
Interconnected Adaptive Responses: A Way Out for Cancer Cells to Avoid Cellular Demise


Abstract
:Simple Summary
Abstract
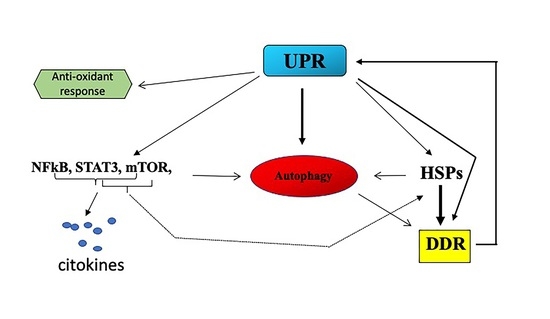
{kind=link}
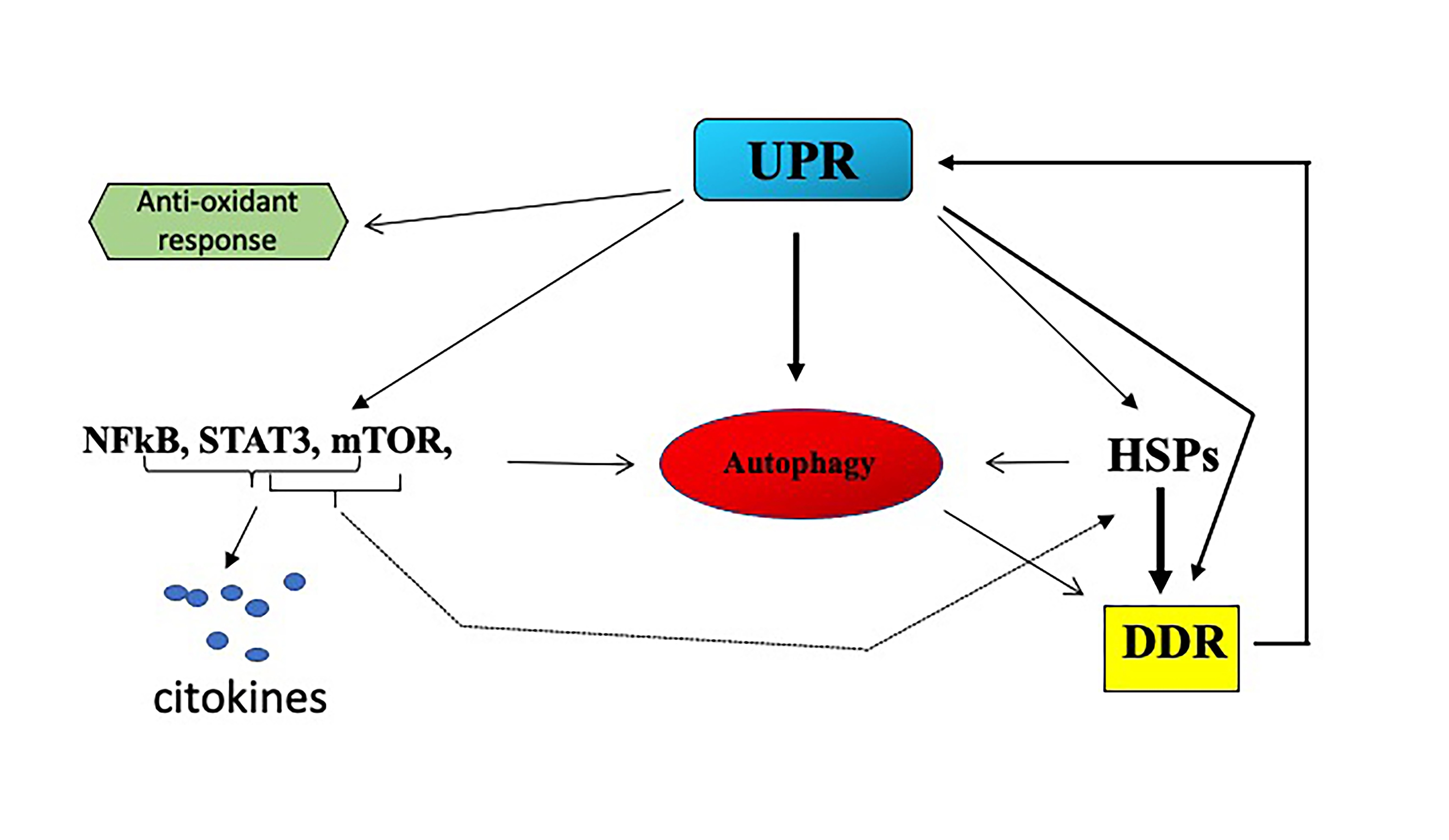
{kind=link}
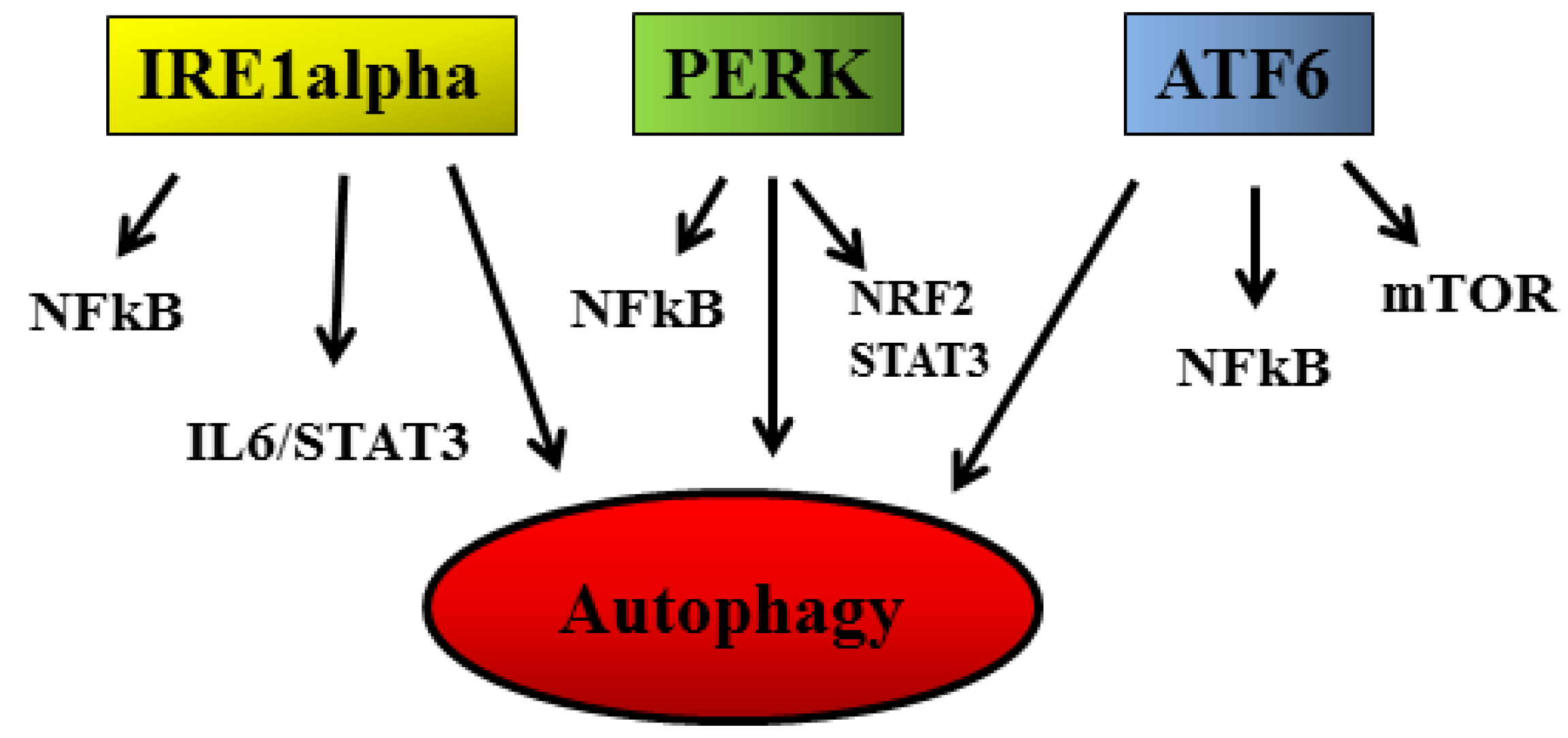
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Stress Response Pathways and Cancer Survival
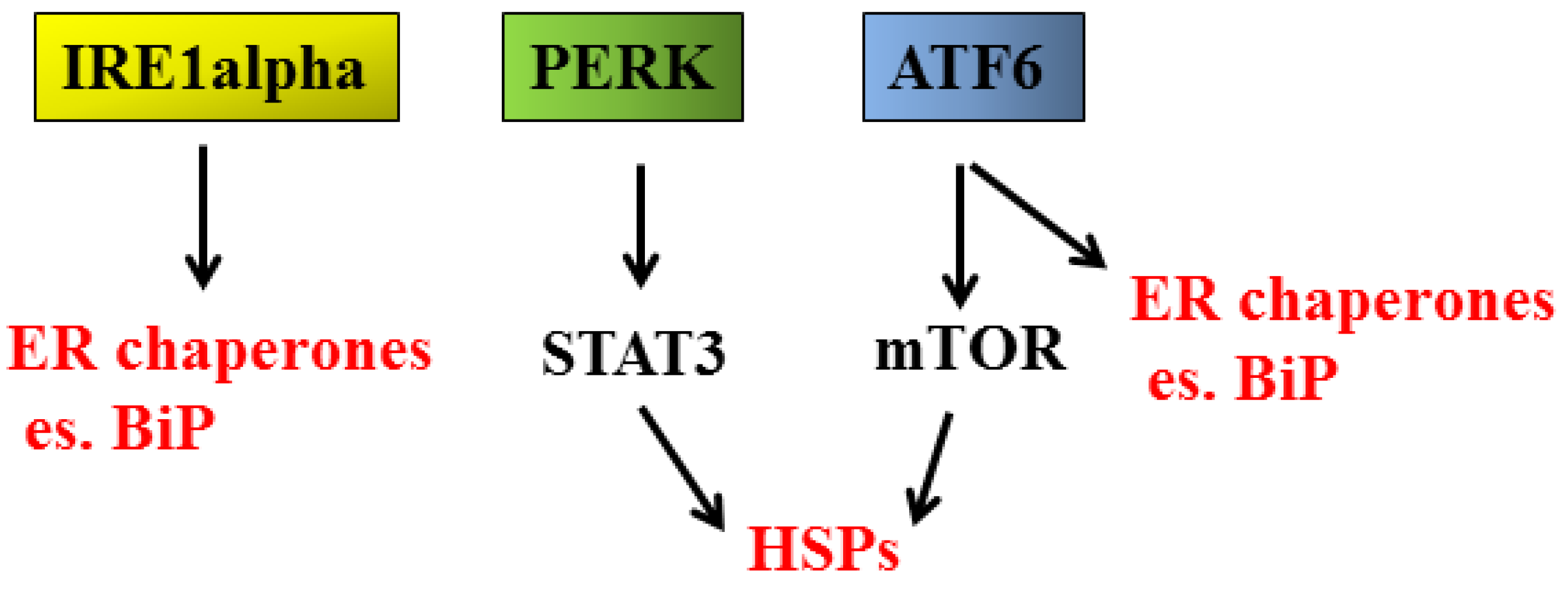
2.1. UPR and Cancer
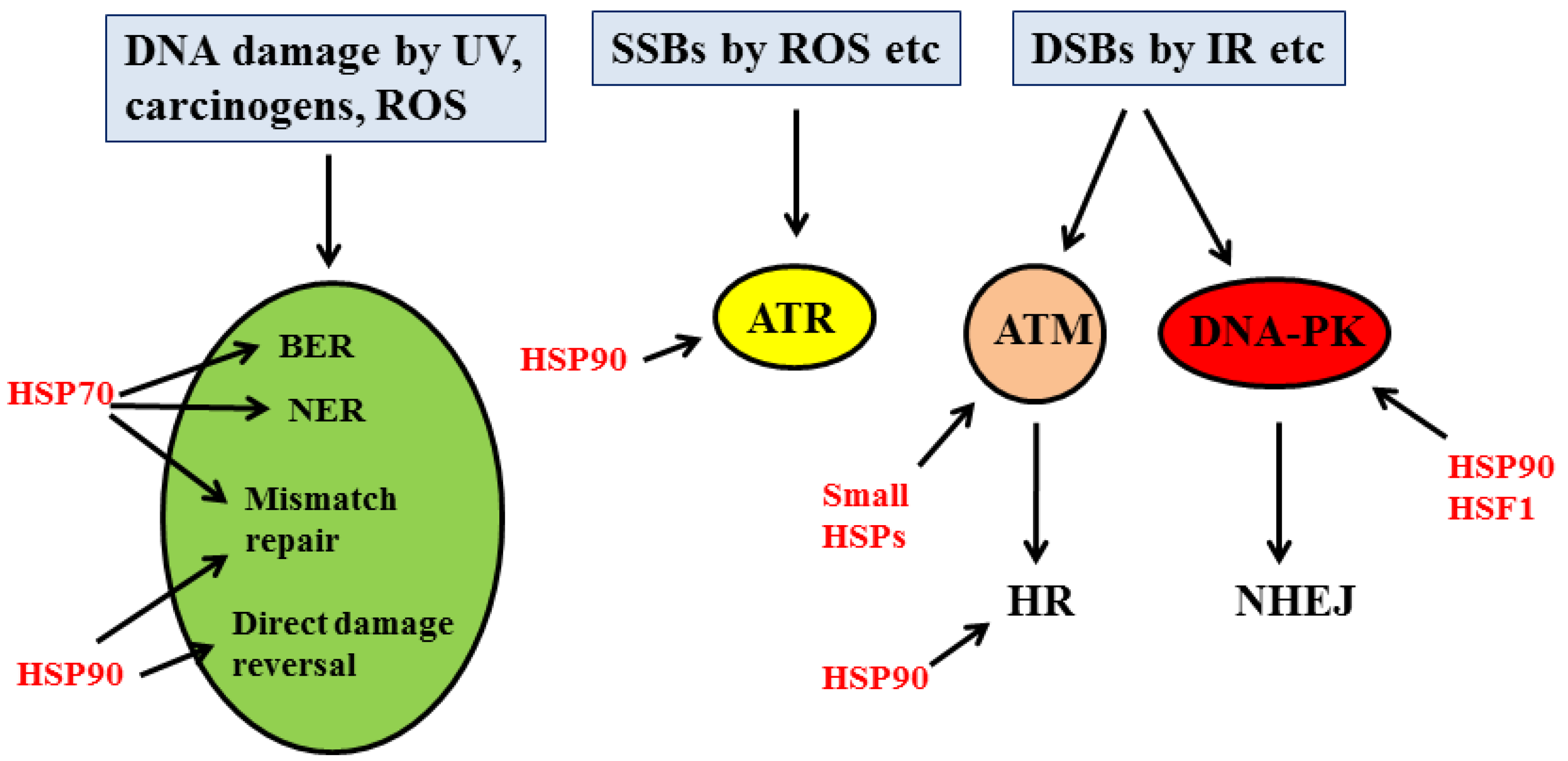
2.2. DDR and Cancer
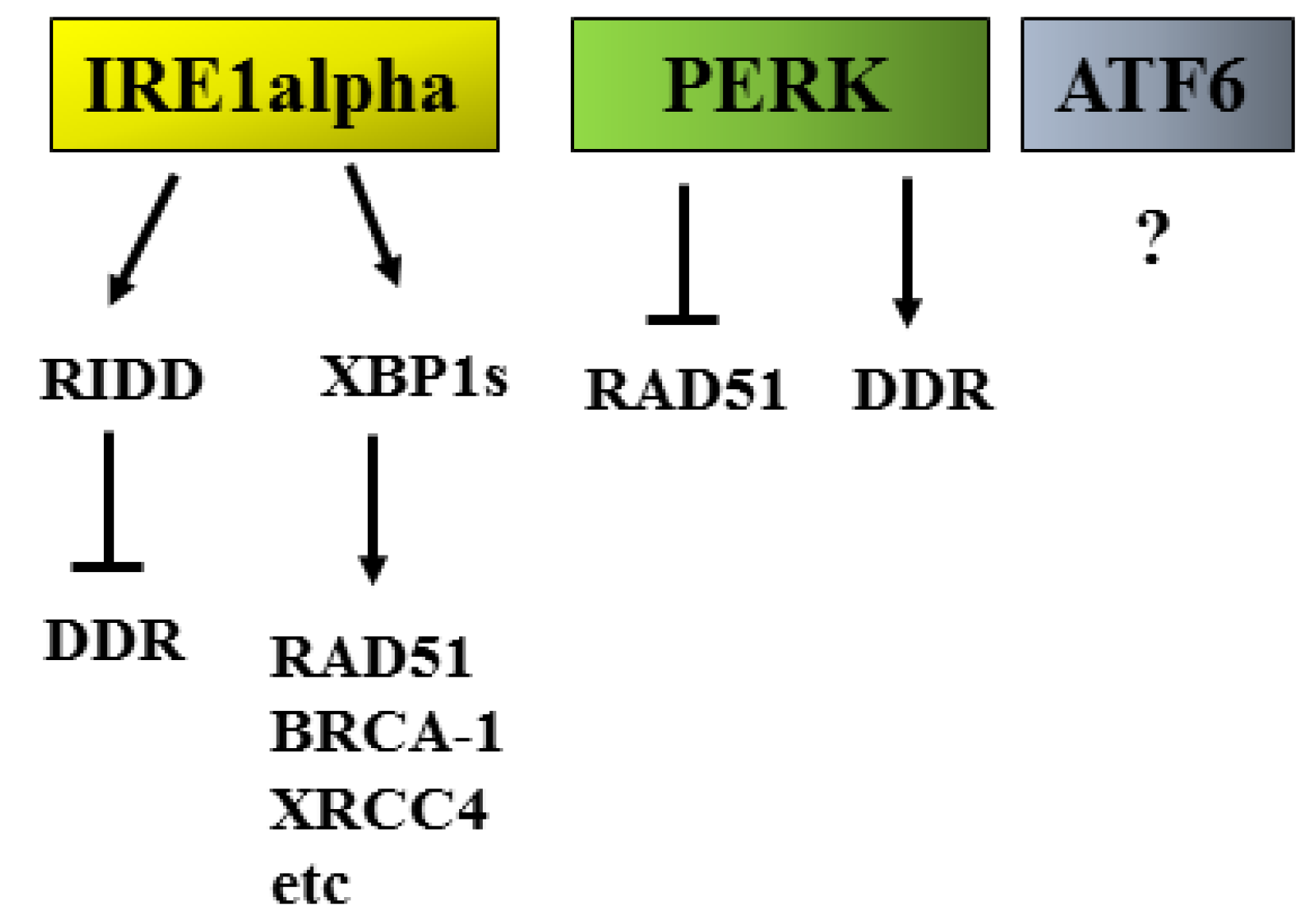
2.3. UPR/DDR Interplay and Cancer
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Gonzalez-Quiroz, M.; Blondel, A.; Sagredo, A.; Hetz, C.; Chevet, E.; Pedeux, R. When Endoplasmic Reticulum Proteostasis Meets the DNA Damage Response. Trends Cell Biol. 2020, 30, 881–891. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharm. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Davenport, E.L.; Moore, H.E.; Dunlop, A.S.; Sharp, S.Y.; Workman, P.; Morgan, G.J.; Davies, F.E. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood 2007, 110, 2641–2649. [Google Scholar] [CrossRef] [Green Version]
- Walczak, A.; Gradzik, K.; Kabzinski, J.; Przybylowska-Sygut, K.; Majsterek, I. The Role of the ER-Induced UPR Pathway and the Efficacy of Its Inhibitors and Inducers in the Inhibition of Tumor Progression. Oxid. Med. Cell Longev. 2019, 2019, 5729710. [Google Scholar] [CrossRef] [Green Version]
- Ojha, R.; Amaravadi, R.K. Targeting the unfolded protein response in cancer. Pharm. Res. 2017, 120, 258–266. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [Green Version]
