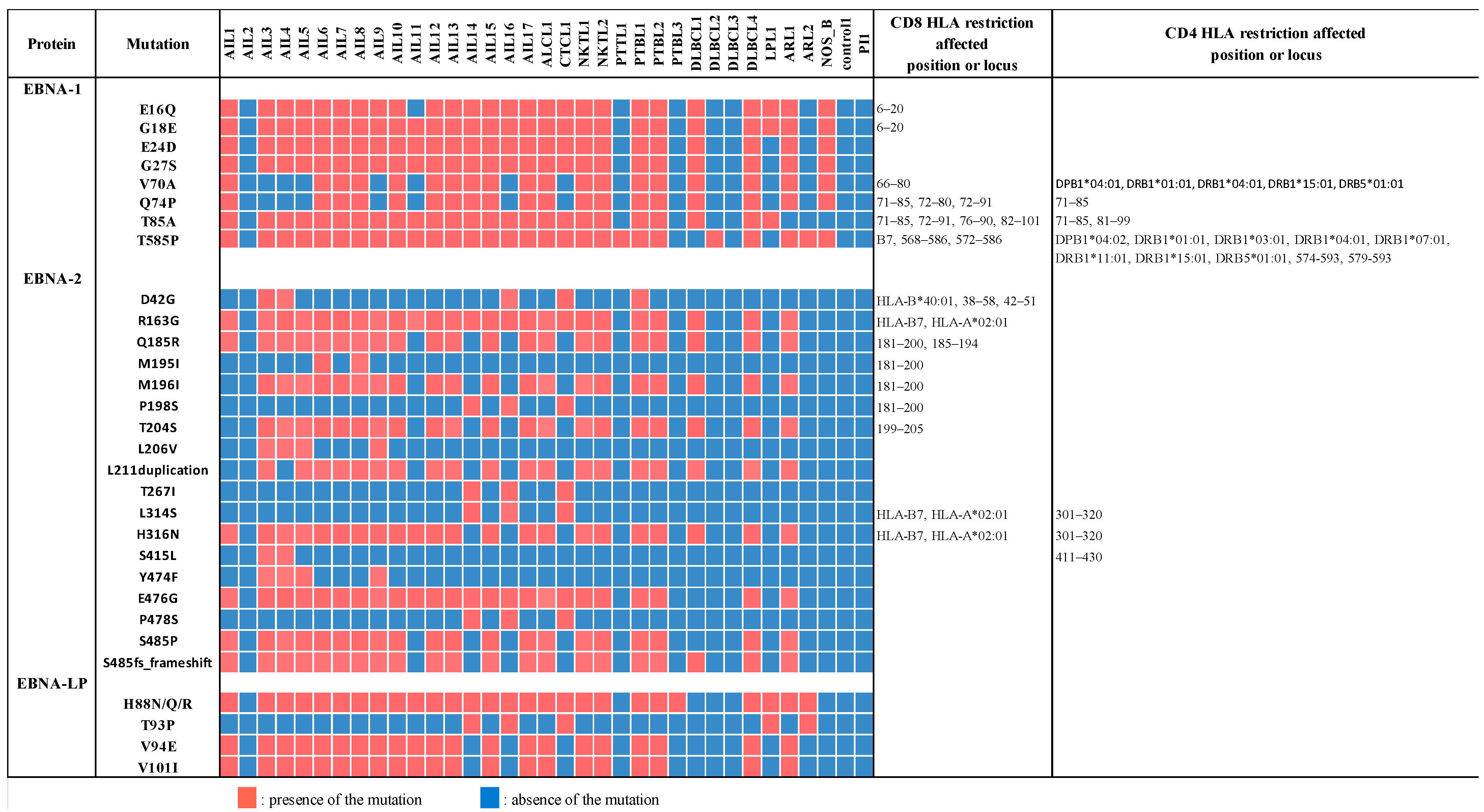
3.7. EBNA-1, EBNA-2, and Mainly EBNA-LP Genes Were the Most Mutated among Latency Genes
3.8. Two Tegument Genes, BNRF1 and BBRF2, Were Especially Mutated in AITL Biopsies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Young, L.S.; Murray, P.G. Epstein-barr virus and oncogenesis: From latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeoch, D.J.; Gatherer, D. Lineage structures in the genome sequences of three epstein-barr virus strains. Virology 2007, 359, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Romero-Masters, J.C.; Huebner, S.M.; Ohashi, M.; Bristol, J.A.; Benner, B.E.; Barlow, E.A.; Turk, G.L.; Nelson, S.E.; Baiu, D.C.; Van Sciver, N.; et al. B Cells infected with type 2 epstein-barr virus (EBV) have increased NFATc1/NFATc2 activity and enhanced lytic gene expression in comparison to type 1 EBV infection. PLoS Pathog. 2020, 16, e1008365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, S.; Bridges, R.; Wegner, F.; Venturini, C.; Palser, A.; Middeldorp, J.M.; Cohen, J.I.; Lorenzetti, M.A.; Bassano, I.; White, R.E.; et al. Sequence variation of epstein-barr virus: Viral types, geography, codon usage, and diseases. J. Virol. 2018, 92, e01132-18. [Google Scholar] [CrossRef] [Green Version]
- Hämmerl, L.; Colombet, M.; Rochford, R.; Ogwang, D.M.; Parkin, D.M. The burden of burkitt lymphoma in Africa. Infect. Agents Cancer 2019, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Mahdavifar, N.; Ghoncheh, M.; Mohammadian-Hafshejani, A.; Khosravi, B.; Salehiniya, H. Epidemiology and inequality in the incidence and mortality of nasopharynx cancer in Asia. Osong Public Health Res. Perspect. 2016, 7, 360–372. [Google Scholar] [CrossRef] [Green Version]
- Kutok, J.L.; Wang, F. Spectrum of epstein-barr virus-associated diseases. Annu. Rev. Pathol. 2006, 1, 375–404. [Google Scholar] [CrossRef]
- Palser, A.L.; Grayson, N.E.; White, R.E.; Corton, C.; Correia, S.; Ba Abdullah, M.M.; Watson, S.J.; Cotten, M.; Arrand, J.R.; Murray, P.G.; et al. Genome diversity of epstein-barr virus from multiple tumor types and normal infection. J. Virol. 2015, 89, 5222–5237. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.M.; Yu, K.J.; Mbulaiteye, S.M.; Hildesheim, A.; Bhatia, K. The extent of genetic diversity of epstein-barr virus and its geographic and disease patterns: A need for reappraisal. Virus Res. 2009, 143, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-H.; Lin, X.; Shumilov, A.; Bernhardt, K.; Feederle, R.; Poirey, R.; Kopp-Schneider, A.; Pereira, B.; Almeida, R.; Delecluse, H.-J. The biological properties of different epstein-barr virus strains explain their association with various types of cancers. Oncotarget 2017, 8, 10238–10254. [Google Scholar] [CrossRef] [Green Version]
- Hui, K.F.; Chan, T.F.; Yang, W.; Shen, J.J.; Lam, K.P.; Kwok, H.; Sham, P.C.; Tsao, S.W.; Kwong, D.L.; Lung, M.L.; et al. High risk epstein-barr virus variants characterized by distinct polymorphisms in the EBER locus are strongly associated with nasopharyngeal carcinoma. Int. J. Cancer 2019, 144, 3031–3042. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Burassakarn, A.; Kang, Y.; Iizasa, H.; Yoshiyama, H. A single nucleotide polymorphism in the BART promoter region of epstein-barr virus isolated from nasopharyngeal cancer cells. Biochem. Biophys. Res. Commun. 