
Cancer-Induced Metabolic Rewiring of Tumor Endothelial Cells



Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Blood Vessel Formation
1.2. Tumor Vasculature
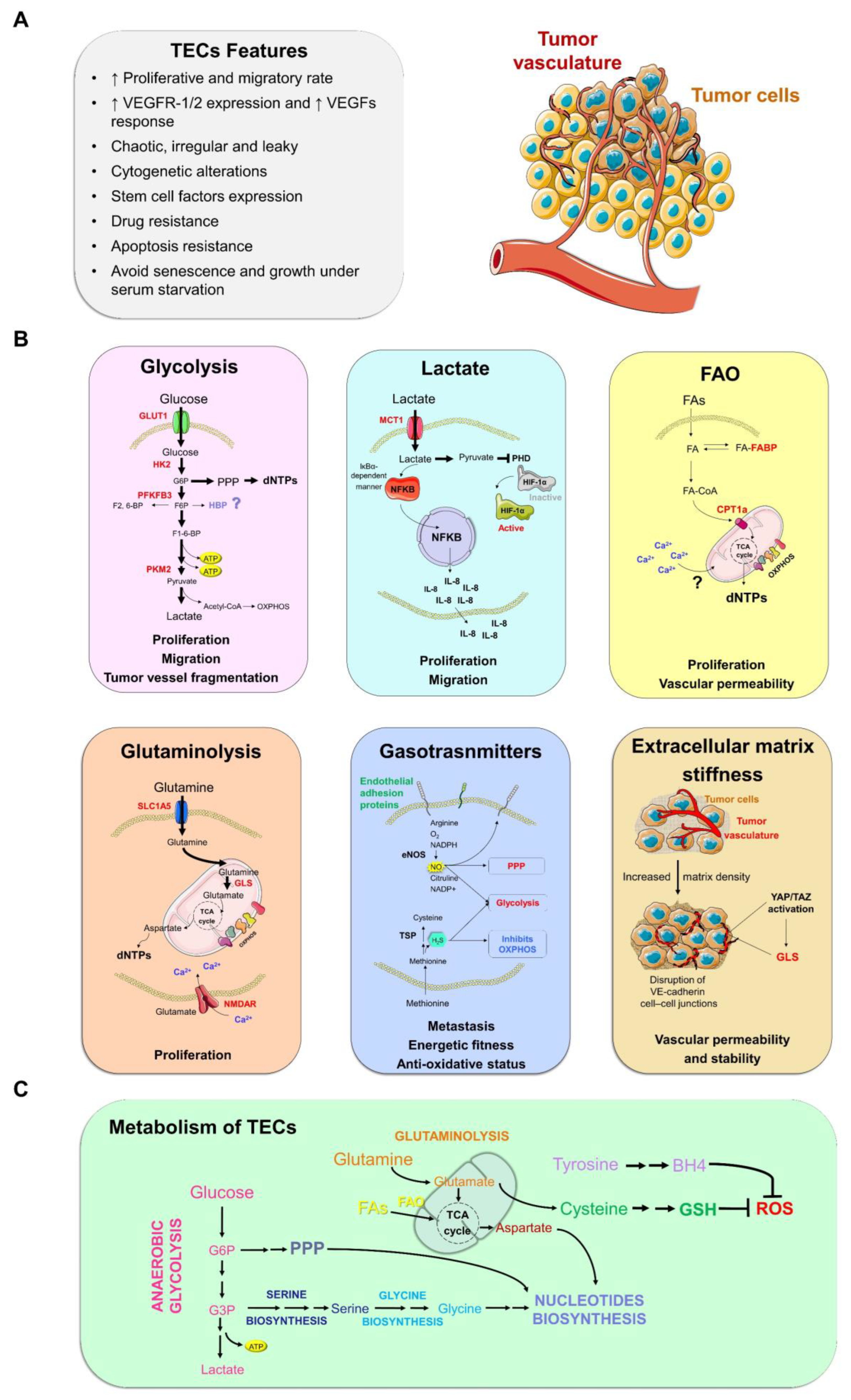
1.3. Molecular Aspects of Tumor Endothelial Cells (TECs)
2. Endothelial Metabolism during Angiogenesis
3. Rewiring of Endothelial Metabolism in Cancer
3.1. Glycolysis

{kind=link}
| Metabolic Pathway | Molecular Mechanism | Tumor Type | Reference |
|---|---|---|---|
| Glycolysis | PFKFB3 inhibition or haploinsufficiency normalize tumor vessel reducing glycolysis. | Melanoma, pancreatic tumor. | [11,44] |
| Glycolytic genes over-expression. | NSCLC, Melanoma. | [29,43] | |
| Lactate | VEGF-A induces carbonic anhydrase 2 (CAII), which reduces lactate acidosis in the tumor environment and enhances TEC survival. | Melanoma. | [49] |
| Cancer-produced lactate enter ECs by monocarboxylate transporter (MCT-1) and fuels angiogenesis through NF-κB pathway, leading to the autocrine stimulation of IL-8. | Colorectal, breast cancer. | [50] | |
| Within TEC, lactate can be converted into pyruvate, which exerts a negative feedback on PHD2 triggering angiogenesis by HIF1α activation. | Pulmonary Lewis lung carcinoma. | [51] | |
| OXPHOS | Mitochondrial respiration is necessary to sustain tumor angiogenesis. | Colorectal cancer, melanoma. | [46] |
| OXPHOS genes over-expression. | NSCLC. | [29] | |
| Hypoxia | EC-specific HIF1α deletion prevents autocrine signaling of VEGF, leading to reduction of vasculature. | Pulmonary Lewis lung carcinoma. | [52] |
| PHD2 haploinsufficiency in ECs reduces glycolysis normalizing tumor vasculature. | Melanoma. | [53] | |
| H2S | CBS silencing and pharmacological inhibition of CTH in cancer cells, the major H2S producers, reduce tumor angiogenesis. | Breast cancer. | [54] |
| CTH knockdown reduces lymphangiogenesis. | Prostate cancer. | [55] | |
| CBS silencing reduces vasculature. | Colon cancer. | [55] | |
| ECM stiffness | Pharmacological inhibition of the stiffness of the ECM reduces tumor angiogenesis. | Breast cancer. | [56] |
| EC-specific knockout of YAP/TAZ results in impaired tumor and tumor vessel growth. | Melanoma. | [57] |
3.2. Lactate
3.3. Fatty Acid Oxidation
3.4. Non-Essential Amino Acids
3.4.1. Glutamine–Glutamate
3.4.2. Aspartate–Asparagine
3.4.3. Serine
3.4.4. Glycine
3.4.5. Cysteine
3.4.6. Tyrosine
3.5. Metabolic Interactions between Tumor Tissue and Endothelial Cells
3.6. Gasotransmitters: Signals in the Air
3.7. Extracellular Matrix Stiffness and Angiogenesis
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, N.; Chen, H.; Davis-Smyth, T.; Gerber, H.P.; Nguyen, T.N.; Peers, D.; Chisholm, V.; Hillan, K.J.; Schwall, R.H. Vascular endothelial growth factor is essential for corpus luteum angiogenesis. Nat. Med. 1998, 4, 336–340. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquière, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Cantelmo, A.R.; Conradi, L.-C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.-A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Sobierajska, K.; Ciszewski, W.M.; Sacewicz-Hofman, I.; Niewiarowska, J. Endothelial Cells in the Tumor Microenvironment. In Tumor Microenvironment: Non-Hematopoietic Cells; Birbrair, A., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 71–86. ISBN 978-3-030-37184-5. [Google Scholar]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Siemann, D.W. The Unique Characteristics of Tumor Vasculature and Preclinical Evidence for its Selective Disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev. 2011, 37, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Aird, W.C. Endothelial Cell Heterogeneity. Cold Spring Harb Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Frieboes, H.B.; McDougall, S.R.; Chaplain, M.A.J.; Cristini, V.; Lowengrub, J. The effect of interstitial pressure on tumor growth: Coupling with the blood and lymphatic vascular systems. J. Theor. Biol. 2013, 320, 131–151. [Google Scholar] [CrossRef] [Green Version]