- Cirone, M.; Di Renzo, L.; Lotti, L.V.; Conte, V.; Trivedi, P.; Santarelli, R.; Gonnella, R.; Frati, L.; Faggioni, A. Activation of dendritic cells by tumor cell death. Oncoimmunology 2012, 1, 1218–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Benedetti, R.; Piconese, S.; Pulcinelli, F.M.; Timperio, A.M.; Romeo, M.A.; Masuelli, L.; Mattei, M.; Bei, R.; D’Orazi, G.; et al. PGE2 Released by Pancreatic Cancer Cells Undergoing ER Stress Transfers the Stress to DCs Impairing Their Immune Function. Mol. Cancer 2021, 20, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Pan, H.; Wang, J.; Wang, T.; Huo, X.; Ma, Y.; Lu, Z.; Sun, B.; Jiang, H. Unfolded protein response in colorectal cancer. Cell Biosci. 2021, 11, 26. [Google Scholar] [CrossRef]
- Chen, S.; Chen, J.; Hua, X.; Sun, Y.; Cui, R.; Sha, J.; Zhu, X. The emerging role of XBP1 in cancer. Biomed. Pharm. 2020, 127, 110069. [Google Scholar] [CrossRef]
- Liu, C.Y.; Hsu, C.C.; Huang, T.T.; Lee, C.H.; Chen, J.L.; Yang, S.H.; Jiang, J.K.; Chen, W.S.; Lee, K.D.; Teng, H.W. ER stress-related ATF6 upregulates CIP2A and contributes to poor prognosis of colon cancer. Mol. Oncol. 2018, 12, 1706–1717. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Allen, D.; Seo, J. ER Stress Activates the TOR Pathway through Atf6. J. Mol. Signal. 2018, 13, 1. [Google Scholar] [CrossRef]
- Song, M.; Wang, C.; Yang, H.; Chen, Y.; Feng, X.; Li, B.; Fan, H. P-STAT3 Inhibition Activates Endoplasmic Reticulum Stress-Induced Splenocyte Apoptosis in Chronic Stress. Front. Physiol. 2020, 11, 680. [Google Scholar] [CrossRef]
- Malovrh, E.; Volkers, M. Crosstalk Between the Endoplasmic Reticulum and mTOR Signaling. Circ. Res. 2019, 124, 9–11. [Google Scholar] [CrossRef]