2019, 520, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yao, Y.; Chen, H.; Zhang, S.; Cao, S.-M.; Zhang, Z.; Luo, B.; Liu, Z.; Li, Z.; Xiang, T.; et al. Genome sequencing analysis identifies epstein-barr virus subtypes associated with high risk of nasopharyngeal carcinoma. Nat. Genet. 2019, 51, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the world health organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Vose, J.; Armitage, J.; Weisenburger, D. International T-cell lymphoma project international peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [CrossRef]
- Federico, M.; Rudiger, T.; Bellei, M.; Nathwani, B.N.; Luminari, S.; Coiffier, B.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Savage, K.J.; et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: Analysis of the international peripheral T-cell lymphoma project. J. Clin. Oncol. 2013, 31, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Lunning, M.A.; Vose, J.M. Angioimmunoblastic T-cell lymphoma: The many-faced lymphoma. Blood 2017, 129, 1095–1102. [Google Scholar] [CrossRef]
- Mourad, N.; Mounier, N.; Brière, J.; Raffoux, E.; Delmer, A.; Feller, A.; Meijer, C.J.L.M.; Emile, J.-F.; Bouabdallah, R.; Bosly, A.; et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the groupe d’etude des lymphomes de l’adulte (GELA) trials. Blood 2008, 111, 4463–4470. [Google Scholar] [CrossRef]
- Piccaluga, P.P.; Agostinelli, C.; Califano, A.; Carbone, A.; Fantoni, L.; Ferrari, S.; Gazzola, A.; Gloghini, A.; Righi, S.; Rossi, M.; et al. Gene expression analysis of angioimmunoblastic lymphoma indicates derivation from T follicular helper cells and vascular endothelial growth factor deregulation. Cancer Res. 2007, 67, 10703–10710. [Google Scholar] [CrossRef] [Green Version]
- De Leval, L.; Rickman, D.S.; Thielen, C.; de Reynies, A.; Huang, Y.-L.; Delsol, G.; Lamant, L.; Leroy, K.; Brière, J.; Molina, T.; et al. The gene expression profile of nodal peripheral t-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood 2007, 109, 4952–4963. [Google Scholar] [CrossRef]
- Tan, B.T.; Warnke, R.A.; Arber, D.A. The frequency of B- and T-cell gene rearrangements and epstein-barr virus in T-cell lymphomas: A comparison between angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, unspecified with and without associated B-cell proliferations. J. Mol. Diagn. 2006, 8, 466–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.L.; Hodges, E.; Quin, C.T.; McCarthy, K.P.; Wright, D.H. Frequent T and B Cell oligoclones in histologically and immunophenotypically characterized angioimmunoblastic lymphadenopathy. Am. J. Pathol. 2000, 156, 661–669. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.N.; Tran, N.T.B.; Nguyen, T.P.X.; Ngo, T.N.M.; Lai, D.V.; Deel, C.D.; Hassell, L.A.; Vuong, H.G. Clinicopathological implications of RHOA mutations in angioimmunoblastic T-cell lymphoma: A meta-analysis. Clin. Lymphoma Myeloma Leuk. 2021, 21, 431–438. [Google Scholar] [CrossRef]
- Willemsen, M.; Abdul Hamid, M.; Winkens, B.; Zur Hausen, A. Mutational heterogeneity of angioimmunoblastic T-cell lymphoma indicates distinct lymphomagenic pathways. Blood Cancer J. 2018, 8, 6. [Google Scholar] [CrossRef]