- Baluk, P.; Hashizume, H.; McDonald, D.M. Cellular abnormalities of blood vessels as targets in cancer. Curr. Opin. Genet. Dev. 2005, 15, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Brown, L.F.; Senger, D.R.; Lanir, N.; Van de Water, L.; Dvorak, A.M.; Dvorak, H.F. Pathogenesis of tumor stroma generation: A critical role for leaky blood vessels and fibrin deposition. Biochim. Biophys. Acta 1989, 948, 305–326. [Google Scholar] [CrossRef]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.-C.; Totsuka, Y.; Klagsbrun, M.; et al. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Hida, Y.; Amin, D.N.; Flint, A.F.; Panigrahy, D.; Morton, C.C.; Klagsbrun, M. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004, 64, 8249–8255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, K.; Ohga, N.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Ishikawa, S.; Maishi, N.; Akino, T.; Kondoh, M.; Matsuda, A.; et al. Tumor endothelial cells acquire drug resistance by MDR1 up-regulation via VEGF signaling in tumor microenvironment. Am. J. Pathol. 2012, 180, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.-Q.; Sun, H.-C.; Zhang, W.; Zhu, X.-D.; Zhuang, P.-Y.; Zhang, J.-B.; Wang, L.; Wu, W.-Z.; Qin, L.-X.; Tang, Z.-Y. Human hepatocellular carcinoma tumor-derived endothelial cells manifest increased angiogenesis capability and drug resistance compared with normal endothelial cells. Clin. Cancer Res. 2009, 15, 4838–4846. [Google Scholar] [CrossRef] [Green Version]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef] [Green Version]
- Hamada, K.; Sasaki, T.; Koni, P.A.; Natsui, M.; Kishimoto, H.; Sasaki, J.; Yajima, N.; Horie, Y.; Hasegawa, G.; Naito, M.; et al. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 2005, 19, 2054–2065. [Google Scholar] [CrossRef] [Green Version]
- Goveia, J.; Rohlenova, K.; Taverna, F.; Treps, L.; Conradi, L.-C.; Pircher, A.; Geldhof, V.; de Rooij, L.P.M.H.; Kalucka, J.; Sokol, L.; et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell 2020, 37, 21–36.e13. [Google Scholar] [CrossRef] [PubMed]
- De Smet, F.; Segura, I.; De Bock, K.; Hohensinner, P.J.; Carmeliet, P. Mechanisms of vessel branching: Filopodia on endothelial tip cells lead the way. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalucka, J.; de Rooij, L.P.M.H.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.-A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e20. [Google Scholar] [CrossRef]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Brüning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 2010, 12, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Moya, I.M.; Umans, L.; Maas, E.; Pereira, P.N.G.; Beets, K.; Francis, A.; Sents, W.; Robertson, E.J.; Mummery, C.L.; Huylebroeck, D.; et al. Stalk Cell Phenotype Depends on Integration of Notch and Smad1/5 Signaling Cascades. Dev. Cell 2012, 22, 501–514. [Google Scholar] [CrossRef] [Green Version]
- Diebold, L.P.; Gil, H.J.; Gao, P.; Martinez, C.A.; Weinberg, S.E.; Chandel, N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 2019, 1, 158–171. [Google Scholar] [CrossRef]
- Eelen, G.; Dubois, C.; Cantelmo, A.R.; Goveia, J.; Brüning, U.; DeRan, M.; Jarugumilli, G.; van Rijssel, J.; Saladino, G.; Comitani, F.; et al. Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature 2018, 561, 63–69. [Google Scholar] [CrossRef]
- Krützfeldt, A.; Spahr, R.; Mertens, S.; Siegmund, B.; Piper, H.M. Metabolism of exogenous substrates by coronary endothelial cells in culture. J. Mol. Cell Cardiol. 1990, 22, 1393–1404. [Google Scholar] [CrossRef]