- Dong, G.; Liu, Y.; Zhang, L.; Huang, S.; Ding, H.F.; Dong, Z. mTOR contributes to ER stress and associated apoptosis in renal tubular cells. Am. J. Physiol. Ren. Physiol. 2015, 308, F267–F274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirone, M. Cancer cells dysregulate PI3K/AKT/mTOR pathway activation to ensure their survival and proliferation: Mimicking them is a smart strategy of gammaherpesviruses. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Gilardini Montani, M.S.; Granato, M.; Garufi, A.; Faggioni, A.; D’Orazi, G. Autophagy manipulation as a strategy for efficient anticancer therapies: Possible consequences. J. Exp. Clin. Cancer Res. 2019, 38, 262. [Google Scholar] [CrossRef] [PubMed]
- Zada, S.; Hwang, J.S.; Ahmed, M.; Lai, T.H.; Pham, T.M.; Elashkar, O.; Kim, D.R. Cross talk between autophagy and oncogenic signaling pathways and implications for cancer therapy. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188565. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A.; Thamm, D.H.; Gustafson, D.L. Autophagy and cancer therapy. Mol. Pharm. 2014, 85, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 2016, 1, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Cirone, M. ER Stress, UPR Activation and the Inflammatory Response to Viral Infection. Viruses 2021, 13, 798. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, N.R.; Rodvold, J.; Sepulveda, H.; Rossi, S.; Drew, A.F.; Zanetti, M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6561–6566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Liu, Y.; Chang, A. Heat shock response relieves ER stress. EMBO J. 2008, 27, 1049–1059. [Google Scholar] [CrossRef] [Green Version]
- Jee, H. Size dependent classification of heat shock proteins: A mini-review. J. Exerc. Rehabil. 2016, 12, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.D.; Prince, T.; Gong, J.; Calderwood, S.K. mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS ONE 2012, 7, e39679. [Google Scholar] [CrossRef]