- Timmins, M.A.; Wagner, S.D.; Ahearne, M.J. The new biology of PTCL-NOS and AITL: Current status and future clinical impact. Br. J. Haematol. 2020, 189, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Tari, G.; Lemonnier, F.; Morschhauser, F. Epigenetic focus on angioimmunoblastic T-cell lymphoma: Pathogenesis and treatment. Curr. Opin. Oncol. 2021, 33, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, F.; Gaulard, P.; de Leval, L. New insights in the pathogenesis of T-cell lymphomas. Curr. Opin. Oncol. 2018, 30, 277–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donner, I.; Katainen, R.; Kaasinen, E.; Aavikko, M.; Sipilä, L.J.; Pukkala, E.; Aaltonen, L.A. Candidate susceptibility variants in angioimmunoblastic T-cell lymphoma. Fam. Cancer 2019, 18, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Liu, P. No survival improvement for patients with angioimmunoblastic T-cell lymphoma over the past two decades: A population-based study of 1207 cases. PLoS ONE 2014, 9, e92585. [Google Scholar] [CrossRef] [Green Version]
- Anagnostopoulos, I.; Hummel, M.; Finn, T.; Tiemann, M.; Korbjuhn, P.; Dimmler, C.; Gatter, K.; Dallenbach, F.; Parwaresch, M.R.; Stein, H. Heterogeneous epstein-barr virus infection patterns in peripheral T-cell lymphoma of angioimmunoblastic lymphadenopathy type. Blood 1992, 80, 1804–1812. [Google Scholar] [CrossRef] [Green Version]
- Willenbrock, K.; Bräuninger, A.; Hansmann, M.-L. Frequent occurrence of B-cell lymphomas in angioimmunoblastic T-cell lymphoma and proliferation of epstein-barr virus-infected cells in early cases. Br. J. Haematol. 2007, 138, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.; Dorion, P. Angioimmunoblastic T-cell lymphoma presenting with an acute serologic epstein-barr virus profile. Hematol. Rep. 2015, 7, 5893. [Google Scholar] [CrossRef] [Green Version]
- Zettl, A.; Lee, S.-S.; Rüdiger, T.; Starostik, P.; Marino, M.; Kirchner, T.; Ott, M.; Müller-Hermelink, H.K.; Ott, G. Epstein-barr virus-associated B-cell lymphoproliferative disorders in angloimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, unspecified. Am. J. Clin. Pathol. 2002, 117, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; McKenna, R.W.; Hoang, M.P.; Collins, R.H.; Kroft, S.H. Composite angioimmunoblastic T-cell lymphoma and diffuse large B-cell lymphoma: A case report and review of the literature. Am. J. Clin. Pathol. 2002, 118, 848–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla-Martinez, L.; Fend, F.; Moguel, L.R.; Spilove, L.; Beaty, M.W.; Kingma, D.W.; Raffeld, M.; Jaffe, E.S. Peripheral T-cell lymphoma with reed-sternberg-like cells of B-cell phenotype and genotype associated with epstein-barr virus infection. Am. J. Surg. Pathol. 1999, 23, 1233–1240. [Google Scholar] [CrossRef]
- Dunleavy, K.; Wilson, W.H. Angioimmunoblastic T-cell lymphoma: Immune modulation as a therapeutic strategy. Leuk. Lymphoma 2007, 48, 449–451. [Google Scholar] [CrossRef]