- Veys, K.; Fan, Z.; Ghobrial, M.; Bouché, A.; García-Caballero, M.; Vriens, K.; Conchinha, N.V.; Seuwen, A.; Schlegel, F.; Gorski, T.; et al. Role of the GLUT1 Glucose Transporter in Postnatal CNS Angiogenesis and Blood-Brain Barrier Integrity. Circ. Res. 2020, 127, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Goveia, J.; García-Caballero, M.; Subramanian, A.; Kalucka, J.; Treps, L.; Falkenberg, K.D.; de Rooij, L.P.M.H.; Zheng, Y.; Lin, L.; et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab. 2020, 31, 862–877.e14. [Google Scholar] [CrossRef] [PubMed]
- Conradi, L.-C.; Brajic, A.; Cantelmo, A.R.; Bouché, A.; Kalucka, J.; Pircher, A.; Brüning, U.; Teuwen, L.-A.; Vinckier, S.; Ghesquière, B.; et al. Tumor vessel disintegration by maximum tolerable PFKFB3 blockade. Angiogenesis 2017, 20, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Wilhelm, K.; Dubrac, A.; Tung, J.K.; Alves, T.C.; Fang, J.S.; Xie, Y.; Zhu, J.; Chen, Z.; De Smet, F.; et al. FGF-dependent metabolic control of vascular development. Nature 2017, 545, 224–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutelle, O.; Hornig-Do, H.-T.; Witt, A.; Andree, M.; Schiffmann, L.M.; Piekarek, M.; Brinkmann, K.; Seeger, J.M.; Liwschitz, M.; Miwa, S.; et al. Embelin inhibits endothelial mitochondrial respiration and impairs neoangiogenesis during tumor growth and wound healing. EMBO Mol. Med. 2014, 6, 624–639. [Google Scholar] [CrossRef]
- Facchinello, N.; Astone, M.; Audano, M.; Oberkersch, R.E.; Spizzotin, M.; Calura, E.; Marques, M.; Crisan, M.; Mitro, N.; Santoro, M.M. Oxidative pentose phosphate pathway controls vascular mural cell coverage by regulating extracellular matrix composition. Nat. Metab. 2022, 4, 123–140. [Google Scholar] [CrossRef]
- Moiz, B.; Garcia, J.; Basehore, S.; Sun, A.; Li, A.; Padmanabhan, S.; Albus, K.; Jang, C.; Sriram, G.; Clyne, A.M. 13C Metabolic Flux Analysis Indicates Endothelial Cells Attenuate Metabolic Perturbations by Modulating TCA Activity. Metabolites 2021, 11, 226. [Google Scholar] [CrossRef]
- Annan, D.A.; Maishi, N.; Soga, T.; Dawood, R.; Li, C.; Kikuchi, H.; Hojo, T.; Morimoto, M.; Kitamura, T.; Alam, M.T.; et al. Carbonic anhydrase 2 (CAII) supports tumor blood endothelial cell survival under lactic acidosis in the tumor microenvironment. Cell Commun. Signal. 2019, 17, 169. [Google Scholar] [CrossRef] [Green Version]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef]
- Bruning, U.; Morales-Rodriguez, F.; Kalucka, J.; Goveia, J.; Taverna, F.; Queiroz, K.C.S.; Dubois, C.; Cantelmo, A.R.; Chen, R.; Loroch, S.; et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 2018, 28, 866–880.e15. [Google Scholar] [CrossRef] [Green Version]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.S.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, N.-H.; Basu, D.; Samovski, D.; Pietka, T.A.; Peche, V.S.; Willecke, F.; Fang, X.; Yu, S.-Q.; Scerbo, D.; Chang, H.R.; et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Investig. 2018, 128, 4329–4342. [Google Scholar] [CrossRef] [Green Version]
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 2010, 464, 917–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cifarelli, V.; Appak-Baskoy, S.; Peche, V.S.; Kluzak, A.; Shew, T.; Narendran, R.; Pietka, K.M.; Cella, M.; Walls, C.W.; Czepielewski, R.; et al. Visceral obesity and insulin resistance associate with CD36 deletion in lymphatic endothelial cells. Nat. Commun. 2021, 12, 3350. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C—Dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smathers, R.L.; Petersen, D.R. The human fatty acid-binding protein family: Evolutionary divergences and functions. Hum. Genom. 2011, 5, 170. [Google Scholar] [CrossRef]
- Elmasri, H.; Karaaslan, C.; Teper, Y.; Ghelfi, E.; Weng, M.; Ince, T.A.; Kozakewich, H.; Bischoff, J.; Cataltepe, S. Fatty acid binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J. 2009, 23, 3865–3873. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Kawagishi, H.; Yan, Y.; Liu, J.; Wells, Q.S.; Edmunds, L.R.; Fergusson, M.M.; Yu, Z.-X.; Rovira, I.I.; Brittain, E.L.; et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol. Cell 2018, 69, 689–698.e7. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.J.; Nam, J.-K.; Kim, J.-H.; Choi, S.-H.; Lee, Y.-J. Endothelial-to-mesenchymal transition in anticancer therapy and normal tissue damage. Exp. Mol. Med. 2020, 52, 781–792. [Google Scholar] [CrossRef]