- Stephanou, A.; Latchman, D.S. Transcriptional regulation of the heat shock protein genes by STAT family transcription factors. Gene Expr. 1999, 7, 311–319. [Google Scholar]
- Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Santarelli, R.; D’Orazi, G.; Cirone, M. STAT3 and mutp53 Engage a Positive Feedback Loop Involving HSP90 and the Mevalonate Pathway. Front. Oncol. 2020, 10, 1102. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314.e7. [Google Scholar] [CrossRef] [Green Version]
- Granato, M.; Gilardini Montani, M.S.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Apigenin, by activating p53 and inhibiting STAT3, modulates the balance between pro-apoptotic and pro-survival pathways to induce PEL cell death. J. Exp. Clin. Cancer Res. 2017, 36, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijink, A.M.; Krajewska, M.; van Vugt, M.A. The DNA damage response during mitosis. Mutat. Res. 2013, 750, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boesch, P.; Weber-Lotfi, F.; Ibrahim, N.; Tarasenko, V.; Cosset, A.; Paulus, F.; Lightowlers, R.N.; Dietrich, A. DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim. Biophys. Acta 2011, 1813, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandsma, I.; Fleuren, E.D.G.; Williamson, C.T.; Lord, C.J. Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin. Investig. Drugs 2017, 26, 1341–1355. [Google Scholar] [CrossRef]
- Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Arena, A.; Maretto, M.; Bassetti, E.; Caiazzo, R.; D’Orazi, G.; Cirone, M. Anticancer effect of AZD2461 PARP inhibitor against colon cancer cells carrying wt or dysfunctional p53. Exp. Cell Res. 2021, 408, 112879. [Google Scholar] [CrossRef]