- Van Dongen, J.J.M.; Langerak, A.W.; Brüggemann, M.; Evans, P.A.S.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; García-Sanz, R.; et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 genome project data processing subgroup the sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. DeepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, snpeff: SNPs in the genome of drosophila melanogaster strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Tarbouriech, N.; Buisson, M.; Géoui, T.; Daenke, S.; Cusack, S.; Burmeister, W.P. Structural genomics of the epstein-barr virus. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 1276–1285. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Kwok, H.; Tong, A.H.Y.; Lin, C.H.; Lok, S.; Farrell, P.J.; Kwong, D.L.W.; Chiang, A.K.S. Genomic sequencing and comparative analysis of epstein-barr virus genome isolated from primary nasopharyngeal carcinoma biopsy. PLoS ONE 2012, 7, e36939. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, P.-P.; Tung, C.-L.; Chan, A.B.-W.; Liao, J.-B.; Wang, J.-S.; Tseng, H.-H.; Su, H.-H.; Chang, K.-C.; Chang, C.-C. EBV viral load in tumor tissue is an important prognostic indicator for nasal NK/T-cell lymphoma. Am. J. Clin. Pathol. 2007, 128, 579–584. [Google Scholar] [CrossRef]
- Gutiérrez, M.I.; Raj, A.; Spangler, G.; Sharma, A.; Hussain, A.; Judde, J.G.; Tsao, S.W.; Yuen, P.W.; Joab, I.; Magrath, I.T.; et al. Sequence variations in EBNA-1 may dictate restriction of tissue distribution of epstein-barr virus in normal and tumour cells. J. Gen. Virol. 1997, 78 Pt 7, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- He, H.-P.; Luo, M.; Cao, Y.-L.; Lin, Y.-X.; Zhang, H.; Zhang, X.; Ou, J.-Y.; Yu, B.; Chen, X.; Xu, M.; et al. Structure of epstein-barr virus tegument protein complex BBRF2-BSRF1 reveals its potential role in viral envelopment. Nat. Commun. 2020, 11, 5405. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Attygalle, A.D.; Chuang, S.-S.; Diss, T.; Ye, H.; Liu, H.; Hamoudi, R.A.; Munson, P.; Bacon, C.M.; Dogan, A.; et al. Angioimmunoblastic T-cell lymphoma: Histological progression associates with EBV and HHV6B viral load. Br. J. Haematol. 2007, 138, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M.; Zajac, B.; Henle, G.; Henle, W. Morphological and virological investigations on cultured burkitt tumor lymphoblasts (Strain Raji). J. Natl. Cancer Inst. 1966, 37, 547–559. [Google Scholar]
- Gaulard, P.; de Leval, L. The microenvironment in T-cell lymphomas: Emerging themes. Semin. Cancer Biol. 2014, 24, 49–60. [Google Scholar] [CrossRef]
- Weiss, L.M.; Jaffe, E.S.; Liu, X.F.; Chen, Y.Y.; Shibata, D.; Medeiros, L.J. Detection and localization of epstein-barr viral genomes in angioimmunoblastic lymphadenopathy and angioimmunoblastic lymphadenopathy-like lymphoma. Blood 1992, 79, 1789–1795. [Google Scholar] [CrossRef] [Green Version]
- Bayda, N.; Tilloy, V.; Chaunavel, A.; Bahri, R.; Halabi, M.A.; Feuillard, J.; Jaccard, A.; Ranger-Rogez, S. Comprehensive epstein-barr virus transcriptome by RNA-sequencing in angioimmunoblastic t cell lymphoma (AITL) and other lymphomas. Cancers 2021, 13, 610. [Google Scholar] [CrossRef]
- Gru, A.A.; Haverkos, B.H.; Freud, A.G.; Hastings, J.; Nowacki, N.B.; Barrionuevo, C.; Vigil, C.E.; Rochford, R.; Natkunam, Y.; Baiocchi, R.A.; et al. The epstein-barr virus (EBV) in T cell and NK cell lymphomas: Time for a reassessment. Curr. Hematol. Malig. Rep. 2015, 10, 456–467. [Google Scholar] [CrossRef]
- Kwok, H.; Wu, C.W.; Palser, A.L.; Kellam, P.; Sham, P.C.; Kwong, D.L.W.; Chiang, A.K.S. Genomic diversity of epstein-barr virus genomes isolated from primary nasopharyngeal carcinoma biopsy samples. J. Virol. 2014, 88, 10662–10672. [Google Scholar] [CrossRef] [Green Version]