- Oberkersch, R.E.; Santoro, M.M. Role of amino acid metabolism in angiogenesis. Vasc. Pharmacol. 2019, 112, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Leighton, B.; Curi, R.; Hussein, A.; Newsholme, E.A. Maximum activities of some key enzymes of glycolysis, glutaminolysis, Krebs cycle and fatty acid utilization in bovine pulmonary endothelial cells. FEBS Lett. 1987, 225, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Polet, F.; Feron, O. Endothelial cell metabolism and tumour angiogenesis: Glucose and glutamine as essential fuels and lactate as the driving force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Brüning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef]
- Sanchez, E.L.; Carroll, P.A.; Thalhofer, A.B.; Lagunoff, M. Latent KSHV Infected Endothelial Cells Are Glutamine Addicted and Require Glutaminolysis for Survival. PLoS Pathog. 2015, 11, e1005052. [Google Scholar] [CrossRef] [Green Version]
- Hargadon, K.M. Dysregulation of TGFβ1 Activity in Cancer and Its Influence on the Quality of Anti-Tumor Immunity. J. Clin. Med. 2016, 5, 76. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Deng, Y.; Li, X.; Ning, Y.; Lin, X.; Guo, S.; Chen, M.; Han, M. Glutaminolysis Was Induced by TGF-β1 through PP2Ac Regulated Raf-MEK-ERK Signaling in Endothelial Cells. PLoS ONE 2016, 11, e0162658. [Google Scholar] [CrossRef]
- Oberkersch, R.E.; Pontarin, G.; Astone, M.; Spizzotin, M.; Arslanbaeva, L.; Tosi, G.; Panieri, E.; Ricciardi, S.; Allega, M.F.; Brossa, A.; et al. Aspartate metabolism in endothelial cells activates the mTORC1 pathway to initiate translation during angiogenesis. Dev. Cell 2022, 57, 1241–1256.e8. [Google Scholar] [CrossRef]
- Camp, C.R.; Yuan, H. GRIN2D/GluN2D NMDA Receptor: Unique Features and Its Contribution to Pediatric Developmental and Epileptic Encephalopathy. Eur. Paediatr. Neurol. 2020, 24, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, H.J.M.; Wragg, J.W.; Ward, S.; Heath, V.L.; Ismail, T.; Bicknell, R. Glutamate dependent NMDA receptor 2D is a novel angiogenic tumour endothelial marker in colorectal cancer. Oncotarget 2016, 7, 20440–20454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Ndrepepa, G. Aspartate aminotransferase and cardiovascular disease—A narrative review. JLPM 2020, 6, 1–17. [Google Scholar] [CrossRef]
- Exertier, P.; Javerzat, S.; Wang, B.; Franco, M.; Herbert, J.; Platonova, N.; Winandy, M.; Pujol, N.; Nivelles, O.; Ormenese, S.; et al. Impaired angiogenesis and tumor development by inhibition of the mitotic kinesin Eg5. Oncotarget 2013, 4, 2302–2316. [Google Scholar] [CrossRef]
- Rajcevic, U.; Petersen, K.; Knol, J.C.; Loos, M.; Bougnaud, S.; Klychnikov, O.; Li, K.W.; Pham, T.V.; Wang, J.; Miletic, H.; et al. iTRAQ-based proteomics profiling reveals increased metabolic activity and cellular cross-talk in angiogenic compared with invasive glioblastoma phenotype. Mol. Cell. Proteom. 2009, 8, 2595–2612. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.B.; Geck, R.C.; Toker, A. Metabolic pathway alterations in microvascular endothelial cells in response to hypoxia. PLoS ONE 2020, 15, e0232072. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 2018, 27, 428–438.e5. [Google Scholar] [CrossRef] [Green Version]