- Gao, H.; Xue, J.; Zhou, L.; Lan, J.; He, J.; Na, F.; Yang, L.; Deng, L.; Lu, Y. Bevacizumab radiosensitizes non-small cell lung cancer xenografts by inhibiting DNA double-strand break repair in endothelial cells. Cancer Lett. 2015, 365, 79–88. [Google Scholar] [CrossRef]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur. J. Med. Chem. 2022, 230, 114109. [Google Scholar] [CrossRef]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage repair in cancer: From mechanisms to applications. Ann. Transl. Med. 2020, 8, 1685. [Google Scholar] [CrossRef]
- Sottile, M.L.; Nadin, S.B. Heat shock proteins and DNA repair mechanisms: An updated overview. Cell Stress Chaperones 2018, 23, 303–315. [Google Scholar] [CrossRef]
- Zhang, Z.; Jing, J.; Ye, Y.; Chen, Z.; Jing, Y.; Li, S.; Hong, W.; Ruan, H.; Liu, Y.; Hu, Q.; et al. Characterization of the dual functional effects of heat shock proteins (HSPs) in cancer hallmarks to aid development of HSP inhibitors. Genome Med. 2020, 12, 101. [Google Scholar] [CrossRef] [PubMed]
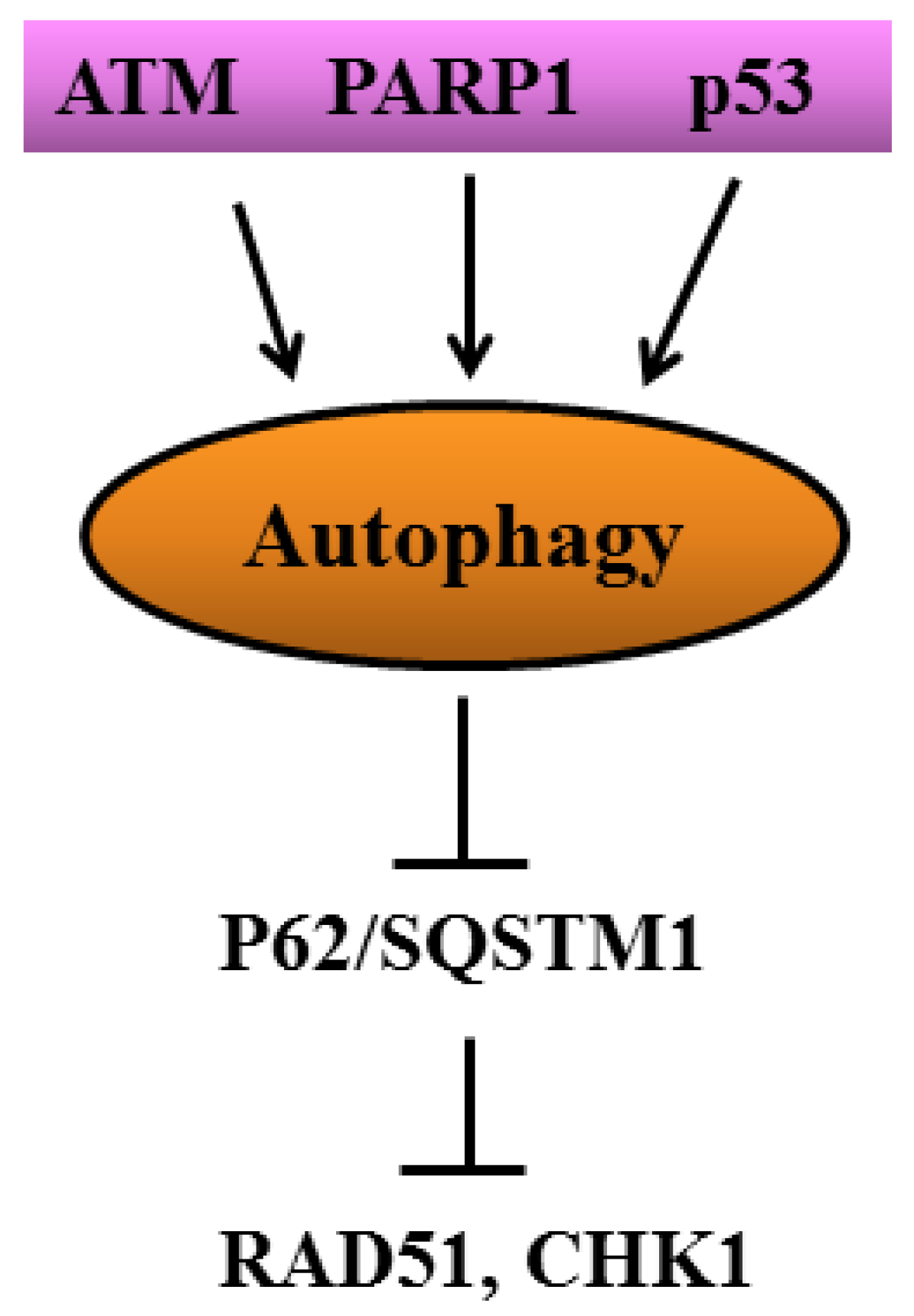
- Gomes, L.R.; Menck, C.F.M.; Leandro, G.S. Autophagy Roles in the Modulation of DNA Repair Pathways. Int. J. Mol. Sci. 2017, 18, 2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA Damage Response and Autophagy: A Meaningful Partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martin, P.; Saito, T.; Komatsu, M. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, G.; Carroll, B.; Sarallah, R.; Correia-Melo, C.; Ogrodnik, M.; Nelson, G.; Otten, E.G.; Manni, D.; Antrobus, R.; Morgan, B.A.; et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 2016, 12, 1917–1930. [Google Scholar] [CrossRef]
- Gilardini Montani, M.S.; Tarquini, G.; Santarelli, R.; Gonnella, R.; Romeo, M.A.; Benedetti, R.; Arena, A.; Faggioni, A.; Cirone, M. p62/SQSTM1 promotes mitophagy and activates the NRF2-mediated antioxidant and anti-inflammatory response restraining EBV-driven B lymphocyte proliferation. Carcinogenesis 2022, 43, 277–287. [Google Scholar] [CrossRef]