- Bräuninger, A.; Spieker, T.; Willenbrock, K.; Gaulard, P.; Wacker, H.H.; Rajewsky, K.; Hansmann, M.L.; Küppers, R. Survival and clonal expansion of mutating “forbidden” (immunoglobulin receptor-deficient) epstein-barr virus-infected b cells in angioimmunoblastic T cell lymphoma. J. Exp. Med. 2001, 194, 927–940. [Google Scholar] [CrossRef]
- Lei, H.; Li, T.; Hung, G.-C.; Li, B.; Tsai, S.; Lo, S.-C. Identification and characterization of EBV genomes in spontaneously immortalized human peripheral blood B lymphocytes by NGS technology. BMC Genomics 2013, 14, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borozan, I.; Zapatka, M.; Frappier, L.; Ferretti, V. Analysis of epstein-barr virus genomes and expression profiles in gastric adenocarcinoma. J. Virol. 2018, 92, e01239-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, S.; Palser, A.; Elgueta Karstegl, C.; Middeldorp, J.M.; Ramayanti, O.; Cohen, J.I.; Hildesheim, A.; Fellner, M.D.; Wiels, J.; White, R.E.; et al. Natural variation of epstein-barr virus genes, proteins, and primary microRNA. J. Virol. 2017, 91, e00375-17. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yang, W.; Pan, Y.; Ji, J.; Lu, Z.; Ke, Y. Genome-wide analysis of epstein-barr virus (EBV) isolated from EBV-associated gastric carcinoma (EBVaGC). Oncotarget 2016, 7, 4903–4914. [Google Scholar] [CrossRef] [Green Version]
- Deakyne, J.S.; Malecka, K.A.; Messick, T.E.; Lieberman, P.M. Structural and functional basis for an EBNA1 hexameric ring in epstein-barr virus episome maintenance. J. Virol. 2017, 91, e01046-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-Y.; Chien, Y.-C.; Jan, J.-S.; Chueh, C.-M.; Lin, J.-C. Consistent sequence variation of epstein-barr virus nuclear antigen 1 in primary tumor and peripheral blood cells of patients with nasopharyngeal carcinoma. Clin. Cancer Res. 2002, 8, 2586–2590. [Google Scholar] [PubMed]
- Bhatia, K.; Raj, A.; Guitierrez, M.I.; Judde, J.G.; Spangler, G.; Venkatesh, H.; Magrath, I.T. Variation in the sequence of epstein barr virus nuclear antigen 1 in normal peripheral blood lymphocytes and in burkitt’s lymphomas. Oncogene 1996, 13, 177–181. [Google Scholar] [PubMed]
- Wang, X.; Wang, Y.; Wu, G.; Chao, Y.; Sun, Z.; Luo, B. Sequence analysis of epstein-barr virus EBNA-2 gene coding amino acid 148–487 in nasopharyngeal and gastric carcinomas. Virol. J. 2012, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Wang, X.; Strong, M.J.; Concha, M.; Baddoo, M.; Xu, G.; Baribault, C.; Fewell, C.; Hulme, W.; Hedges, D.; et al. Whole-genome sequencing of the akata and mutu epstein-barr virus strains. J. Virol. 2013, 87, 1172–1182. [Google Scholar] [CrossRef] [Green Version]
- Ba Abdullah, M.M.; Palermo, R.D.; Palser, A.L.; Grayson, N.E.; Kellam, P.; Correia, S.; Szymula, A.; White, R.E. Heterogeneity of the epstein-barr virus (EBV) major internal repeat reveals evolutionary mechanisms of EBV and a functional defect in the prototype EBV strain B95-8. J. Virol. 2017, 91, e00920-17. [Google Scholar] [CrossRef] [Green Version]