- Lomelino, C.L.; Andring, J.T.; McKenna, R.; Kilberg, M.S. Asparagine synthetase: Function, structure, and role in disease. J. Biol. Chem. 2017, 292, 19952–19958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, M.N.; Butterworth, E.A.; Kilberg, M.S. Asparagine synthetase: Regulation by cell stress and involvement in tumor biology. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E789–E799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egler, R.A.; Ahuja, S.P.; Matloub, Y. L-asparaginase in the treatment of patients with acute lymphoblastic leukemia. J. Pharmacol. Pharmacother. 2016, 7, 62–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.R.; Mattaini, K.R.; Dennstedt, E.A.; Nguyen, A.A.; Sivanand, S.; Reilly, M.F.; Meeth, K.; Muir, A.; Darnell, A.M.; Bosenberg, M.W.; et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 2019, 29, 1410–1421.e4. [Google Scholar] [CrossRef]
- Maralani, M.N.; Movahedian, A.; Javanmard, S.H. Antioxidant and cytoprotective effects of L-Serine on human endothelial cells. Res. Pharm. Sci. 2012, 7, 209–215. [Google Scholar]
- Vandekeere, S.; Dubois, C.; Kalucka, J.; Sullivan, M.R.; García-Caballero, M.; Goveia, J.; Chen, R.; Diehl, F.F.; Bar-Lev, L.; Souffreau, J.; et al. Serine Synthesis via PHGDH Is Essential for Heme Production in Endothelial Cells. Cell Metab. 2018, 28, 573–587.e13. [Google Scholar] [CrossRef] [Green Version]
- Ocaña, M.C.; Yang, C.; Bernal, M.; Martínez-Poveda, B.; Vu, H.S.; Cárdenas, C.; DeBerardinis, R.J.; Quesada, A.R.; Medina, M.Á. The anti-angiogenic compound dimethyl fumarate inhibits the serine synthesis pathway and increases glycolysis in endothelial cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tsuji-Tamura, K.; Sato, M.; Fujita, M.; Tamura, M. Glycine exerts dose-dependent biphasic effects on vascular development of zebrafish embryos. Biochem. Biophys. Res. Commun. 2020, 527, 539–544. [Google Scholar] [CrossRef]
- Amin, K.; Li, J.; Chao, W.R.; Dewhirst, M.W.; Haroon, Z.A. Dietary glycine inhibits angiogenesis during wound healing and tumor growth. Cancer Biol. Ther. 2003, 2, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Bruns, H.; Kazanavicius, D.; Schultze, D.; Saeedi, M.A.; Yamanaka, K.; Strupas, K.; Schemmer, P. Glycine inhibits angiogenesis in colorectal cancer: Role of endothelial cells. Amino Acids 2016, 48, 2549–2558. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Murdoch, C.E.; Xu, H.; Shi, H.; Duan, D.D.; Ahmed, A.; Gu, Y. Vascular endothelial growth factor signaling requires glycine to promote angiogenesis. Sci. Rep. 2017, 7, 14749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ikejima, K.; Honda, H.; Kitamura, T.; Takei, Y.; Sato, N. Glycine prevents apoptosis of rat sinusoidal endothelial cells caused by deprivation of vascular endothelial growth factor. Hepatology 2000, 32, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Ren, W.; Yang, G.; Duan, J.; Huang, X.; Fang, R.; Li, C.; Li, T.; Yin, Y.; Hou, Y.; et al. L-Cysteine metabolism and its nutritional implications. Mol. Nutr. Food Res. 2016, 60, 134–146. [Google Scholar] [CrossRef]
- Giuffrè, A.; Vicente, J.B. Hydrogen Sulfide Biochemistry and Interplay with Other Gaseous Mediators in Mammalian Physiology. Oxid. Med. Cell. Longev. 2018, 2018, 6290931. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Londono, M.; Lee, J.-I.; Hu, M.; Yu, A.F. Enzymes and metabolites of cysteine metabolism in nonhepatic tissues of rats show little response to changes in dietary protein or sulfur amino acid levels. J. Nutr. 2002, 132, 3369–3378. [Google Scholar] [CrossRef] [Green Version]
- Okumura, N.; Inoue, R.; Kakutani, K.; Nakahara, M.; Kinoshita, S.; Hamuro, J.; Koizumi, N. Corneal Endothelial Cells Have an Absolute Requirement for Cysteine for Survival. Cornea 2017, 36, 988–994. [Google Scholar] [CrossRef]
- Miura, K.; Ishii, T.; Sugita, Y.; Bannai, S. Cystine uptake and glutathione level in endothelial cells exposed to oxidative stress. Am. J. Physiol. 1992, 262, C50–C58. [Google Scholar] [CrossRef]