- Yamamori, T.; Meike, S.; Nagane, M.; Yasui, H.; Inanami, O. ER stress suppresses DNA double-strand break repair and sensitizes tumor cells to ionizing radiation by stimulating proteasomal degradation of Rad51. FEBS Lett. 2013, 587, 3348–3353. [Google Scholar] [CrossRef] [Green Version]
- Gonnella, R.; Guttieri, L.; Gilardini Montani, M.S.; Santarelli, R.; Bassetti, E.; D’Orazi, G.; Cirone, M. Zinc Supplementation Enhances the Pro-Death Function of UPR in Lymphoma Cells Exposed to Radiation. Biology 2022, 11, 132. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; van der Kogel, A.J.; Sweep, F.C.; Span, P.N. The PERK/ATF4/LAMP3-arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response. Radiother. Oncol. 2013, 108, 415–421. [Google Scholar] [CrossRef]
- Bolland, H.; Ma, T.S.; Ramlee, S.; Ramadan, K.; Hammond, E.M. Links between the unfolded protein response and the DNA damage response in hypoxia: A systematic review. Biochem. Soc. Trans. 2021, 49, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.J.; Yang, J.W.; Park, J.H.; Choi, E.S.; Lim, C.S.; Lee, S.; Han, C.Y. Endoplasmic Reticulum Stress Increases DUSP5 Expression via PERK-CHOP Pathway, Leading to Hepatocyte Death. Int. J. Mol. Sci. 2019, 20, 4369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.J.; Chen, S.; Wu, P.; Hu, C.S.; Thorne, R.F.; Luo, C.M.; Hersey, P.; Zhang, X.D. Inhibition of MEK blocks GRP78 up-regulation and enhances apoptosis induced by ER stress in gastric cancer cells. Cancer Lett. 2009, 274, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Yan, J.; Tang, D. Extracellular signal-regulated kinases modulate DNA damage response—A contributing factor to using MEK inhibitors in cancer therapy. Curr. Med. Chem. 2011, 18, 5476–5482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, S.; Campbell, C.; Venkitaraman, A.R.; Esposito, A. Pulsatile MAPK Signaling Modulates p53 Activity to Control Cell Fate Decisions at the G2 Checkpoint for DNA Damage. Cell Rep. 2020, 30, 2083–2093.