- Masud, H.M.A.A.; Yanagi, Y.; Watanabe, T.; Sato, Y.; Kimura, H.; Murata, T. Epstein-barr virus BBRF2 is required for maximum infectivity. Microorganisms 2019, 7, 705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, K.; Thikmyanova, N.; Wojcechowskyj, J.A.; Delecluse, H.-J.; Lieberman, P.M. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 2011, 7, e1002376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Deng, Z.; Vladimirova, O.; Wiedmer, A.; Lu, F.; Lieberman, P.M.; Patel, D.J. Structural basis underlying viral hijacking of a histone chaperone complex. Nat. Commun. 2016, 7, 12707. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Age at Diagnosis | Pathology According to WHO Criteria (2016) | Survival after Diagnosis (Years) |
|---|---|---|---|---|
| AIL1 | F | 73 | Angioimmunoblastic T-cell lymphoma | 1 |
| AIL2 | M | 62 | Angioimmunoblastic T-cell lymphoma | 10 |
| AIL3 | M | 80 | Angioimmunoblastic T-cell lymphoma | 2 |
| AIL4 | M | 55 | Angioimmunoblastic T-cell lymphoma | 2 |
| AIL5 | F | 66 | Angioimmunoblastic T-cell lymphoma | 4.5 |
| AIL6 | F | 84 | Angioimmunoblastic T-cell lymphoma | 1 |
| AIL7 | F | 59 | Angioimmunoblastic T-cell lymphoma | 4 |
| AIL8 | F | 52 | Angioimmunoblastic T-cell lymphoma | 11 |
| AIL9 | F | 76 | Angioimmunoblastic T-cell lymphoma | 1 |
| AIL10 | F | 53 | Angioimmunoblastic T-cell lymphoma | >16 |
| AIL11 | M | 59 | Angioimmunoblastic T-cell lymphoma | 10 |
| AIL12 | F | 56 | Angioimmunoblastic T-cell lymphoma | 11 |
| AIL13 | F | 78 | Angioimmunoblastic T-cell lymphoma | >10 |
| AIL14 | M | 62 | Angioimmunoblastic T-cell lymphoma | 3 |
| AIL15 | M | 67 | Angioimmunoblastic T-cell lymphoma | 3.5 |
| AIL16 | M | 50 | Angioimmunoblastic T-cell lymphoma | 1 |
| AIL17 | F | 84 | Angioimmunoblastic T-cell lymphoma | 8 |
| AIL18 | F | 67 | Angioimmunoblastic T-cell lymphoma | >10 |
| ALCL1 | F | 73 | Anaplastic large T cell lymphoma | 3.5 |
| CTCL1 | F | 73 | Cutaneous T cell lymphoma | 1 |
| NK/TL1 | F | 51 | NK/T cell lymphoma | >21 |
| NK/TL2 | M | 64 | NK/T cell lymphoma | >9 |
| PTTL1 | M | 63 | Post-transplant T lymphoma | 1.5 |
| PTBL1 | M | 28 | Post-transplant B lymphoma | >5 |
| PTBL2 | F | 63 | Post-transplant B lymphoma | <1 |
| PTBL3 | F | 65 | Post-transplant B lymphoma | >10 |
| PTBL4 | F | 52 | Post-transplant B lymphoma | >15 |
| DLBCL1 | F | 74 | Diffuse large B-cell lymphoma | 2.5 |
| DLBCL2 | M | 77 | Diffuse large B-cell lymphoma | <1 |
| DLBCL3 | F | 59 | Diffuse large B-cell lymphoma | 4.5 |
| DLBCL4 | F | 31 | Diffuse large B-cell lymphoma | 10 |
| LPL1 | F | 18 | Lymphoplasmacytic lymphoma | >7 |
| ARL1 | F | 89 | Age related lymphoma | <1 |
| ARL2 | F | 76 | Age related lymphoma | >8 |
| Ct1 | F | 73 | Reactive adenopathy | |
| PI | M | 13 | EBV primary infection |
| Sample | Number of Reads | Mapping to EBV (%) | Mapping to hg19 (%) | Mean Depth | Genome Coverage 10× (%) | EBNA-2,3 EBV-1/EBNA-2,3 EBV-2 Read Numbers |
|---|---|---|---|---|---|---|
| AIL1 | 73,730 | 97.86 | 6.76 | 68 | 97.1 | 6.85 |
| AIL2 | 105,196 | 97.03 | 7.24 | 82 | 95.5 | 6.15 |
| AIL3 | 23,145,280 | 96.89 | 3.38 | 9,235 | 99.9 | 6.99 |
| AIL4 | 59,822 | 98.18 | 12.11 | 46 | 96.8 | 7.07 |
| AIL5 | 128,648 | 97.73 | 7.13 | 100 | 98.5 | 5.57 |
| AIL6 | 13,342,518 | 98.28 | 22.04 | 4,426 | 99.8 | 8.24 |
| AIL7 | 922,026 | 98.73 | 5.4 | 779 | 99.8 | 6.18 |
| AIL8 | 9,076,710 | 97.99 | 29.91 | 2,702 | 99.8 | 8.38 |