- Lopes-Coelho, F.; Martins, F.; Hipólito, A.; Mendes, C.; Sequeira, C.O.; Pires, R.F.; Almeida, A.M.; Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J. The Activation of Endothelial Cells Relies on a Ferroptosis-Like Mechanism: Novel Perspectives in Management of Angiogenesis and Cancer Therapy. Front. Oncol. 2021, 11, 656229. [Google Scholar] [CrossRef]
- Puris, E.; Gynther, M.; Auriola, S.; Huttunen, K.M. L-Type amino acid transporter 1 as a target for drug delivery. Pharm. Res. 2020, 37, 88. [Google Scholar] [CrossRef]
- Quan, L.; Ohgaki, R.; Hara, S.; Okuda, S.; Wei, L.; Okanishi, H.; Nagamori, S.; Endou, H.; Kanai, Y. Amino acid transporter LAT1 in tumor-associated vascular endothelium promotes angiogenesis by regulating cell proliferation and VEGF-A-dependent mTORC1 activation. J. Exp. Clin. Cancer Res. 2020, 39, 266. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yeung, S.-C.J.; Liu, S.; Qdaisat, A.; Jiang, D.; Liu, W.; Cheng, Z.; Liu, W.; Wang, H.; Li, L.; et al. Cyst(e)ine in nutrition formulation promotes colon cancer growth and chemoresistance by activating mTORC1 and scavenging ROS. Signal Transduct. Target. Ther. 2021, 6, 188. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; White, A.C.; Fanburg, B.L. Modulation of transforming growth factor-beta 1 antiproliferative effects on endothelial cells by cysteine, cystine, and N-acetylcysteine. J. Clin. Investig. 1992, 90, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Azabdaftari, A.; van der Giet, M.; Schuchardt, M.; Hennermann, J.B.; Plöckinger, U.; Querfeld, U. The cardiovascular phenotype of adult patients with phenylketonuria. Orphanet J. Rare Dis. 2019, 14, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermida-Ameijeiras, A.; Crujeiras, V.; Roca, I.; Calvo, C.; Leis, R.; Couce, M.-L. Arterial stiffness assessment in patients with phenylketonuria. Medicine 2017, 96, e9322. [Google Scholar] [CrossRef]
- Sugiyama, T.; Levy, B.D.; Michel, T. Tetrahydrobiopterin recycling, a key determinant of endothelial nitric-oxide synthase-dependent signaling pathways in cultured vascular endothelial cells. J. Biol. Chem. 2009, 284, 12691–12700. [Google Scholar] [CrossRef] [Green Version]
- Heikal, L.; Starr, A.; Hussein, D.; Prieto-Lloret, J.; Aaronson, P.; Dailey, L.A.; Nandi, M. l-Phenylalanine Restores Vascular Function in Spontaneously Hypertensive Rats Through Activation of the GCH1-GFRP Complex. JACC Basic Transl. Sci. 2018, 3, 366–377. [Google Scholar] [CrossRef]
- Lau, A.N.; Heiden, M.G.V. Metabolism in the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2020, 4, 26. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.J.; Amendola, C.R.; Hollinshead, K.E.R.; Yu, Q.; Yamamoto, K.; Encarnación-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.W.; Biancur, D.E.; et al. Selective Alanine Transporter Utilization Creates a Targetable Metabolic Niche in Pancreatic Cancer. Cancer Discov. 2020, 10, 1018–1037. [Google Scholar] [CrossRef]
- Vecchio, E.; Caiazza, C.; Mimmi, S.; Avagliano, A.; Iaccino, E.; Brusco, T.; Nisticò, N.; Maisano, D.; Aloisio, A.; Quinto, I.; et al. Metabolites Profiling of Melanoma Interstitial Fluids Reveals Uridine Diphosphate as Potent Immune Modulator Capable of Limiting Tumor Growth. Front. Cell Dev. Biol. 2021, 9, 730726. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.-L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acevedo-Acevedo, S.; Millar, D.C.; Simmons, A.D.; Favreau, P.; Cobra, P.F.; Skala, M.; Palecek, S.P. Metabolomics revealed the influence of breast cancer on lymphatic endothelial cell metabolism, metabolic crosstalk, and lymphangiogenic signaling in co-culture. Sci. Rep. 2020, 10, 21244. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; De Giorgio, F.; Kopecka, J.; Genova, T.; Fiorito, V.; Allocco, A.L.; Bertino, F.; Chiabrando, D.; Mussano, F.; Altruda, F.; et al. Endothelial Heme Dynamics Drive Cancer Cell Metabolism by Shaping the Tumor Microenvironment. Biomedicines 2021, 9, 1557. [Google Scholar] [CrossRef] [PubMed]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [Green Version]