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, K.; Arakawa, H.; Nakamura, Y. Dual-specificity phosphatase 5 (DUSP5) as a direct transcriptional target of tumor suppressor p53. Oncogene 2003, 22, 5586–5591. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Lynch, C.; Medeiros, B.C.; Liedtke, M.; Bam, R.; Tam, A.B.; Yang, Z.; Alagappan, M.; Abidi, P.; Le, Q.T.; et al. Identification of Doxorubicin as an Inhibitor of the IRE1alpha-XBP1 Axis of the Unfolded Protein Response. Sci. Rep. 2016, 6, 33353. [Google Scholar] [CrossRef]
- Dufey, E.; Bravo-San Pedro, J.M.; Eggers, C.; Gonzalez-Quiroz, M.; Urra, H.; Sagredo, A.I.; Sepulveda, D.; Pihan, P.; Carreras-Sureda, A.; Hazari, Y.; et al. Genotoxic stress triggers the activation of IRE1alpha-dependent RNA decay to modulate the DNA damage response. Nat. Commun. 2020, 11, 2401. [Google Scholar] [CrossRef]
- Arena, A.; Romeo, M.A.; Benedetti, R.; Montani, M.S.G.; Cirone, M. The impairment of DDR reduces XBP1s, further increasing DNA damage, and triggers autophagy via PERK/eIF2alpha in MM and IRE1alpha/JNK1/2 in PEL cells. Biochem. Biophys. Res. Commun. 2022, 613, 19–25. [Google Scholar] [CrossRef]
- Gong, C.; Yang, Z.; Zhang, L.; Wang, Y.; Gong, W.; Liu, Y. Quercetin suppresses DNA double-strand break repair and enhances the radiosensitivity of human ovarian cancer cells via p53-dependent endoplasmic reticulum stress pathway. OncoTargets Ther. 2018, 11, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Vakifahmetoglu-Norberg, H.; Norberg, E.; Perdomo, A.B.; Olsson, M.; Ciccosanti, F.; Orrenius, S.; Fimia, G.M.; Piacentini, M.; Zhivotovsky, B. Caspase-2 