| AIL9 | 34,991,168 | 98.67 | 11.49 | 13,180 | 99.9 | 8.59 |
| AIL10 | 27,435,252 | 98.99 | 9.46 | 10,600 | 99.9 | 7.80 |
| AIL11 | 2,827,228 | 94.83 | 50.35 | 582 | 98 | 12.48 |
| AIL12 | 38,416,258 | 99.2 | 7.46 | 15,199 | 99.9 | 8.46 |
| AIL13 | 74,846 | 96.91 | 13.01 | 58 | 96.8 | 6.03 |
| AIL14 | 1,231,958 | 98.89 | 5.03 | 1,056 | 99.8 | 6.21 |
| AIL15 | 1,047,872 | 98.84 | 5.28 | 875 | 99.7 | 6.41 |
| AIL16 | 101,308 | 98.16 | 10.22 | 77 | 98.3 | 5.78 |
| AIL17 | 53,864,512 | 99 | 7.72 | 21,194 | 99.9 | 9.21 |
| AIL18 | 12,827,446 | 97.91 | 20.05 | 4,210 | 99.8 | 0.39 |
| DLBCL1 | 2,529,424 | 94.2 | 53.57 | 485 | 93.5 | 8.52 |
| DLBCL2 | 27,943,648 | 98.3 | 5.38 | 22,975 | 98.3 | 5.57 |
| DLBCL3 | 20,223,716 | 98.06 | 5.55 | 16,076 | 98.2 | 6.28 |
| DLBCL4 | 393,996 | 98.4 | 6.34 | 316 | 99.3 | 6.42 |
| LPL1 | 102,642 | 96.7 | 10.26 | 75 | 97.7 | 3.57 |
| PTBL1 | 4,050,570 | 98.34 | 5.02 | 3,184 | 99.8 | 6.26 |
| PTBL2 | 2,588,074 | 98.48 | 4.64 | 2,139 | 99.8 | 6.45 |
| PTBL3 | 828,392 | 97 | 6.4 | 630 | 99.8 | 14.44 |
| PTLB4 | 624,062 | 98.42 | 5.07 | 513 | 99.7 | 6.08 |
| ARL1 | 328,098 | 78.44 | 5.95 | 159 | 98.6 | 7.83 |
| ARL2 | 1,772,596 | 98.26 | 5.47 | 1,435 | 99.5 | 2.21 |
| PTTL1 | 1,777,550 | 75.57 | 39.28 | 437 | 99.4 | 7.05 |
| ALCL1 | 1,021,930 | 98.38 | 5.83 | 116 | 99 | 6.40 |
| CTCL1 | 18,578,118 | 98.63 | 5.32 | 15,039 | 96.1 | 5.60 |
| NK/TL1 | 342,062 | 98.16 | 6.1 | 170 | 98.9 | 7.12 |
| NK/TL2 | 6,742,818 | 98.43 | 4.74 | 5,689 | 99.9 | 6.52 |
| Control1 | 841,766 | 77.41 | 7.86 | 495 | 99.4 | 6.24 |
| PI1 | 13,1045,844 | 96.05 | 2.61 | 604 | 99.9 | 2.33 |
| B95.8 | 17,718,578 | 98.6 | 7.33 | 15,283 | 98.5 | 6.13 |
| Mec04 | 715,980 | 97.99 | 6.87 | 619 | 99.8 | 6.60 |
| Raji | 1,820,758 | 97.63 | 6.66 | 1,460 | 98.4 | 4.90 |
| Jijoye | 3,813,610 | 98.15 | 6.43 | 3,124 | 98.3 | 0.39 |
| Namalwa | 944,054 | 97.56 | 5.58 | 780 | 96.2 | 0.39 |
| P3HR1 | 99,174 | 80.61 | 11.67 | 69 | 92 | 0.30 |
| SNK6 | 1,733,840 | 98.05 | 7.04 | 1,487 | 96.7 | 0.41 |
| Sample | Contigs > 1000 | Total Length of Contigs > 1000 bp | Contig Number | Total Length | Largest Contig | GC% | N50 Size | GenBank Accession Number |
|---|---|---|---|---|---|---|---|---|
| AIL1 | 9 | 142,548 | 11 | 143,842 | 52,398 | 57.81 | 36,930 | MH837512 |
| AIL2 | 19 | 144,946 | 33 | 154,536 | 32,793 | 58.14 | 9,443 | MH837513 |
| AIL5 | 24 | 153,365 | 41 | 164,471 | 22,826 | 58.8 | 11,438 | MH837514 |
| AIL7 | 11 | 172,444 | 12 | 173,126 | 50,944 | 59.45 | 43,411 | MH837515 |
| AIL13 | 29 | 152,897 | 47 | 164,966 | 35,094 | 58.74 | 8,301 | MH837516 |
| AIL14 | 4 | 169,984 | 7 | 171,612 | 80,073 | 59.53 | 73,612 | MH837517 |
| AIL15 | 5 | 170,653 | 5 | 170,653 | 81,106 | 59.5 | 71,452 | MH837518 |
| AIL16 | 13 | 147,983 | 21 | 153,913 | 55,163 | 57.89 | 44,505 | MH837519 |
| CTCL1 | 8 | 162,594 | 12 | 165,345 | 43,990 | 59.14 | 43,099 | MH837521 |
| NK/TL2 | 3 | 170,356 | 6 | 172,153 | 96,347 | 59.53 | 96,347 | MH837524 |
| PTBL1 | 5 | 171,454 | 6 | 172,258 | 71,791 | 59.59 | 50,232 | MH837525 |
| PTBL2 | 4 | 171,267 | 4 | 171,267 | 80,971 | 59.16 | 73,531 | MH837526 |
| PTBL3 | 9 | 169,947 | 13 | 172,617 | 63,345 | 59.38 | 28,613 | MH837527 |
| PTBL4 | 12 | 174,524 | 13 | 175,108 | 49,504 | 59.46 | 39,096 | MH837528 |