- Szabo, C. Gasotransmitters in cancer: From pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 2016, 15, 185–203. [Google Scholar] [CrossRef] [Green Version]
- Kroll, J.; Waltenberger, J. VEGF-A Induces Expression of eNOS and iNOS in Endothelial Cells via VEGF Receptor-2 (KDR). Biochem. Biophys. Res. Commun. 1998, 252, 743–746. [Google Scholar] [CrossRef]
- Ha, J.M.; Jin, S.Y.; Lee, H.S.; Shin, H.K.; Lee, D.H.; Song, S.H.; Kim, C.D.; Bae, S.S. Regulation of retinal angiogenesis by endothelial nitric oxide synthase signaling pathway. Korean J. Physiol. Pharmacol. 2016, 20, 533–538. [Google Scholar] [CrossRef] [Green Version]
- Namba, T.; Koike, H.; Murakami, K.; Aoki, M.; Makino, H.; Hashiya, N.; Ogihara, T.; Kaneda, Y.; Kohno, M.; Morishita, R. Angiogenesis Induced by Endothelial Nitric Oxide Synthase Gene Through Vascular Endothelial Growth Factor Expression in a Rat Hindlimb Ischemia Model. Circulation 2003, 108, 2250–2257. [Google Scholar] [CrossRef] [Green Version]
- Arnold, W.P.; Mittal, C.K.; Katsuki, S.; Murad, F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. USA 1977, 74, 3203–3207. [Google Scholar] [CrossRef] [Green Version]
- Thibeault, S.; Rautureau, Y.; Oubaha, M.; Faubert, D.; Wilkes, B.C.; Delisle, C.; Gratton, J.-P. S-Nitrosylation of β-Catenin by eNOS-Derived NO Promotes VEGF-Induced Endothelial Cell Permeability. Mol. Cell 2010, 39, 468–476. [Google Scholar] [CrossRef]
- Paik, J.-Y.; Lee, K.-H.; Ko, B.-H.; Choe, Y.S.; Choi, Y.; Kim, B.-T. Nitric oxide stimulates 18F-FDG uptake in human endothelial cells through increased hexokinase activity and GLUT1 expression. J. Nucl. Med. 2005, 46, 365–370. [Google Scholar] [PubMed]
- Siragusa, M.; Thöle, J.; Bibli, S.; Luck, B.; Loot, A.E.; de Silva, K.; Wittig, I.; Heidler, J.; Stingl, H.; Randriamboavonjy, V.; et al. Nitric oxide maintains endothelial redox homeostasis through PKM2 inhibition. EMBO J. 2019, 38, e100938. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Mikami, Y.; Kimura, Y.; Nagahara, N.; Kimura, H. Vascular Endothelium Expresses 3-Mercaptopyruvate Sulfurtransferase and Produces Hydrogen Sulfide. J. Biochem. 2009, 146, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Erdélyi, K.; Ditrói, T.; Johansson, H.J.; Czikora, Á.; Balog, N.; Silwal-Pandit, L.; Ida, T.; Olasz, J.; Hajdú, D.; Mátrai, Z.; et al. Reprogrammed transsulfuration promotes basal-like breast tumor progression via realigning cellular cysteine persulfidation. Proc. Natl. Acad. Sci. USA 2021, 118, e2100050118. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Huang, J.-T.; Chen, W.-L.; Wang, R.-H.; Kao, M.-C.; Pan, Y.-R.; Chan, S.-H.; Tsai, K.-W.; Kung, H.-J.; Lin, K.-T.; et al. Dysregulation of cystathionine γ-lyase promotes prostate cancer progression and metastasis. EMBO Rep. 2019, 20, e45986. [Google Scholar] [CrossRef]
- Szabo, C.; Coletta, C.; Chao, C.; Módis, K.; Szczesny, B.; Papapetropoulos, A.; Hellmich, M.R. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12474–12479. [Google Scholar] [CrossRef] [Green Version]
- Longchamp, A.; Mirabella, T.; Arduini, A.; MacArthur, M.R.; Das, A.; Treviño-Villarreal, J.H.; Hine, C.; Ben-Sahra, I.; Knudsen, N.H.; Brace, L.E.; et al. Amino Acid Restriction Triggers Angiogenesis via GCN2/ATF4 Regulation of VEGF and H2S Production. Cell 2018, 173, 117–129.e14. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.-P.; Ferrara, N.; Johnson, R.S. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 2004, 6, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, M.; Dettori, D.; Leite de Oliveira, R.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.-M.; Lanahan, A.A.; Pollard, P.; Ruiz de Almodovar, C.; et al. Heterozygous Deficiency of PHD2 Restores Tumor Oxygenation and Inhibits Metastasis via Endothelial Normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Klomp, J.; Hyun, J.; Klomp, J.E.; Pajcini, K.; Rehman, J.; Malik, A.B. Comprehensive transcriptomic profiling reveals SOX7 as an early regulator of angiogenesis in hypoxic human endothelial cells. J. Biol. Chem. 2020, 295, 4796–4808. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordeleau, F.; Mason, B.N.; Lollis, E.M.; Mazzola, M.; Zanotelli, M.R.; Somasegar, S.; Califano, J.P.; Montague, C.; LaValley, D.J.; Huynh, J.; et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, 492–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Agarwal, S. Mechanical Signals Activate Vascular Endothelial Growth Factor Receptor-2 To Upregulate Endothelial Cell Proliferation during Inflammation. J. Immunol. 2010, 185, 1215–1221. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, X.; Liu, Y.; Singhal, M.; Gürkaşlar, C.; Valls, A.F.; Lei, Y.; Hu, W.; Schermann, G.; Adler, H.; et al. STAT3-YAP/TAZ signaling in endothelial cells promotes tumor angiogenesis. Sci. Signal. 2021, 14, eabj8393. [Google Scholar] [CrossRef]
- Wang, X.; Freire Valls, A.; Schermann, G.; Shen, Y.; Moya, I.M.; Castro, L.; Urban, S.; Solecki, G.M.; Winkler, F.; Riedemann, L.; et al. YAP/TAZ Orchestrate VEGF Signaling during Developmental Angiogenesis. Dev. Cell 2017, 42, 462–478.e7. [Google Scholar] [CrossRef] [Green Version]
- Sakabe, M.; Fan, J.; Odaka, Y.; Liu, N.; Hassan, A.; Duan, X.; Stump, P.; Byerly, L.; Donaldson, M.; Hao, J.; et al. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc. Natl. Acad. Sci. USA 2017, 114, 10918–10923. [Google Scholar] [CrossRef] [Green Version]
- Reid, S.E.; Kay, E.J.; Neilson, L.J.; Henze, A.-T.; Serneels, J.; McGhee, E.J.; Dhayade, S.; Nixon, C.; Mackey, J.B.G.; Santi, A.; et al. Tumor matrix stiffness promotes metastatic cancer cell interaction with the endothelium. EMBO J. 2017, 36, 2373–2389. [Google Scholar] [CrossRef]
- Cox, A.G.; Tsomides, A.; Yimlamai, D.; Hwang, K.L.; Miesfeld, J.; Galli, G.G.; Fowl, B.H.; Fort, M.; Ma, K.Y.; Sullivan, M.R.; et al. Yap regulates glucose utilization and sustains nucleotide synthesis to enable organ growth. EMBO J. 2018, 37, e100294. [Google Scholar] [CrossRef]
- Liu, C.; Li, M.; Dong, Z.-X.; Jiang, D.; Li, X.; Lin, S.; Chen, D.; Zou, X.; Zhang, X.-D.; Luker, G.D. Heterogeneous microenvironmental stiffness regulates pro-metastatic functions of breast cancer cells. Acta Biomater. 2021, 131, 326–340. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.-Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.; Hsu, P.-P.; Chen, B.P.; Yuan, S.; Usami, S.; Shyy, J.Y.-J.; Li, Y.-S.; Chien, S. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc. Natl. Acad. Sci. USA 2000, 97, 9385–9389. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.-C.; Yeh, Y.-T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.-L.; Li, Y.-S.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Hu, X.; He, W.; Zhao, Y.; Hao, S.; Wu, Q.; Li, S.; Zhang, S.; Shi, M. Fluid shear stress and tumor metastasis. Am. J. Cancer Res. 2018, 8, 763–777. [Google Scholar]
- Huang, Q.; Li, S.; Hu, X.; Sun, M.; Wu, Q.; Dai, H.; Tan, Y.; Sun, F.; Wang, C.; Rong, X.; et al. Shear stress activates ATOH8 via autocrine VEGF promoting glycolysis dependent-survival of colorectal cancer cells in the circulation. J. Exp. Clin. Cancer Res. 2020, 39, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lidonnici, J.; Santoro, M.M.; Oberkersch, R.E. Cancer-Induced Metabolic Rewiring of Tumor Endothelial Cells. Cancers 2022, 14, 2735. https://doi.org/10.3390/cancers14112735
Lidonnici J, Santoro MM, Oberkersch RE. Cancer-Induced Metabolic Rewiring of Tumor Endothelial Cells. Cancers. 2022; 14(11):2735. https://doi.org/10.3390/cancers14112735
Chicago/Turabian StyleLidonnici, Jacopo, Massimo M. Santoro, and Roxana E. Oberkersch. 2022. "Cancer-Induced Metabolic Rewiring of Tumor Endothelial Cells" Cancers 14, no. 11: 2735. https://doi.org/10.3390/cancers14112735