promotes cytoskeleton protein degradation during apoptotic cell death. Cell Death Dis. 2013, 4, e940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Sicari, D.; Fantuz, M.; Bellazzo, A.; Valentino, E.; Apollonio, M.; Pontisso, I.; Di Cristino, F.; Dal Ferro, M.; Bicciato, S.; Del Sal, G.; et al. Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene 2019, 38, 6184–6195. [Google Scholar] [CrossRef]
- Arena, A.; Gilardini Montani, M.S.; Romeo, M.A.; Benedetti, R.; Gaeta, A.; Cirone, M. DNA damage triggers an interplay between wtp53 and c-Myc affecting lymphoma cell proliferation and Kaposi sarcoma herpesvirus replication. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119168. [Google Scholar] [CrossRef] [PubMed]
- Arena, A.; Romeo, M.A.; Benedetti, R.; Gilardini Montani, M.S.; Cirone, M. Targeting c-Myc Unbalances UPR towards Cell Death and Impairs DDR in Lymphoma and Multiple Myeloma Cells. Biomedicines 2022, 10, 731. [Google Scholar] [CrossRef] [PubMed]
- Nastasi, C.; Mannarino, L.; D’Incalci, M. DNA Damage Response and Immune Defense. Int. J. Mol. Sci. 2020, 21, 7504. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Orazi, G.; Cirone, M. Interconnected Adaptive Responses: A Way Out for Cancer Cells to Avoid Cellular Demise. Cancers 2022, 14, 2780. https://doi.org/10.3390/cancers14112780
D’Orazi G, Cirone M. Interconnected Adaptive Responses: A Way Out for Cancer Cells to Avoid Cellular Demise. Cancers. 2022; 14(11):2780. https://doi.org/10.3390/cancers14112780
Chicago/Turabian StyleD’Orazi, Gabriella, and Mara Cirone. 2022. "Interconnected Adaptive Responses: A Way Out for Cancer Cells to Avoid Cellular Demise" Cancers 14, no. 11: 2780. https://doi.org/10.3390/cancers14112780