| DLBCL2 | 10 | 163,503 | 15 | 166,737 | 66,384 | 59.36 | 57,091 | MH837522 |
| DLBCL4 | 17 | 171,309 | 20 | 173,272 | 36,271 | 59.36 | 11,510 | MH837523 |
| ARL2 | 8 | 169,962 | 11 | 172,116 | 71,378 | 59.34 | 28,847 | MH837520 |
| Sample | EBV (Copies/μg DNA) | EBV (Copies/Cell) | Latence (L) or Reactivation (R) |
|---|---|---|---|
| AIL1 | 281,630 | 0.081 | L |
| AIL2 | 413,060 | 0.082 | L |
| AIL3 | 13,380 | 0.008 | L |
| AIL4 | 150.324 | 0.348 | L |
| AIL5 | 1,092,700 | 12,074.033 | R |
| AIL6 | 31,130 | 0.002 | L |
| AIL7 | 3,846,140 | 4.498 | L |
| AIL8 | 60,490 | 0.003 | L |
| AIL9 | 82,314 | 0.01 | L |
| AIL10 | 156,480 | 0.012 | L |
| AIL11 | 33,950 | 0.004 | L |
| AIL12 | 59,830 | 0.005 | L |
| AIL13 | 189,538 | 0.016 | L |
| AIL14 | 2,713,100 | 0.185 | L |
| AIL15 | 3,363,280 | 0.33 | L |
| AIL16 | 372,724 | 7.499 | L |
| AIL17 | 60,740 | 0.007 | L |
| AIL18 | 40,080 | 0.006 | L |
| ALCL1 | 141,168 | 0.065 | L |
| CTCL1 | 299,914,110 | 413.674 | R |
| NK/TL1 | 688,300 | 0.531 | L |
| NK/TL2 | 22,046,280 | 27.731 | R |
| PTTL1 | 74,270 | 0.117 | L |
| PTBL1 | 18,916,830 | 34.084 | R |
| PTBL2 | 17,616,830 | 16.698 | L |
| PTBL3 | 5,774,190 | 6.451 | L |
| PTLB4 | 7,021,700 | 9.002 | L |
| DLBCL1 | 44,870 | 0.01 | L |
| DLBCL2 | 105,109,230 | 12.012 | L |
| DLBCL3 | 106,311,737 | 556.606 | R |
| DLBCL4 | 122,317 | 0.15 | L |
| LPL1 | 363,043 | 0.637 | L |
| ARL1 | 110,690 | 17.295 | L |
| ARL2 | 21,677,340 | 58.986 | R |
| Gene | Protein Function or Location | AIL | Other Lymphomas | p |
|---|---|---|---|---|
| EBNA-LP | Latency protein | 4.76 | 3.06 | 0.0042417244 |
| BKRF3 | Replication protein | 1.00 | 0.56 | 0.0049754187 |
| BBRF2 | Tegument protein | 1.00 | 0.63 | 0.0108844758 |
| BORF2 | Replication protein | 0.35 | 1.31 | 0.0133170812 |
| BBLF2/BBLF3 | Replication protein | 2.82 | 1.88 | 0.0214000908 |
| EBNA-2 | Latency protein | 9.35 | 6.31 | 0.0295629871 |
| BORF1 | Capsid protein | 1.24 | 1.81 | 0.0329743486 |
| BNRF1 | Tegument protein | 7.82 | 6.13 | 0.0393795760 |
| BMRF1 | Replication protein | 0.12 | 0.44 | 0.0406785170 |
| EBNA-1 | Latency protein | 15.76 | 13.44 | 0.0411169198 |
| BALF3 | DNA cleavage and packaging protein | 0.18 | 0.75 | 0.0419918591 |
| BBLF4 | Replication protein | 4.82 | 4.13 | 0.0427272346 |
| BFRF3 | Capsid protein | 0.94 | 1.13 | 0.0447940279 |
| BGRF1/BDRF1 | DNA cleavage and packaging protein | 0.35 | 0.06 | 0.0452271888 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahri, R.; Boyer, F.; Halabi, M.A.; Chaunavel, A.; Feuillard, J.; Jaccard, A.; Ranger-Rogez, S. Epstein-Barr Virus (EBV) Is Mostly Latent and Clonal in Angioimmunoblastic T Cell Lymphoma (AITL). Cancers 2022, 14, 2899. https://doi.org/10.3390/cancers14122899
Bahri R, Boyer F, Halabi MA, Chaunavel A, Feuillard J, Jaccard A, Ranger-Rogez S. Epstein-Barr Virus (EBV) Is Mostly Latent and Clonal in Angioimmunoblastic T Cell Lymphoma (AITL). Cancers. 2022; 14(12):2899. https://doi.org/10.3390/cancers14122899
Chicago/Turabian StyleBahri, Racha, François Boyer, Mohamad Adnan Halabi, Alain Chaunavel, Jean Feuillard, Arnaud Jaccard, and Sylvie Ranger-Rogez. 2022. "Epstein-Barr Virus (EBV) Is Mostly Latent and Clonal in Angioimmunoblastic T Cell Lymphoma (AITL)" Cancers 14, no. 12: 2899. https://doi.org/10.3390/cancers14122899